

Understanding the Exchange Interaction between Paramagnetic Metal Ions and Radical Ligands: DFT and Ab Initio Study on Semiquinonato Cu(II) Complexes


Abstract
:1. Introduction
2. Results and Discussion
2.1. General Considerations
2.2. Performance of Computational Methods
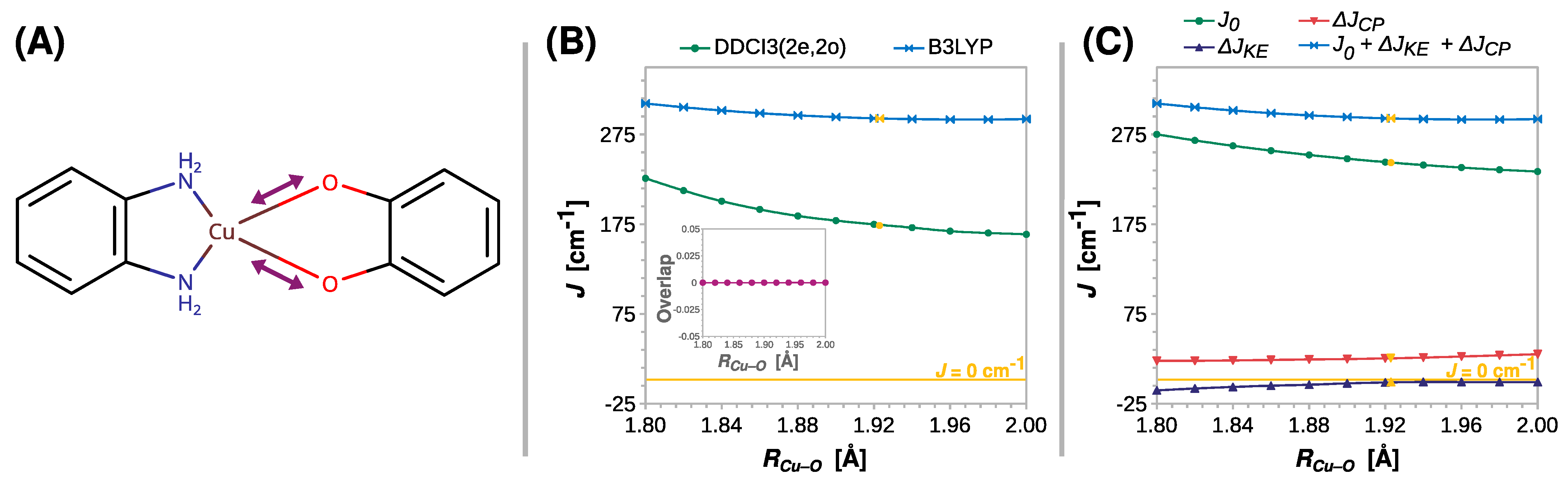
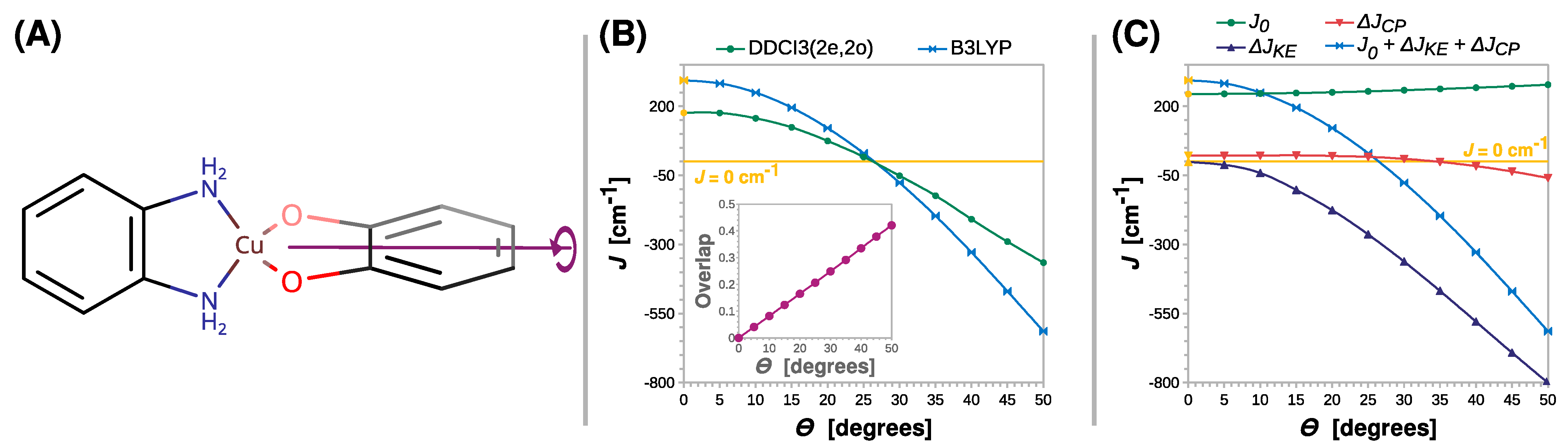
2.3. Magnetostructural Correlations
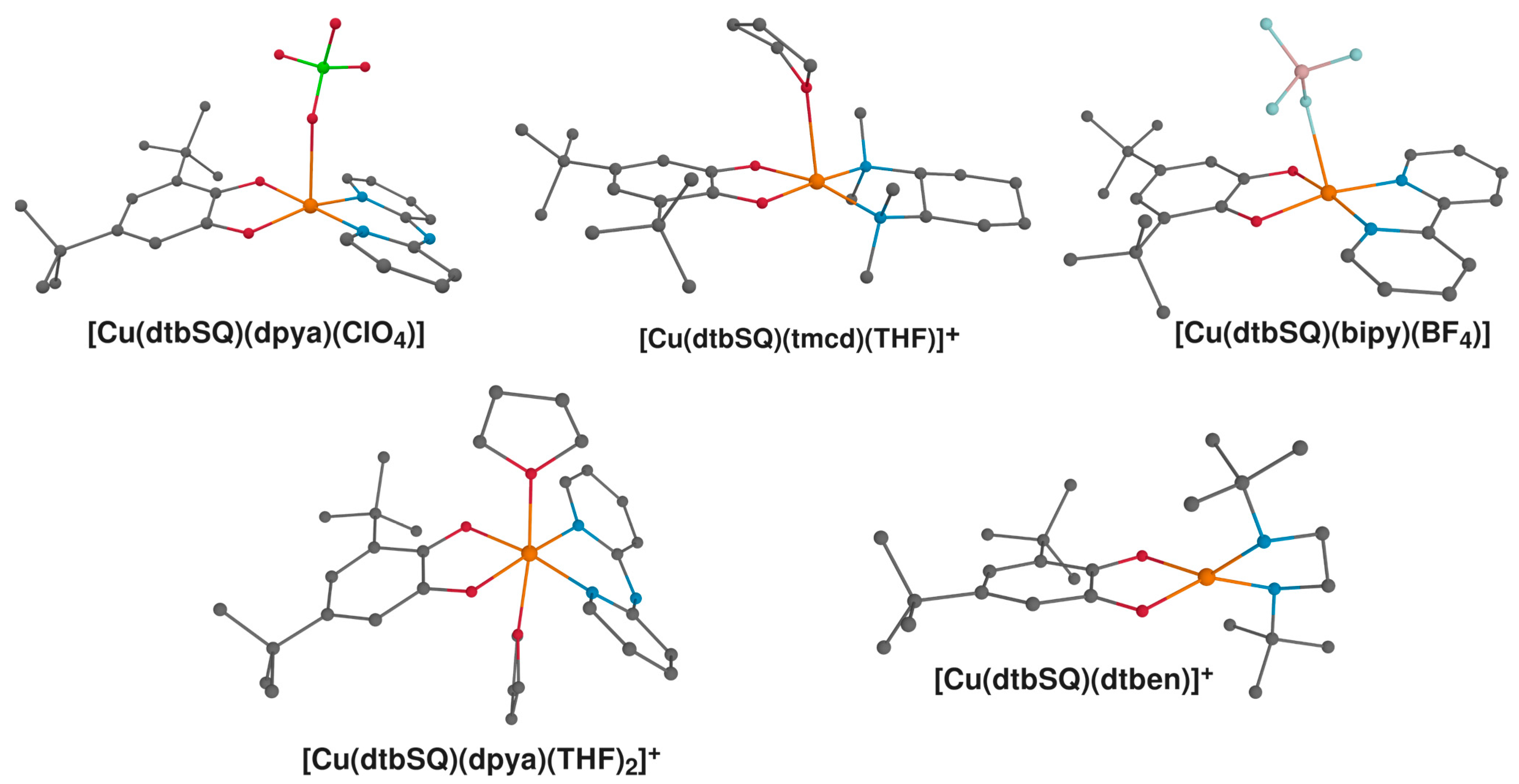
2.4. Comparison with Experiment
3. Methods and Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pierpont, C.G.; Buchanan, R.M. Transition Metal Complexes of O-Benzoquinone, o-Semiquinone, and Catecholate Ligands. Coord. Chem. Rev. 1981, 38, 45–87. [Google Scholar] [CrossRef]
- Pierpont, C.G.; Lange, C.W. The Chemistry of Transition Metal Complexes Containing Catechol and Semiquinone Ligands. Prog. Inorg. Chem. 2007, 41, 331–442. [Google Scholar]
- Mederos, A.; Domínguez, S.; Hernández-Molina, R.; Sanchiz, J.; Brito, F. Coordinating Ability of Phenylenediamines. Coord. Chem. Rev. 1999, 193–195, 913–939. [Google Scholar] [CrossRef]
- Pierpont, C. Unique Properties of Transition Metal Quinone Complexes of the MQ3 Series. Coord. Chem. Rev. 2001, 219–221, 415–433. [Google Scholar] [CrossRef]
- Poddel’sky, A.I.; Cherkasov, V.K.; Abakumov, G.A. Transition Metal Complexes with Bulky 4,6-Di-Tert-Butyl-N-Aryl(Alkyl)-o-Iminobenzoquinonato Ligands: Structure, EPR and Magnetism. Coord. Chem. Rev. 2009, 253, 291–324. [Google Scholar] [CrossRef]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Beyer, K.; Filippou, V.; Záliš, S. Charge and Spin Coupling in Copper Compounds with Hemilabile Noninnocent Ligands—Ambivalence in Three Dimensions. Coord. Chem. Rev. 2018, 355, 173–179. [Google Scholar] [CrossRef]
- Rajput, A.; Sharma, A.K.; Barman, S.K.; Saha, A.; Mukherjee, R. Valence Tautomerism and Delocalization in Transition Metal Complexes of o-Amidophenolates and Other Redox-Active Ligands. Some Recent Results. Coord. Chem. Rev. 2020, 414, 213240. [Google Scholar] [CrossRef]
- Ershova, I.V.; Piskunov, A.V.; Cherkasov, V.K. Complexes of Diamagnetic Cations with Radical Anion Ligands. Russ. Chem. Rev. 2020, 89, 1157–1183. [Google Scholar] [CrossRef]
- Pashanova, K.I.; Poddel’sky, A.I.; Piskunov, A.v. Complexes of “Late” Transition Metals of the 3d Row Based on Functionalized o-Iminobenzoquinone Type Ligands: Interrelation of Molecular and Electronic Structure, Magnetic Behaviour. Coord. Chem. Rev. 2022, 459, 214399. [Google Scholar] [CrossRef]
- Kaim, W.; Schwederski, B. Non-Innocent Ligands in Bioinorganic Chemistry—An Overview. Coord. Chem. Rev. 2010, 254, 1580–1588. [Google Scholar] [CrossRef]
- Heims, F.; Ray, K. Multiple Spin Scenarios in Transition-Metal Complexes Involving Redox Non-Innocent Ligands. In Spin States in Biochemistry and Inorganic Chemistry; John Wiley & Sons, Ltd.: Oxford, UK, 2015; pp. 229–262. [Google Scholar]
- Stubbe, J.; van der Donk, W.A. Protein Radicals in Enzyme Catalysis. Chem. Rev. 1998, 98, 705–762. [Google Scholar] [CrossRef] [PubMed]
- Storr, T.; Verma, P.; Pratt, R.C.; Wasinger, E.C.; Shimazaki, Y.; Stack, T.D.P. Defining the Electronic and Geometric Structure of One-Electron Oxidized Copper−Bis-Phenoxide Complexes. J. Am. Chem. Soc. 2008, 130, 15448–15459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, J.W. Free Radical Catalysis by Galactose Oxidase. Chem. Rev. 2003, 103, 2347–2364. [Google Scholar] [CrossRef]
- Kaim, W. The Chemistry and Biochemistry of the Copper–Radical Interaction. Dalton Trans. 2003, 761–768. [Google Scholar] [CrossRef]
- Kodera, M.; Kawata, T.; Kano, K.; Tachi, Y.; Itoh, S.; Kojo, S. Mechanism for Aerobic Oxidation of 3,5-Di-Tert-Butylcatechol to 3,5-Di-Tert-Butyl-o-Benzoquinone Catalyzed by Di-μ-Hydroxo-Dicopper(II) Complexes of Peralkylated Ethylelnediamine Ligands. Bull. Chem. Soc. Jpn 2003, 76, 1957–1964. [Google Scholar] [CrossRef]
- Verma, P.; Weir, J.; Mirica, L.; Stack, T.D.P. Tale of a Twist: Magnetic and Optical Switching in Copper(II) Semiquinone Complexes. Inorg. Chem. 2011, 50, 9816–9825. [Google Scholar] [CrossRef]
- Liu, J.; Lorraine, S.C.; Dolinar, B.S.; Hoover, J.M. Aerobic Oxidation Reactivity of Well-Defined Cobalt(II) and Cobalt(III) Aminophenol Complexes. Inorg. Chem. 2022, 61, 6008–6016. [Google Scholar] [CrossRef]
- Lecarme, L.; Kochem, A.; Chiang, L.; Moutet, J.; Berthiol, F.; Philouze, C.; Leconte, N.; Storr, T.; Thomas, F. Electronic Structure and Reactivity of One-Electron-Oxidized Copper(II) Bis(Phenolate)–Dipyrrin Complexes. Inorg. Chem. 2018, 57, 9708–9719. [Google Scholar] [CrossRef]
- Lyaskovskyy, V.; de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Sutradhar, M.; Pombeiro, A.J.L.; da Silva, J.A.L. Water Oxidation with Transition Metal Catalysts with Non-Innocent Ligands and Its Mechanisms. Coord. Chem. Rev. 2021, 439, 213911. [Google Scholar] [CrossRef]
- Vostrikova, K.E. High-Spin Molecules Based on Metal Complexes of Organic Free Radicals. Coord. Chem. Rev. 2008, 252, 1409–1419. [Google Scholar] [CrossRef]
- Preuss, K.E. Metal-Radical Coordination Complexes of Thiazyl and Selenazyl Ligands. Coord. Chem. Rev. 2015, 289–290, 49–61. [Google Scholar] [CrossRef]
- Matsuoka, R.; Yoshimoto, T.; Kitagawa, Y.; Kusamoto, T. Structural and Magnetic Studies on Nickel(II) and Cobalt(II) Complexes with Polychlorinated Diphenyl(4-Pyridyl)Methyl Radical Ligands. Molecules 2021, 26, 5596. [Google Scholar] [CrossRef]
- Ito, S.; Yoshitake, T.; Ishida, T. Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions. Molecules 2022, 27, 4930. [Google Scholar] [CrossRef]
- Demir, S.; Jeon, I.R.; Long, J.R.; Harris, T.D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [Google Scholar] [CrossRef] [Green Version]
- Chakarawet, K.; Harris, T.D.; Long, J.R. Semiquinone Radical-Bridged M2 (M = Fe, Co, Ni) Complexes with Strong Magnetic Exchange Giving Rise to Slow Magnetic Relaxation. Chem. Sci. 2020, 11, 8196–8203. [Google Scholar] [CrossRef]
- Himmel, H.J. Valence Tautomerism in Copper Coordination Chemistry. Inorg. Chim. Acta 2018, 481, 56–68. [Google Scholar] [CrossRef]
- Sundaresan, S.; Diehl, M.; Carrella, L.M.; Rentschler, E. Triggering of Valence Tautomeric Transitions in Dioxolene-Based Cobalt Complexes Influenced by Ligand Substituents, Co-Ligands, and Anions. Magnetochemistry 2022, 8, 109. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Kondo, M.; Fujita, T.; Namiki, K.; Murata, M.; Nishihara, H. Thioacetyl-Terminated Ferrocene-Anthraquinone Conjugates: Synthesis, Photo- and Electrochemical Properties Triggered by Protonation-Induced Intramolecular Electron Transfer. Molecules 2010, 15, 150–163. [Google Scholar] [CrossRef]
- Fleming, C.; Chung, D.; Ponce, S.; Brook, D.J.R.; DaRos, J.; Das, R.; Ozarowski, A.; Stoian, S.A. Valence Tautomerism in a Cobalt-Verdazyl Coordination Compound. Chem. Commun. 2020, 56, 4400–4403. [Google Scholar] [CrossRef] [PubMed]
- Ferrando-Soria, J.; Vallejo, J.; Castellano, M.; Martínez-Lillo, J.; Pardo, E.; Cano, J.; Castro, I.; Lloret, F.; Ruiz-García, R.; Julve, M. Molecular Magnetism, Quo Vadis? A Historical Perspective from a Coordination Chemist Viewpoint☆. Coord. Chem. Rev. 2017, 339, 17–103. [Google Scholar] [CrossRef]
- Kishishita, S.; Okajima, T.; Kim, M.; Yamaguchi, H.; Hirota, S.; Suzuki, S.; Kuroda, S.; Tanizawa, K.; Mure, M. Role of Copper Ion in Bacterial Copper Amine Oxidase: Spectroscopic and Crystallographic Studies of Metal-Substituted Enzymes. J. Am. Chem. Soc. 2003, 125, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guo, H.; Zhou, J.; Wang, Y.; Yan, H.; Jin, R.; Tang, Y. Evodiamine and Rutaecarpine as Potential Anticancer Compounds: A Combined Computational Study. Int. J. Mol. Sci. 2022, 23, 11513. [Google Scholar] [CrossRef]
- Muzalevskiy, V.M.; Sizova, Z.A.; Nechaev, M.S.; Nenajdenko, V.G. Acid-Switchable Synthesis of Trifluoromethylated Triazoles and Isoxazoles via Reaction of CF3-Ynones with NaN3: DFT Study of the Reaction Mechanism. Int. J. Mol. Sci. 2022, 23, 14522. [Google Scholar] [CrossRef]
- Huang, H.; Hu, G.; Hu, C.; Fan, X. Enhanced Hydrogen Evolution Reactivity of T’-Phase Tungsten Dichalcogenides (WS2, WSe2, and WTe2) Materials: A DFT Study. Int. J. Mol. Sci. 2022, 23, 11727. [Google Scholar] [CrossRef]
- Krupka, K.M.; Pocheć, M.; Panek, J.J.; Jezierska, A. Comprehensive Empirical Model of Substitution—Influence on Hydrogen Bonding in Aromatic Schiff Bases. Int. J. Mol. Sci. 2022, 23, 12439. [Google Scholar] [CrossRef]
- Domagała, M.; Jabłoński, M.; Dubis, A.T.; Zabel, M.; Pfitzner, A.; Palusiak, M. Testing of Exchange-Correlation Functionals of DFT for a Reliable Description of the Electron Density Distribution in Organic Molecules. Int. J. Mol. Sci. 2022, 23, 14719. [Google Scholar] [CrossRef]
- Witwicki, M.; Walencik, P.K.; Jezierska, J. How Accurate Is Density Functional Theory in Predicting Spin Density? An Insight from the Prediction of Hyperfine Coupling Constants. J. Mol. Model. 2020, 26, 10. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density Functional Theory Is Straying from the Path toward the Exact Functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Wojtkowiak, K.; Michalczyk, M.; Zierkiewicz, W.; Jezierska, A.; Panek, J.J. Chalcogen Bond as a Factor Stabilizing Ligand Conformation in the Binding Pocket of Carbonic Anhydrase IX Receptor Mimic. Int. J. Mol. Sci. 2022, 23, 13701. [Google Scholar] [CrossRef] [PubMed]
- Witwicki, M.; Jezierska, J. Protonated O-Semiquinone Radical as a Mimetic of the Humic Acids Native Radicals: A DFT Approach to the Molecular Structure and EPR Properties. Geochim. Cosmochim. Acta 2012, 86, 384–391. [Google Scholar] [CrossRef]
- Witwicki, M.; Lewińska, A.; Ozarowski, A. O-Semiquinone Radical Anion Isolated as an Amorphous Porous Solid. Phys. Chem. Chem. Phys. 2021, 23, 17408–17419. [Google Scholar] [CrossRef] [PubMed]
- Ritacco, I.; Russo, N.; Sicilia, E. DFT Investigation of the Mechanism of Action of Organoiridium(III) Complexes As Anticancer Agents. Inorg. Chem. 2015, 54, 10801–10810. [Google Scholar] [CrossRef] [PubMed]
- Lewińska, A.; Kulbacka, J.; Domżał-Kędzia, M.; Witwicki, M. Antiradical Properties of N-Oxide Surfactants—Two in One. Int. J. Mol. Sci. 2021, 22, 8040. [Google Scholar] [CrossRef]
- Amić, A.; Mastiľák Cagardová, D. DFT Study of the Direct Radical Scavenging Potency of Two Natural Catecholic Compounds. Int. J. Mol. Sci. 2022, 23, 14497. [Google Scholar] [CrossRef]
- Jerzykiewicz, M.; Ćwieląg-Piasecka, I.; Witwicki, M.; Jezierski, A. EPR Spin Trapping and DFT Studies on Structure of Active Antioxidants in Biogycerol. Chem. Phys. Lett. 2010, 497, 135–141. [Google Scholar] [CrossRef]
- Amić, A.; Cagardová, D.M. Mactanamide and Lariciresinol as Radical Scavengers and Fe2+ Ion Chelators—A DFT Study. Phytochemistry 2022, 204, 113442. [Google Scholar] [CrossRef]
- Amić, A.; Milenković, D.; Marković, Z.; Cagardová, D.; Rodríguez-Guerra Pedregal, J.; Dimitrić Marković, J.M. Impact of the Phenolic O–H vs. C-Ring C–H Bond Cleavage on the Antioxidant Potency of Dihydrokaempferol. New J. Chem. 2021, 45, 7977–7986. [Google Scholar] [CrossRef]
- Amić, A.; Lučić, B.; Stepanić, V.; Marković, Z.; Marković, S.; Dimitrić Marković, J.M.; Amić, D. Free Radical Scavenging Potency of Quercetin Catecholic Colonic Metabolites: Thermodynamics of 2H+/2e− Processes. Food Chem. 2017, 218, 144–151. [Google Scholar] [CrossRef]
- Cong, Y.; Zhai, Y.; Chen, X.; Li, H. The Accuracy of Semi-Empirical Quantum Chemistry Methods on Soot Formation Simulation. Int. J. Mol. Sci. 2022, 23, 13371. [Google Scholar] [CrossRef] [PubMed]
- Ćwieląg-Piasecka, I.; Witwicki, M.; Jerzykiewicz, M.; Jezierska, J. Can Carbamates Undergo Radical Oxidation in the Soil Environment? A Case Study on Carbaryl and Carbofuran. Environ. Sci. Technol. 2017, 51, 14124–14134. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Prediction of Molecular Properties and Molecular Spectroscopy with Density Functional Theory: From Fundamental Theory to Exchange-Coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Sikdar, Y.; Modak, R.; Bose, D.; Banerjee, S.; Bieńko, D.; Zierkiewicz, W.; Bieńko, A.; das Saha, K.; Goswami, S. Doubly Chloro Bridged Dimeric Copper(II) Complex: Magneto-Structural Correlation and Anticancer Activity. Dalton Trans. 2015, 44, 8876–8888. [Google Scholar] [CrossRef]
- Baffert, C.; Orio, M.; Pantazis, D.A.; Duboc, C.; Blackman, A.G.; Blondin, G.; Neese, F.; Deronzier, A.; Collomb, M.-N. Trinuclear Terpyridine Frustrated Spin System with a MnIV3O4 Core: Synthesis, Physical Characterization, and Quantum Chemical Modeling of Its Magnetic Properties. Inorg. Chem. 2009, 48, 10281–10288. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, D.A.; Petrenko, T.; Neese, F. Magnetic and Spectroscopic Properties of Mixed Valence Manganese (III,IV) Dimers: A Systematic Study Using Broken Symmetry Density Functional Theory. Inorg. Chem. 2009, 48, 7251–7260. [Google Scholar] [CrossRef]
- Singh, S.K.; Tibrewal, N.K.; Rajaraman, G. Density Functional Studies on Dinuclear {NiIIGdIII} and Trinuclear {NiIIGdIIINiII} Complexes: Magnetic Exchange and Magneto-Structural Maps. Dalton Trans. 2011, 40, 10897. [Google Scholar] [CrossRef]
- Rajaraman, G.; Totti, F.; Bencini, A.; Caneschi, A.; Sessoli, R.; Gatteschi, D. Density Functional Studies on the Exchange Interaction of a Dinuclear Gd(Iii)–Cu(Ii) Complex: Method Assessment, Magnetic Coupling Mechanism and Magneto-Structural Correlations. Dalton Trans. 2009, 17, 3153–3161. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Krewald, V.; Orio, M.; Neese, F. Theoretical Magnetochemistry of Dinuclear Manganese Complexes: Broken Symmetry Density Functional Theory Investigation on the Influence of Bridging Motifs on Structure and Magnetism. Dalton Trans. 2010, 39, 4959. [Google Scholar] [CrossRef]
- Buvaylo, E.A.; Kokozay, V.N.; Makhankova, V.G.; Melnyk, A.K.; Korabik, M.; Witwicki, M.; Skelton, B.W.; Vassilyeva, O.Y. Synthesis, Characterization, and Magnetic Properties of a Series of Copper(II) Chloride Complexes of Pyridyliminebenzoic Acids. Eur. J. Inorg. Chem. 2018, 2018, 1603–1619. [Google Scholar] [CrossRef]
- Oms, O.; Rota, J.; Norel, L.; Calzado, C.J.; Rousselière, H.; Train, C.; Robert, V. Beyond Kahn’s Model: Substituent and Heteroatom Influence on Exchange Interaction in a Metal-Verdazyl Complex. Eur. J. Inorg. Chem. 2010, 2010, 5373–5378. [Google Scholar] [CrossRef]
- Zueva, E.M.; Petrova, M.M.; Shamsieva, A.V.; Trigulova, K.R.; Musina, E.I.; Fayzullin, R.R.; Bogomyakov, A.S.; Ovcharenko, V.I.; Karasik, A.A. Insight into the Influence of Terminal Ligands on Magnetic Exchange Coupling in a Series of Dimeric Copper(II) Acetate Adducts. Int. J. Quantum Chem. 2020, 120, e26145. [Google Scholar] [CrossRef]
- Bolvin, H.; Wagner, F.R. Case of a Strong Antiferromagnetic Exchange Coupling Induced by Spin Polarization of a Mn–Mn Partial Single Bond. Inorg. Chem. 2012, 51, 7112–7118. [Google Scholar] [CrossRef] [PubMed]
- Montenegro-Pohlhammer, N.; Urzúa-Leiva, R.; Páez-Hernández, D.; Cárdenas-Jirón, G. Spin-Filter Transport and Magnetic Properties in a Binuclear Cu(II) Expanded Porphyrin Based Molecular Junction. Dalton Trans. 2019, 48, 8418–8426. [Google Scholar] [CrossRef] [PubMed]
- Maurice, R.; Sivalingam, K.; Ganyushin, D.; Guihéry, N.; de Graaf, C.; Neese, F. Theoretical Determination of the Zero-Field Splitting in Copper Acetate Monohydrate. Inorg. Chem. 2011, 50, 6229–6236. [Google Scholar] [CrossRef] [PubMed]
- Coulaud, E.; Malrieu, J.-P.; Guihéry, N.; Ferré, N. Additive Decomposition of the Physical Components of the Magnetic Coupling from Broken Symmetry Density Functional Theory Calculations. J. Chem. Theory Comput. 2013, 9, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Ferré, N.; Guihéry, N.; Malrieu, J.-P. Spin Decontamination of Broken-Symmetry Density Functional Theory Calculations: Deeper Insight and New Formulations. Phys. Chem. Chem. Phys. 2015, 17, 14375–14382. [Google Scholar] [CrossRef]
- David, G.; Wennmohs, F.; Neese, F.; Ferré, N. Chemical Tuning of Magnetic Exchange Couplings Using Broken-Symmetry Density Functional Theory. Inorg. Chem. 2018, 57, 12769–12776. [Google Scholar] [CrossRef]
- Terencio, T.; Bastardis, R.; Suaud, N.; Maynau, D.; Bonvoisin, J.; Malrieu, J.P.; Calzado, C.J.; Guihéry, N. Physical Analysis of the Through-Ligand Long-Distance Magnetic Coupling: Spin-Polarization versus Anderson Mechanism. Phys. Chem. Chem. Phys. 2011, 13, 12314. [Google Scholar] [CrossRef] [Green Version]
- Pantazis, D.A. Assessment of Double-Hybrid Density Functional Theory for Magnetic Exchange Coupling in Manganese Complexes. Inorganics 2019, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Teyar, B.; Belkhiri, L.; Costuas, K.; Boucekkine, A.; Meyer, K. Electronic Structure and Magnetic Properties of Dioxo-Bridged Diuranium Complexes with Diamond-Core Structural Motifs: A Relativistic DFT Study. Inorg. Chem. 2016, 55, 2870–2881. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.-C.; Zheng, Y.-Z. Methods and Models of Theoretical Calculation for Single-Molecule Magnets. Magnetochemistry 2021, 7, 107. [Google Scholar] [CrossRef]
- Fujii, T.; Kitagawa, Y.; Ikenaga, K.; Tada, H.; Era, I.; Nakano, M. Theoretical Study on Magnetic Interaction in Pyrazole-Bridged Dinuclear Metal Complex: Possibility of Intramolecular Ferromagnetic Interaction by Orbital Counter-Complementarity. Magnetochemistry 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Pantazis, D.A. Meeting the Challenge of Magnetic Coupling in a Triply-Bridged Chromium Dimer: Complementary Broken-Symmetry Density Functional Theory and Multireference Density Matrix Renormalization Group Perspectives. J. Chem. Theory Comput. 2019, 15, 938–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benediktsson, B.; Bjornsson, R. Analysis of the Geometric and Electronic Structure of Spin-Coupled Iron–Sulfur Dimers with Broken-Symmetry DFT: Implications for FeMoco. J. Chem. Theory Comput. 2022, 18, 1437–1457. [Google Scholar] [CrossRef]
- Jung, J.; Puget, M.; Cador, O.; Bernot, K.; Calzado, C.J.; le Guennic, B. Analysis of the Magnetic Exchange Interactions in Yttrium(III) Complexes Containing Nitronyl Nitroxide Radicals. Inorg. Chem. 2017, 56, 6788–6801. [Google Scholar] [CrossRef]
- Zapata-Rivera, J.; Calzado, C.J. Magnetostructural Relationships in [Ni(Dmit) 2]—Radical Anions. Dalton Trans. 2021, 50, 6620–6630. [Google Scholar] [CrossRef]
- Starikova, A.A.; Minkin, V.I. Adducts of Transition Metal Complexes with Redox-Active Ligands: The Structure and Spin-State-Switching Rearrangements. Russ. Chem. Rev. 2018, 87, 1049–1079. [Google Scholar] [CrossRef]
- Minkin, V.I.; Starikova, A.A. Molecular Design of the Valence Tautomeric Mixed-Ligand Adducts of CoII Diketonates with Redox-Active Ligands. Mendeleev Commun. 2015, 25, 83–92. [Google Scholar] [CrossRef]
- Kaim, W.; Paretzki, A. Interacting Metal and Ligand Based Open Shell Systems: Challenges for Experiment and Theory. Coord. Chem. Rev. 2017, 344, 345–354. [Google Scholar] [CrossRef]
- Ansari, A.; Ansari, M.; Singha, A.; Rajaraman, G. Interplay of Electronic Cooperativity and Exchange Coupling in Regulating the Reactivity of Diiron(IV)-Oxo Complexes towards C−H and O−H Bond Activation. Chem. Eur. J. 2017, 23, 10110–10125. [Google Scholar] [CrossRef]
- Chen, P.; Solomon, E.I. O2 Activation by Binuclear Cu Sites: Noncoupled versus Exchange Coupled Reaction Mechanisms. Proc. Natl. Acad. Sci. USA 2004, 101, 13105–13110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weldon, B.T.; Wheeler, D.E.; Kirby, J.P.; McCusker, J.K. Bimolecular Electron and Energy Transfer Reactivity of Exchange-Coupled Dinuclear Iron(III) Complexes. Inorg. Chem. 2001, 40, 6802–6812. [Google Scholar] [CrossRef]
- Davidson, R.A.; Hao, J.; Rheingold, A.L.; Miller, J.S. High Spin Ground State Copper(II) and Nickel(II) Complexes Possessing the 3,5-Di-Tert-Butyl-1,2-Semiquinonate Radical Anion. Polyhedron 2017, 133, 348–357. [Google Scholar] [CrossRef]
- Harmalker, S.; Jones, S.E.; Sawyer, D.T. Electrochemical and Spectroscopic Studies of 3,5-Di-Tert-Butylcatecholato and 3,5-Di-Tert-Butyl-o-Semiquinonato Complexes of Copper(II). Inorg. Chem. 1983, 22, 2790–2794. [Google Scholar] [CrossRef]
- Thompson, J.S.; Calabrese, J.C. Synthesis, Spectroscopy, and Structures of Copper(II)-3,5-Di-Tert-Butyl-o-Semiquinone Complexes. Inorg. Chem. 1985, 24, 3167–3171. [Google Scholar] [CrossRef]
- Kahn, O.; Prins, R.; Reedijk, J.; Thompson, J.S. Orbital Symmetries and Magnetic Interaction between Copper(II) Ions and the o-Semiquinone Radical. Magnetic Studies of (Di-2-Pyridylamine)(3,5-Di-Tert-Butyl-o-Semiquinonato)Copper(II) Perchlorate and Bis(Bis(3,5-Di-Tert-Butyl-o-Semiquinonato)Copper(II)). Inorg. Chem. 1987, 26, 3557–3561. [Google Scholar] [CrossRef]
- Dooley, D.M.; McGuirl, M.A.; Brown, D.E.; Turowski, P.N.; Mclntire, W.S.; Knowles, P.F. A Cu(I)-Semiquinone State in Substrate-Reduced Amine Oxidases. Nature 1991, 349, 262–264. [Google Scholar] [CrossRef]
- Rall, J.; Kaim, W. Ligand-Controlled Oxidation State Ambivalence in Copper–Quinone Complexes. Replacement of N-Donor by S-Donor Ligands Favours the Copper(I)–Semiquinone over the Copper(II)–Catecholate Form. J. Chem. Soc. Faraday Trans. 1994, 90, 2905–2908. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; Lee, H.; Shultz, D.A.; Sorace, L. Ferromagnetically Coupled Bis(Semiquinone) Ligand Enforces High-Spin Ground States in Bis-Metal Complexes. Inorg. Chem. 2001, 40, 408–411. [Google Scholar] [CrossRef]
- Dei, A.; Gatteschi, D.; Pardi, L.; Barra, A.L.; Brunel, L.C. Millimeter Band EPR Spectra Reveal Large Zero-Field Splittings in Copper(II)—Semiquinonato Complexes. Chem. Phys. Lett. 1990, 175, 589–592. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Cherkasov, V.K.; Nevodchikov, V.I.; Kuropatov, V.A.; Yee, G.T.; Pierpont, C.G. Magnetic Properties and Redox Isomerism for 4,4‘-Bis(Semiquinone) Complexes of Copper. Inorg. Chem. 2001, 40, 2434–2436. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Verani, C.N.; Bill, E.; Bothe, E.; Weyhermüller, T.; Wieghardt, K. Electronic Structure of Bis(0-Iminobenzosemiquinonato)Metal Complexes (Cu, Ni, Pd). The Art of Establishing Physical Oxidation States in Transition-Metal Complexes Containing Radical Ligands. J. Am. Chem. Soc. 2001, 123, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, V.I.; Gorelik, E.V.; Fokin, S.V.; Romanenko, G.V.; Ikorskii, V.N.; Krashilina, A.V.; Cherkasov, V.K.; Abakumov, G.A. Ligand Effects on the Ferro- to Antiferromagnetic Exchange Ratio in Bis(o-Semiquinonato)Copper(II). J. Am. Chem. Soc. 2007, 129, 10512–10521. [Google Scholar] [CrossRef]
- Malrieu, J.P.; Caballol, R.; Calzado, C.J.; de Graaf, C.; Guihéry, N. Magnetic Interactions in Molecules and Highly Correlated Materials: Physical Content, Analytical Derivation, and Rigorous Extraction of Magnetic Hamiltonians. Chem. Rev. 2014, 114, 429–492. [Google Scholar] [CrossRef]
- Roemelt, M.; Krewald, V.; Pantazis, D.A. Exchange Coupling Interactions from the Density Matrix Renormalization Group and N -Electron Valence Perturbation Theory: Application to a Biomimetic Mixed-Valence Manganese Complex. J. Chem. Theory Comput. 2018, 14, 166–179. [Google Scholar] [CrossRef]
- Buchanan, J.K.; Dais, T.N.; Plieger, P.G. Computational Studies of the Magneto-Structural Correlations in a Manganese Dimer with Jahn–Teller Distortions. Phys. Chem. Chem. Phys. 2022, 24, 4407–4414. [Google Scholar] [CrossRef]
- Razquin-Bobillo, L.; Pajuelo-Corral, O.; Artetxe, B.; Zabala-Lekuona, A.; Choquesillo-Lazarte, D.; Rodríguez-Diéguez, A.; San Sebastian, E.; Cepeda, J. Combined Experimental and Theoretical Investigation on the Magnetic Properties Derived from the Coordination of 6-Methyl-2-Oxonicotinate to 3d-Metal Ions. Dalton Trans. 2022, 51, 9780–9792. [Google Scholar] [CrossRef]
- Witwicki, M.; Jerzykiewicz, M.; Jaszewski, A.R.; Jezierska, J.; Ozarowski, A. Influence of Pb(II) Ions on the EPR Properties of the Semiquinone Radicals of Humic Acids and Model Compounds: High Field EPR and Relativistic DFT Studies. J. Phys. Chem. A 2009, 113, 14115–14122. [Google Scholar] [CrossRef]
- Ciofini, I.; Reviakine, R.; Arbuznikov, A.; Kaupp, M. Solvent Effects on G-Tensors of Semiquinone Radical Anions: Polarizable Continuum versus Cluster Models. Theor. Chem. Acc. 2004, 111, 132–140. [Google Scholar] [CrossRef]
- Neese, F.; Ames, W.; Christian, G.; Kampa, M.; Liakos, D.G.; Pantazis, D.A.; Roemelt, M.; Surawatanawong, P.; Shengfa, Y.E. Dealing with Complexity in Open-Shell Transition Metal Chemistry from a Theoretical Perspective: Reaction Pathways, Bonding, Spectroscopy, And Magnetic Properties. Adv. Inorg. Chem. 2010, 62, 301–349. [Google Scholar]
- Witwicki, M. Theoretical Characterisation of Phosphinyl Radicals and Their Magnetic Properties: G Matrix. ChemPhysChem. 2015, 16, 1912–1925. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Ashvar, C.S.; Chabalowski, C.F.; Frisch, M.J. Theoretical Calculation of Vibrational Circular Dichroism Spectra. Faraday Discuss 1994, 99, 103. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Bursch, M.; Mewes, J.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, K.; Takahara, Y.; Fueno, T. Ab-Initio Molecular Orbital Studies of Structure and Reactivity of Transition Metal-OXO Compounds. In Applied Quantum Chemistry; Springer: Dordrecht, The Netherlands, 1986; pp. 155–184. [Google Scholar]
- Soda, T.; Kitagawa, Y.; Onishi, T.; Takano, Y.; Shigeta, Y.; Nagao, H.; Yoshioka, Y.; Yamaguchi, K. Ab Initio Computations of Effective Exchange Integrals for H–H, H–He–H and Mn2O2 Complex: Comparison of Broken-Symmetry Approaches. Chem. Phys. Lett. 2000, 319, 223–230. [Google Scholar] [CrossRef]
- Neese, F. Definition of Corresponding Orbitals and the Diradical Character in Broken Symmetry DFT Calculations on Spin Coupled Systems. J. Phys. Chem. Solids 2004, 65, 781–785. [Google Scholar] [CrossRef]
- Neese, F. Importance of Direct Spin−Spin Coupling and Spin-Flip Excitations for the Zero-Field Splittings of Transition Metal Complexes: A Case Study. J. Am. Chem. Soc. 2006, 128, 10213–10222. [Google Scholar] [CrossRef]
- Pipek, J.; Mezey, P.G. A Fast Intrinsic Localization Procedure Applicable for ab initio and Semiempirical Linear Combination of Atomic Orbital Wave Functions. J. Chem. Phys. 1989, 90, 4916–4926. [Google Scholar] [CrossRef]
- Olsen, J. The CASSCF Method: A Perspective and Commentary. Int. J. Quantum Chem. 2011, 111, 3267–3272. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of N-Electron Valence States for Multireference Perturbation Theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-Electron Valence State Perturbation Theory: A Fast Implementation of the Strongly Contracted Variant. Chem. Phys. Lett. 2001, 350, 297–305. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-Electron Valence State Perturbation Theory: A Spinless Formulation and an Efficient Implementation of the Strongly Contracted and of the Partially Contracted Variants. J. Chem. Phys. 2002, 117, 9138–9153. [Google Scholar] [CrossRef]
- Miralles, J.; Castell, O.; Caballol, R.; Malrieu, J.-P. Specific CI Calculation of Energy Differences: Transition Energies and Bond Energies. Chem. Phys. 1993, 172, 33–43. [Google Scholar] [CrossRef]
- Miralles, J.; Daudey, J.-P.; Caballol, R. Variational Calculation of Small Energy Differences. The Singlet-Triplet Gap in [Cu2Cl6]2−. Chem. Phys. Lett. 1992, 198, 555–562. [Google Scholar] [CrossRef]
- Calzado, C.J.; Sanz, J.F.; Malrieu, J.P. Accurate Ab Initio Determination of Magnetic Interactions and Hopping Integrals in La2−xSrxCuO4 Systems. J. Chem. Phys. 2000, 112, 5158–5167. [Google Scholar] [CrossRef]
- Vancoillie, S.; Chalupský, J.; Ryde, U.; Solomon, E.I.; Pierloot, K.; Neese, F.; Rulíšek, L. Multireference Ab Initio Calculations of g Tensors for Trinuclear Copper Clusters in Multicopper Oxidases. J. Phys. Chem. B 2010, 114, 7692–7702. [Google Scholar] [CrossRef] [Green Version]
- Lunghi, A.; Totti, F. The Role of Anisotropic Exchange in Single Molecule Magnets: A CASSCF/NEVPT2 Study of the Fe4 SMM Building Block [Fe2(OCH3)2(Dbm)4] Dimer. Inorganics 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, A.; Ali, M.E. First-Principle Design of Blatter’s Diradicals with Strong Ferromagnetic Exchange Interactions. J. Phys. Chem. C 2019, 123, 15186–15194. [Google Scholar] [CrossRef]
- Zapata-Rivera, J.; Calzado, C. Light-Induced Control of the Spin Distribution on Cu–Dithiolene Complexes: A Correlated Ab Initio Study. Molecules 2019, 24, 1088. [Google Scholar] [CrossRef] [Green Version]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, Approximate and Parallel Hartree–Fock and Hybrid DFT Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree–Fock Exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Weigend, F.; Kattannek, M.; Ahlrichs, R. Approximated Electron Repulsion Integrals: Cholesky Decomposition versus Resolution of the Identity Methods. J. Chem. Phys. 2009, 130, 164106. [Google Scholar] [CrossRef]
- Kollmar, C.; Sivalingam, K.; Helmich-Paris, B.; Angeli, C.; Neese, F. A Perturbation-based Super-CI Approach for the Orbital Optimization of a CASSCF Wave Function. J. Comput. Chem. 2019, 40, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Hartree–Fock Exchange Fitting Basis Sets for H to Rn. J. Comput. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. A Spectroscopy Oriented Configuration Interaction Procedure. J. Chem. Phys. 2003, 119, 9428–9443. [Google Scholar] [CrossRef]
- Neese, F. Sum-over-States Based Multireferenceab Initio Calculation of EPR Spin Hamiltonian Parameters for Transition Metal Complexes. A Case Study. Magn. Reson. Chem. 2004, 42, S187–S198. [Google Scholar] [CrossRef]
- Allouche, A.-R. Gabedit-A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CASSCF(2e,2o) | CASSCF(10e,6o) | CASSCF(18e,10o) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basis Set | B3LYP | PBE0 | TPSSh | without MRPT | SC-NEVPT2 | FIC-NEVPT2 | DDCI3(2,2) | without MRPT | SC-NEVPT2 | FIC-NEVPT2 | without MRPT | SC-NEVPT2 | FIC-NEVPT2 | ||
| [Cu(SQ)(opd)]+ | def2-TZVP | 294 | 271 | 276 | 74 | 73 | 73 | 173 | 75 | 81 | 80 | 76 | 130 | 129 | |
| def2-SVP | 339 | 316 | 311 | 85 | 88 | 88 | 193 | 85 | 93 | 92 | 85 | 124 | 130 | ||
| cc-pVDZ | 298 | 276 | 282 | 79 | 77 | 77 | 169 | 79 | 85 | 84 | 80 | 135 | 137 | ||
| [Cu(SQ)(acac)] | def2-TZVP | 274 | 257 | 269 | 75 | 78 | 78 | 187 | 75 | 86 | 85 | 76 | 123 | 129 | |
| def2-SVP | 300 | 284 | 286 | 85 | 93 | 93 | 174 | 85 | 97 | 94 | 86 | 124 | 132 | ||
| cc-pVDZ | 283 | 265 | 279 | 80 | 83 | 83 | 188 | 80 | 93 | 92 | 81 | 135 | 138 | ||
| [Cu(SQ)(dtc)] | def2-TZVP | 221 | 216 | 215 | 71 | 73 | 73 | 179 | 71 | 81 | 81 | 73 | 124 | 129 | |
| def2-SVP | 239 | 236 | 231 | 81 | 89 | 89 | 184 | 78 | 116 | 113 | 82 | 145 | 153 | ||
| cc-pVDZ | 232 | 225 | 225 | 76 | 78 | 78 | 170 | 74 | 128 | 127 | 74 | 174 | 169 | ||
| [Cu(SQ)(en)]+ | def2-TZVP | 292 | 269 | 274 | 74 | 72 | 72 | 185 | 74 | 75 | 74 | 74 | 106 | 117 | |
| def2-SVP | 337 | 314 | 310 | 85 | 88 | 88 | 199 | 85 | 92 | 92 | 86 | 128 | 135 | ||
| cc-pVDZ | 297 | 274 | 279 | 78 | 76 | 76 | 180 | 79 | 109 | 108 | 78 | 175 | 177 | ||
| [Cu(SQ)(mnt)]− | def2-TZVP | 308 | 292 | 325 | 76 | 83 | 83 | 193 | 76 | 85 | 87 | 77 | 127 | 131 | |
| def2-SVP | 324 | 309 | 338 | 87 | 99 | 99 | 188 | 87 | 103 | 104 | 88 | 138 | 138 | ||
| cc-pVDZ | 330 | 312 | 345 | 82 | 89 | 89 | 186 | 82 | 91 | 91 | 82 | 134 | 137 | ||
| CASSCF(2e,2o) | CASSCF(10e,6o) | CASSCF(18e,10o) | Exptl. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basis Set | B3LYP | PBE0 | TPSSh | without MRPT | SC-NEVPT2 | FIC-NEVPT2 | DDCI3(2,2) | without MRPT | SC-NEVPT2 | FIC-NEVPT2 | without MRPT | SC-NEVPT2 | FIC-NEVPT2 | |||
| [Cu(dtbSQ)(dpya)(ClO4)] | def2-TZVP | 297 | 274 | 295 | 72 | 73 | 74 | - | 75 | 87 | 85 | 74 | 100 | 117 | 140 (a) | |
| def2-SVP | 335 | 309 | 324 | 81 | 86 | 87 | 218 | 84 | 88 | 91 | 83 | 116 | 125 | 140 (a) | ||
| cc-pVDZ | 312 | 301 | 311 | 77 | 78 | 79 | 204 | 82 | 86 | 85 | 81 | 125 | 130 | 140 (a) | ||
| [Cu(dtbSQ)(tmcd)(THF)]+ | def2-TZVP | 241 | 227 | 229 | 69 | 66 | 71 | - | 74 | 73 | 74 | 73 | 108 | 114 | FM (b) | |
| def2-SVP | 265 | 251 | 247 | 77 | 78 | 79 | 246 | 82 | 85 | 85 | 81 | 109 | 114 | FM (b) | ||
| cc-pVDZ | 250 | 234 | 237 | 73 | 70 | 71 | 210 | 77 | 80 | 79 | 77 | 122 | 124 | FM (b) | ||
| [Cu(dtbSQ)(bipy)(BF4)] | def2-TZVP | 297 | 276 | 291 | 79 | 78 | 79 | - | 85 | 86 | 86 | 84 | 119 | 129 | 191 (a) | |
| def2-SVP | 335 | 312 | 321 | 88 | 91 | 92 | 215 | 92 | 95 | 95 | 90 | 120 | 127 | 191 (a) | ||
| cc-pVDZ | 310 | 287 | 304 | 84 | 83 | 84 | 201 | 85 | 88 | 88 | 85 | 131 | 133 | 191 (a) | ||
| [Cu(dtbSQ)(dpya)(THF)2]+ | def2-TZVP | 264 | 244 | 268 | 69 | 69 | 70 | - | 73 | 79 | 80 | 72 | 119 | 121 | 168 (a) | |
| def2-SVP | 295 | 273 | 284 | 77 | 81 | 81 | 210 | 82 | 85 | 87 | 80 | 106 | 112 | 168 (a) | ||
| cc-pVDZ | 275 | 253 | 270 | 73 | 74 | 74 | 188 | 74 | 79 | 80 | 75 | 119 | 121 | 168 (a) | ||
| [Cu(dtbSQ)(dtben)]+ | def2-TZVP | −1589 | −1312 | −1788 | −284 | −560 | −611 | - | −296 | −495 | −519 | −298 | −501 | −516 | −1150 (c) | |
| def2-SVP | −1790 | −1489 | −1933 | −292 | −548 | −553 | −809 | −302 | −493 | −525 | −303 | −506 | −519 | −1150 (c) | ||
| cc-pVDZ | −1639 | −1367 | −1852 | −299 | −605 | −655 | −1140 | −311 | −582 | −608 | −312 | −577 | −593 | −1150 (c) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziółkowska, A.; Witwicki, M. Understanding the Exchange Interaction between Paramagnetic Metal Ions and Radical Ligands: DFT and Ab Initio Study on Semiquinonato Cu(II) Complexes. Int. J. Mol. Sci. 2023, 24, 4001. https://doi.org/10.3390/ijms24044001
Ziółkowska A, Witwicki M. Understanding the Exchange Interaction between Paramagnetic Metal Ions and Radical Ligands: DFT and Ab Initio Study on Semiquinonato Cu(II) Complexes. International Journal of Molecular Sciences. 2023; 24(4):4001. https://doi.org/10.3390/ijms24044001
Chicago/Turabian StyleZiółkowska, Aleksandra, and Maciej Witwicki. 2023. "Understanding the Exchange Interaction between Paramagnetic Metal Ions and Radical Ligands: DFT and Ab Initio Study on Semiquinonato Cu(II) Complexes" International Journal of Molecular Sciences 24, no. 4: 4001. https://doi.org/10.3390/ijms24044001
APA StyleZiółkowska, A., & Witwicki, M. (2023). Understanding the Exchange Interaction between Paramagnetic Metal Ions and Radical Ligands: DFT and Ab Initio Study on Semiquinonato Cu(II) Complexes. International Journal of Molecular Sciences, 24(4), 4001. https://doi.org/10.3390/ijms24044001