

Placental Mitochondrial Function and Dysfunction in Preeclampsia
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Mitochondria in the Healthy Placenta
2.1. Placental Mitochondrial Content, Structure, and Function across Pregnancy
2.2. Mitochondrial Content, Structure, and Function in Distinct Placental Cell Types
3. Preeclampsia
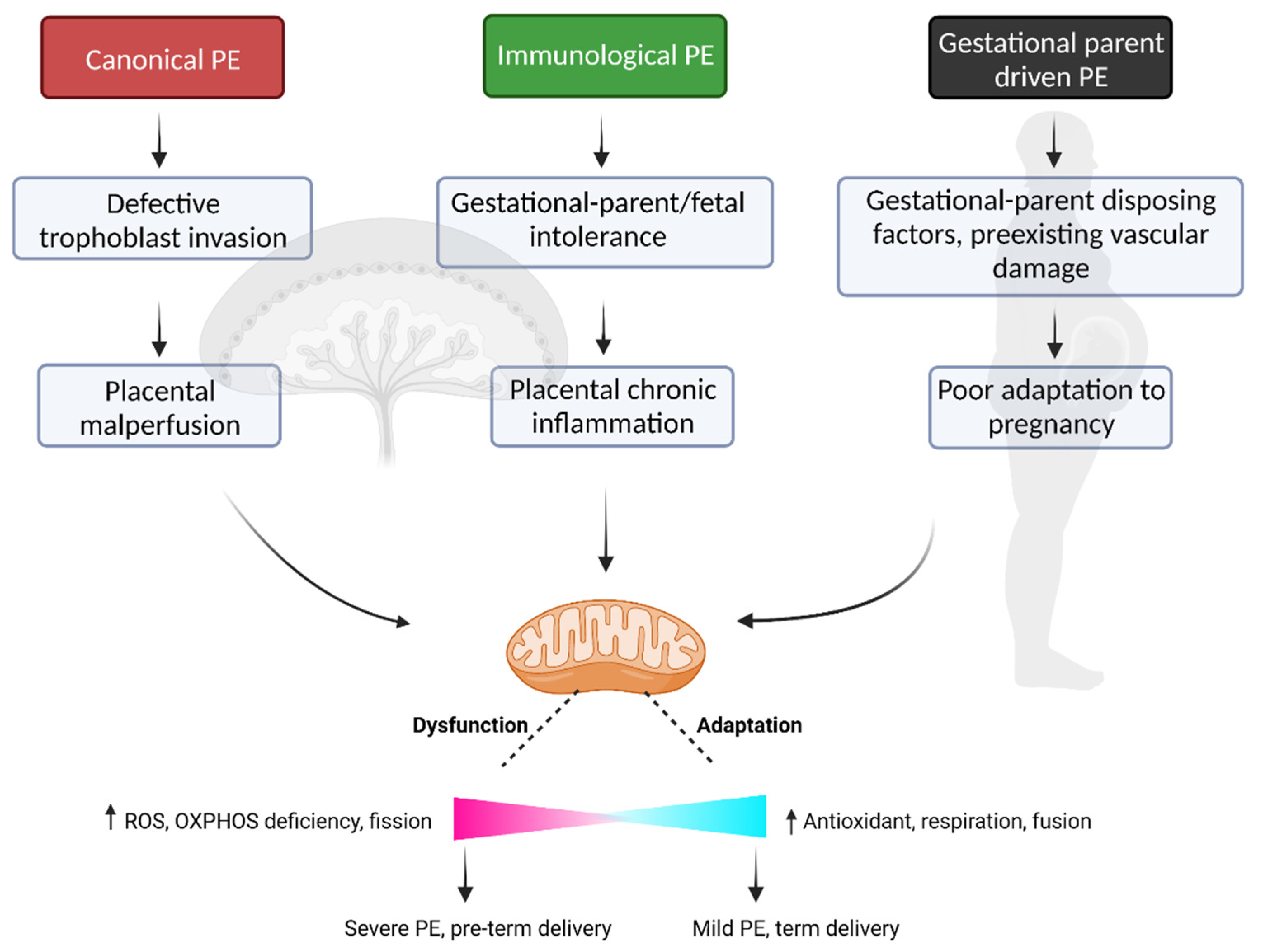
4. Subclasses of Disease in Preeclampsia
4.1. Canonical PE Subclass
4.2. Immunological PE Subclass
4.3. Gestational Parent-Driven PE Subclass
5. Placental Mitochondrial Dysfunction in Preeclampsia
5.1. Mitochondrial Content
5.2. Mitochondrial Structure and Dynamics
5.3. Mitochondrial Function- Respiration and ATP Generation
5.4. Mitochondrial Function—Redox Homeostasis and Apoptosis
6. Mitochondrial Dysfunction—A Point of Convergence across Preeclampsia Subclasses
6.1. Canonical PE Subclass—Ischemia-Reperfusion Induced Mitochondrial Dysfunction
6.2. Immunological PE Subclass—Inflammation Induced Mitochondrial Dysfunction
6.3. Gestational Parent Driven PE—Predisposing Factors and Mitochondrial Dysfunction
7. Potential for Mitochondrial Targeting Therapies in Preeclampsia
7.1. Resveratrol
7.2. Metformin
7.3. MitoQ and MitoTEMPO
7.4. Nicotinamide (NAM)
8. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, M.K.; Harding, J.E.; Bassett, N.S.; Breier, B.H.; Oliver, M.H.; Gallaher, B.H.; Evans, P.C.; Woodall, S.M.; Gluckman, P.D. Fetal growth and placental function. Mol. Cell. Endocrinol. 1998, 140, 115–120. [Google Scholar] [CrossRef]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, S.K.; Campbell, J.P. Placental structure, function and drug transfer. Contin. Educ. Anaesth. Crit. Care Pain 2014, 15, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Aye, I.; Aiken, C.E.; Charnock-Jones, D.S.; Smith, G.C.S. Placental energy metabolism in health and disease-significance of development and implications for preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S928–S944. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell. Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.F.; Myatt, L. Placental mitochondrial dysfunction with metabolic diseases: Therapeutic approaches. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165967. [Google Scholar] [CrossRef]
- Holland, O.; Dekker Nitert, M.; Gallo, L.A.; Vejzovic, M.; Fisher, J.J.; Perkins, A.V. Review: Placental mitochondrial function and structure in gestational disorders. Placenta 2017, 54, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, G.J.; Yung, H.W.; Murray, A.J. Mitochondrial—Endoplasmic reticulum interactions in the trophoblast: Stress and senescence. Placenta 2017, 52, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Constancia, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef] [PubMed]
- Constancia, M.; Angiolini, E.; Sandovici, I.; Smith, P.; Smith, R.; Kelsey, G.; Dean, W.; Ferguson-Smith, A.; Sibley, C.P.; Reik, W.; et al. Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc. Natl. Acad. Sci. USA 2005, 102, 19219–19224. [Google Scholar] [CrossRef] [Green Version]
- Sferruzzi-Perri, A.N.; Vaughan, O.R.; Haro, M.; Cooper, W.N.; Musial, B.; Charalambous, M.; Pestana, D.; Ayyar, S.; Ferguson-Smith, A.C.; Burton, G.J.; et al. An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. FASEB J. 2013, 27, 3928–3937. [Google Scholar] [CrossRef] [PubMed]
- Hay, W.W., Jr. Energy and substrate requirements of the placenta and fetus. Proc. Nutr. Soc. 1991, 50, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, J.J.; Bartho, L.A.; Perkins, A.V.; Holland, O.J. Placental mitochondria and reactive oxygen species in the physiology and pathophysiology of pregnancy. Clin. Exp. Pharmacol. Physiol. 2020, 47, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Bartho, L.A.; Fisher, J.J.; Cuffe, J.S.M.; Perkins, A.V. Mitochondrial transformations in the aging human placenta. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E981–E994. [Google Scholar] [CrossRef] [PubMed]
- Correia, Y.; Scheel, J.; Gupta, S.; Wang, K. Placental mitochondrial function as a driver of angiogenesis and placental dysfunction. Biol. Chem. 2021, 402, 887–909. [Google Scholar] [CrossRef]
- Burton, G.J.; Fowden, A.L. The placenta: A multifaceted, transient organ. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef] [Green Version]
- Jauniaux, E.; Hempstock, J.; Teng, C.; Battaglia, F.C.; Burton, G.J. Polyol concentrations in the fluid compartments of the human conceptus during the first trimester of pregnancy: Maintenance of redox potential in a low oxygen environment. J. Clin. Endocrinol. Metab. 2005, 90, 1171–1175. [Google Scholar] [CrossRef] [Green Version]
- Bloxam, D.L.; Bobinski, P.M. Energy metabolism and glycolysis in the human placenta during ischaemia and in normal labour. Placenta 1984, 5, 381–394. [Google Scholar] [CrossRef]
- Holland, O.J.; Hickey, A.J.R.; Alvsaker, A.; Moran, S.; Hedges, C.; Chamley, L.W.; Perkins, A.V. Changes in mitochondrial respiration in the human placenta over gestation. Placenta 2017, 57, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, D.; Faes, E.; Cos, P.; Briede, J.J.; Gyselaers, W.; Cornette, J.; Gorbanev, Y.; Bogaerts, A.; Spaanderman, M.; Van Craenenbroeck, E.; et al. Oxidative stress in healthy pregnancy and preeclampsia is linked to chronic inflammation, iron status and vascular function. PLoS ONE 2018, 13, e0202919. [Google Scholar] [CrossRef] [Green Version]
- Bardin, N.; Murthi, P.; Alfaidy, N. Normal and pathological placental angiogenesis. Biomed Res. Int. 2015, 2015, 354359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, J.; Bendek, B.; Agamasu, E.; Salafia, C.M.; Mishra, A.; Benfield, N.; Patel, R.; Mikhail, M. Placental Oxidative Status throughout Normal Gestation in Women with Uncomplicated Pregnancies. Obstet. Gynecol. Int. 2015, 2015, 276095. [Google Scholar] [CrossRef] [Green Version]
- Sferruzzi-Perri, A.N.; Higgins, J.S.; Vaughan, O.R.; Murray, A.J.; Fowden, A.L. Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc. Natl. Acad. Sci. USA 2019, 116, 1621–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sesso, A.; Belizario, J.E.; Marques, M.M.; Higuchi, M.L.; Schumacher, R.I.; Colquhoun, A.; Ito, E.; Kawakami, J. Mitochondrial swelling and incipient outer membrane rupture in preapoptotic and apoptotic cells. Anat. Rec. 2012, 295, 1647–1659. [Google Scholar] [CrossRef] [Green Version]
- Huppertz, B.; Frank, H.G.; Reister, F.; Kingdom, J.; Korr, H.; Kaufmann, P. Apoptosis cascade progresses during turnover of human trophoblast: Analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab. Investig. 1999, 79, 1687–1702. [Google Scholar]
- Levy, R.; Nelson, D.M. To be, or not to be, that is the question. Apoptosis in human trophoblast. Placenta 2000, 21, 1–13. [Google Scholar] [CrossRef]
- Kolahi, K.S.; Valent, A.M.; Thornburg, K.L. Cytotrophoblast, Not Syncytiotrophoblast, Dominates Glycolysis and Oxidative Phosphorylation in Human Term Placenta. Sci. Rep. 2017, 7, 42941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De los Rios Castillo, D.; Zarco-Zavala, M.; Olvera-Sanchez, S.; Pardo, J.P.; Juarez, O.; Martinez, F.; Mendoza-Hernandez, G.; Garcia-Trejo, J.J.; Flores-Herrera, O. Atypical cristae morphology of human syncytiotrophoblast mitochondria: Role for complex V. J. Biol. Chem. 2011, 286, 23911–23919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.; Kiriakidou, M.; Strauss, J.F., 3rd. Structural and functional changes in mitochondria associated with trophoblast differentiation: Methods to isolate enriched preparations of syncytiotrophoblast mitochondria. Endocrinology 1997, 138, 2172–2183. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Olvera-Sanchez, S.; Esparza-Perusquia, M.; Gomez-Chang, E.; Flores-Herrera, O. Multiple functions of syncytiotrophoblast mitochondria. Steroids 2015, 103, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.L.; Skepper, J.N.; Jauniaux, E.; Burton, G.J. Susceptibility of human placental syncytiotrophoblastic mitochondria to oxygen-mediated damage in relation to gestational age. J. Clin. Endocrinol. Metab. 1998, 83, 1697–1705. [Google Scholar] [CrossRef]
- Seok, J.; Jun, S.; Lee, J.O.; Kim, G.J. Mitochondrial Dynamics in Placenta-Derived Mesenchymal Stem Cells Regulate the Invasion Activity of Trophoblast. Int. J. Mol. Sci. 2020, 21, 8599. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Jun, S.; Cho, J.; Park, S.; Lee, J.O.; Kim, G.J. Human placenta-derived mesenchymal stem cells induce trophoblast invasion via dynamic effects on mitochondrial function. J. Cell. Physiol. 2021, 236, 6678–6690. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell. Biol. 2014, 16, 992–1003, Erratum in Nat. Cell. Biol. 2014, 16, 1125. [Google Scholar] [CrossRef] [Green Version]
- Hogan, M.C.; Foreman, K.J.; Naghavi, M.; Ahn, S.Y.; Wang, M.; Makela, S.M.; Lopez, A.D.; Lozano, R.; Murray, C.J. Maternal mortality for 181 countries, 1980–2008: A systematic analysis of progress towards Millennium Development Goal 5. Lancet 2010, 375, 1609–1623. [Google Scholar] [CrossRef]
- ACOG Practice Bulletin. No. 202: Gestational Hypertension and Preeclampsia. Obstet. Gynecol. 2019, 133, 1. [Google Scholar] [CrossRef]
- Weiler, J.; Tong, S.; Palmer, K.R. Is fetal growth restriction associated with a more severe maternal phenotype in the setting of early onset pre-eclampsia? A retrospective study. PLoS ONE 2011, 6, e26937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrolia, S.A.; Taran, B.; Kachko, E.; Mor, O.; Beer-Wiesel, R.; Eshkoli, T.; Dukler, D.; Miodownik, S.; Erez, O. Single vs. Recurrent Episodes of Preeclampsia-population–based Epidemiological and Clinical Characteristics. Matern.-Fetal Med. 2021, 3, 190–196. [Google Scholar] [CrossRef]
- Hutchinson, E.S.; Brownbill, P.; Jones, N.W.; Abrahams, V.M.; Baker, P.N.; Sibley, C.P.; Crocker, I.P. Utero-placental haemodynamics in the pathogenesis of pre-eclampsia. Placenta 2009, 30, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Hubel, C.A. The two stage model of preeclampsia: Variations on the theme. Placenta 2009, 30 (Suppl. A), S32–S37. [Google Scholar] [CrossRef] [Green Version]
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: Relationship to clinical outcome. Hypertension 2013, 62, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Aardema, M.W.; Oosterhof, H.; Timmer, A.; van Rooy, I.; Aarnoudse, J.G. Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta 2001, 22, 405–411. [Google Scholar] [CrossRef]
- Khong, T.Y.; De Wolf, F.; Robertson, W.B.; Brosens, I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br. J. Obstet. Gynaecol. 1986, 93, 1049–1059. [Google Scholar] [CrossRef]
- Espinoza, J.; Romero, R.; Mee Kim, Y.; Kusanovic, J.P.; Hassan, S.; Erez, O.; Gotsch, F.; Than, N.G.; Papp, Z.; Jai Kim, C. Normal and abnormal transformation of the spiral arteries during pregnancy. J. Perinat. Med. 2006, 34, 447–458. [Google Scholar] [CrossRef]
- Jones, C.J.; Fox, H. An ultrastructural and ultrahistochemical study of the human placenta in maternal pre-eclampsia. Placenta 1980, 1, 61–76. [Google Scholar] [CrossRef]
- Longtine, M.S.; Chen, B.; Odibo, A.O.; Zhong, Y.; Nelson, D.M. Villous trophoblast apoptosis is elevated and restricted to cytotrophoblasts in pregnancies complicated by preeclampsia, IUGR, or preeclampsia with IUGR. Placenta 2012, 33, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Redman, C.W.; Sargent, I.L.; Staff, A.C. IFPA Senior Award Lecture: Making sense of pre-eclampsia—Two placental causes of preeclampsia? Placenta 2014, 35, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Tannetta, D.; Masliukaite, I.; Vatish, M.; Redman, C.; Sargent, I. Update of syncytiotrophoblast derived extracellular vesicles in normal pregnancy and preeclampsia. J. Reprod. Immunol. 2017, 119, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikuei, P.; Rajaei, M.; Malekzadeh, K.; Nejatizadeh, A.; Mohseni, F.; AtashAbParvar, A. Accuracy of Soluble Endoglin for Diagnosis of Preeclampsia and its Severity. Iran Biomed J. 2017, 21, 312–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of preeclampsia. Hypertens. Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef]
- McCartney, C.P. Pathological Anatomy of Acute Hypertension of Pregnancy. Circulation 1964, 30 (Suppl. 2), 37–42. [Google Scholar] [CrossRef]
- Xiong, X.; Fraser, W.D. Impact of pregnancy-induced hypertension on birthweight by gestational age. Paediatr. Perinat. Epidemiol. 2004, 18, 186–191. [Google Scholar] [CrossRef]
- Redman, C.W.; Staff, A.C. Preeclampsia, biomarkers, syncytiotrophoblast stress, and placental capacity. Am. J. Obstet. Gynecol. 2015, 213 (Suppl. 4), S9.e1–S9.e4. [Google Scholar] [CrossRef]
- Redman, E.K.; Hauspurg, A.; Hubel, C.A.; Roberts, J.M.; Jeyabalan, A. Clinical Course, Associated Factors, and Blood Pressure Profile of Delayed-Onset Postpartum Preeclampsia. Obstet. Gynecol. 2019, 134, 995–1001. [Google Scholar] [CrossRef]
- McElrath, T.F.; Cantonwine, D.E.; Jeyabalan, A.; Doss, R.C.; Page, G.; Roberts, J.M.; Brohman, B.; Zhang, Z.; Rosenblatt, K.P. Circulating microparticle proteins obtained in the late first trimester predict spontaneous preterm birth at less than 35 weeks’ gestation: A panel validation with specific characterization by parity. Am. J. Obstet. Gynecol. 2019, 220, 488.e1–488.e11. [Google Scholar] [CrossRef]
- Powers, R.W.; Roberts, J.M.; Plymire, D.A.; Pucci, D.; Datwyler, S.A.; Laird, D.M.; Sogin, D.C.; Jeyabalan, A.; Hubel, C.A.; Gandley, R.E. Low placental growth factor across pregnancy identifies a subset of women with preterm preeclampsia: Type 1 versus type 2 preeclampsia? Hypertension 2012, 60, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Benton, S.J.; Leavey, K.; Grynspan, D.; Cox, B.J.; Bainbridge, S.A. The clinical heterogeneity of preeclampsia is related to both placental gene expression and placental histopathology. Am. J. Obstet. Gynecol. 2018, 219, 604.e1–604.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.L.; Leavey, K.; Cox, B.J.; Robinson, W.P. Mining DNA methylation alterations towards a classification of placental pathologies. Hum. Mol. Genet. 2018, 27, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavey, K.; Bainbridge, S.A.; Cox, B.J. Large scale aggregate microarray analysis reveals three distinct molecular subclasses of human preeclampsia. PLoS ONE 2015, 10, e0116508. [Google Scholar] [CrossRef] [PubMed]
- Leavey, K.; Grynspan, D.; Cox, B.J. Both “canonical” and “immunological” preeclampsia subtypes demonstrate changes in placental immune cell composition. Placenta 2019, 83, 53–56. [Google Scholar] [CrossRef]
- Roberts, J.M.; Rich-Edwards, J.W.; McElrath, T.F.; Garmire, L.; Myatt, L.; Global Pregnancy, C. Subtypes of Preeclampsia: Recognition and Determining Clinical Usefulness. Hypertension 2021, 77, 1430–1441. [Google Scholar] [CrossRef]
- Conrad, K.P.; Benyo, D.F. Placental cytokines and the pathogenesis of preeclampsia. Am. J. Reprod. Immunol. 1997, 37, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.C.; Cornelius, D.C.; Amaral, L.M.; Faulkner, J.L.; Cunningham, M.W., Jr.; Wallace, K.; LaMarca, B. The role of inflammation in the pathology of preeclampsia. Clin. Sci. 2016, 130, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Moffett, A.; Colucci, F. Uterine NK cells: Active regulators at the maternal-fetal interface. J. Clin. Investig. 2014, 124, 1872–1879. [Google Scholar] [CrossRef] [Green Version]
- Hiby, S.E.; Walker, J.J.; O’Shaughnessy, K.M.; Redman, C.W.; Carrington, M.; Trowsdale, J.; Moffett, A. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J. Exp. Med. 2004, 200, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Triche, E.W.; Harland, K.K.; Field, E.H.; Rubenstein, L.M.; Saftlas, A.F. Maternal-fetal HLA sharing and preeclampsia: Variation in effects by seminal fluid exposure in a case-control study of nulliparous women in Iowa. J. Reprod. Immunol. 2014, 101–102, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Kenny, L.C.; Kell, D.B. Immunological Tolerance, Pregnancy, and Preeclampsia: The Roles of Semen Microbes and the Father. Front. Med. 2017, 4, 239. [Google Scholar] [CrossRef] [Green Version]
- Myatt, L.; Roberts, J.M. Preeclampsia: Syndrome or Disease? Curr. Hypertens. Rep. 2015, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Vangrieken, P.; Al-Nasiry, S.; Bast, A.; Leermakers, P.A.; Tulen, C.B.M.; Schiffers, P.M.H.; van Schooten, F.J.; Remels, A.H.V. Placental Mitochondrial Abnormalities in Preeclampsia. Reprod. Sci. 2021, 28, 2186–2199. [Google Scholar] [CrossRef] [PubMed]
- Holland, O.J.; Cuffe, J.S.M.; Dekker Nitert, M.; Callaway, L.; Kwan Cheung, K.A.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell. Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, D.; Yevale, A.; Naha, R.; Kuthethur, R.; Chakrabarty, S.; Satyamoorthy, K. Mitochondrial DNA copy number variation—A potential biomarker for early onset preeclampsia. Pregnancy Hypertens. 2021, 23, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cushen, S.C.; Ricci, C.A.; Bradshaw, J.L.; Silzer, T.; Blessing, A.; Sun, J.; Zhou, Z.; Scroggins, S.M.; Santillan, M.K.; Santillan, D.A.; et al. Reduced Maternal Circulating Cell-Free Mitochondrial DNA Is Associated With the Development of Preeclampsia. J. Am. Heart Assoc. 2022, 11, e021726. [Google Scholar] [CrossRef]
- Marschalek, J.; Wohlrab, P.; Ott, J.; Wojta, J.; Speidl, W.; Klein, K.U.; Kiss, H.; Pateisky, P.; Zeisler, H.; Kuessel, L. Maternal serum mitochondrial DNA (mtDNA) levels are elevated in preeclampsia—A matched case-control study. Pregnancy Hypertens. 2018, 14, 195–199. [Google Scholar] [CrossRef]
- Qiu, C.; Hevner, K.; Enquobahrie, D.A.; Williams, M.A. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int. J. Mol. Epidemiol. Genet. 2012, 3, 237–244. [Google Scholar]
- McCarthy, C.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef] [Green Version]
- Westrate, L.M.; Drocco, J.A.; Martin, K.R.; Hlavacek, W.S.; MacKeigan, J.P. Mitochondrial morphological features are associated with fission and fusion events. PLoS ONE 2014, 9, e95265. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Guo, X.; Chen, R.; Feng, L. Downregulation of Mitofusin 2 in Placenta Is Related to Preeclampsia. Biomed Res. Int. 2016, 2016, 6323086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.J.; Marsboom, G.; Fang, Y.H.; Toth, P.T.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgado, S.S.; Salgado, M.K.R. Structural changes in pre-eclamptic and eclamptic placentas—An ultrastructural study. J. Coll. Physicians. Surg. Pak. 2011, 21, 482–486. [Google Scholar]
- Muralimanoharan, S.; Maloyan, A.; Mele, J.; Guo, C.; Myatt, L.G.; Myatt, L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 2012, 33, 816–823. [Google Scholar] [CrossRef] [Green Version]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Tsvirkun, D.V.; Vavina, O.V.; Khodzhaeva, Z.S.; Kan, N.E.; Menon, R.; Vysokikh, M.Y.; et al. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci. Rep. 2016, 6, 32410. [Google Scholar] [CrossRef] [Green Version]
- Yung, H.W.; Colleoni, F.; Dommett, E.; Cindrova-Davies, T.; Kingdom, J.; Murray, A.J.; Burton, G.J. Noncanonical mitochondrial unfolded protein response impairs placental oxidative phosphorylation in early-onset preeclampsia. Proc. Natl. Acad. Sci. USA 2019, 116, 18109–18118. [Google Scholar] [CrossRef] [Green Version]
- Ausman, J.; Abbade, J.; Ermini, L.; Farrell, A.; Tagliaferro, A.; Post, M.; Caniggia, I. Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell. Death Dis. 2018, 9, 298. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Benador, I.Y.; Petcherski, A.; Veliova, M.; Benavides, G.A.; Lagarrigue, S.; Caudal, A.; Vergnes, L.; Murphy, A.N.; Karamanlidis, G.; et al. A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 2020, 39, e104073. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell. Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef]
- Gupta, S.; Agarwal, A.; Sharma, R.K. The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet. Gynecol. Surv. 2005, 60, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiktor, H.; Kankofer, M.; Schmerold, I.; Dadak, A.; Lopucki, M.; Niedermuller, H. Oxidative DNA damage in placentas from normal and pre-eclamptic pregnancies. Virchows Arch. 2004, 445, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Llurba, E.; Gratacos, E.; Martin-Gallan, P.; Cabero, L.; Dominguez, C. A comprehensive study of oxidative stress and antioxidant status in preeclampsia and normal pregnancy. Free Radic. Biol. Med. 2004, 37, 557–570. [Google Scholar] [CrossRef]
- Aris, A.; Benali, S.; Ouellet, A.; Moutquin, J.M.; Leblanc, S. Potential biomarkers of preeclampsia: Inverse correlation between hydrogen peroxide and nitric oxide early in maternal circulation and at term in placenta of women with preeclampsia. Placenta 2009, 30, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Walsh, S.W. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta 1998, 19, 581–586. [Google Scholar] [CrossRef]
- Rafeeinia, A.; Tabandeh, A.; Khajeniazi, S.; Marjani, A.J. Serum copper, zinc and lipid peroxidation in pregnant women with preeclampsia in gorgan. Open Biochem. J. 2014, 8, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Shaker, O.G.; Sadik, N.A. Pathogenesis of preeclampsia: Implications of apoptotic markers and oxidative stress. Hum. Exp. Toxicol. 2013, 32, 1170–1178. [Google Scholar] [CrossRef]
- Padmini, E.; Lavanya, S.; Uthra, V. Preeclamptic placental stress and over expression of mitochondrial HSP70. Clin. Chem. Lab. Med. 2009, 47, 1073–1080. [Google Scholar] [CrossRef]
- Wiktor, H.; Kankofer, M. Superoxide dismutase activity in normal and preeclamptic placentas. Ginekol. Pol. 1998, 69, 915–918. [Google Scholar] [PubMed]
- Wu, F.; Tian, F.J.; Lin, Y. Oxidative Stress in Placenta: Health and Diseases. Biomed Res. Int. 2015, 2015, 293271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122 Pt 4, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Allaire, A.D.; Ballenger, K.A.; Wells, S.R.; McMahon, M.J.; Lessey, B.A. Placental apoptosis in preeclampsia. Obstet. Gynecol. 2000, 96, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.N.; Smith, S.C.; To, K.F.; Sahota, D.S.; Baker, P.N. Increased placental apoptosis in pregnancies complicated by preeclampsia. Am. J. Obstet. Gynecol. 2001, 184, 1249–1250. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, N.M.; Ferguson-Smith, A.C.; Burton, G.J. Syncytial knots (Tenney-Parker changes) in the human placenta: Evidence of loss of transcriptional activity and oxidative damage. Am. J. Pathol. 2013, 183, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Skepper, J.N.; Charnock-Jones, D.S.; Burton, G.J. Hypoxia-reoxygenation: A potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ. Res. 2002, 90, 1274–1281. [Google Scholar] [CrossRef]
- Sanchez-Aranguren, L.C.; Prada, C.E.; Riano-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [Green Version]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; LaMarca, B. Role of Mitochondrial Dysfunction and Reactive Oxygen Species in Mediating Hypertension in the Reduced Uterine Perfusion Pressure Rat Model of Preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef]
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengeller, Z.K. AP39, a Modulator of Mitochondrial Bioenergetics, Reduces Antiangiogenic Response and Oxidative Stress in Hypoxia-Exposed Trophoblasts: Relevance for Preeclampsia Pathogenesis. Am. J. Pathol. 2019, 189, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: An implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 2004, 145, 4838–4845. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zou, Y.; Ge, Z.; Zuo, Q.; Huang, S.Y.; Sun, L. A Role of sFlt-1 in Oxidative Stress and Apoptosis in Human and Mouse Pre-Eclamptic Trophoblasts. Biol. Reprod. 2015, 93, 73. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Allison, B.J.; Brain, K.L.; Patey, O.V.; Niu, Y.; Botting, K.J.; Ford, S.G.; Garrud, T.A.; Wooding, P.F.B.; Shaw, C.J.; et al. Chronic Hypoxia in Ovine Pregnancy Recapitulates Physiological and Molecular Markers of Preeclampsia in the Mother, Placenta, and Offspring. Hypertension 2022, 79, 1525–1535. [Google Scholar] [CrossRef]
- Peracoli, J.C.; Rudge, M.V.; Peracoli, M.T. Tumor necrosis factor-alpha in gestation and puerperium of women with gestational hypertension and pre-eclampsia. Am. J. Reprod. Immunol. 2007, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Raghupathy, R. Cytokines as key players in the pathophysiology of preeclampsia. Med. Princ. Pract. 2013, 22 (Suppl. 1), 8–19. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Fiers, W.; Beyaert, R.; Declercq, W.; Vandenabeele, P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 1999, 18, 7719–7730. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Walsh, S.W. TNF alpha concentrations and mRNA expression are increased in preeclamptic placentas. J. Reprod. Immunol. 1996, 32, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Weel, I.C.; Romao-Veiga, M.; Matias, M.L.; Fioratti, E.G.; Peracoli, J.C.; Borges, V.T.; Araujo, J.P., Jr.; Peracoli, M.T. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J. Reprod. Immunol. 2017, 123, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.T.; Cockrell, K.L.; Massey, M.B.; Bennett, W.A.; Granger, J.P. Tumor necrosis factor-alpha-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am. J. Hypertens. 2002, 15 Pt 1, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Feng, L.; Jia, J.; Chen, R.; Yu, J. Upregulation of VEGF by small activating RNA and its implications in preeclampsia. Placenta 2016, 46, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Nakhla, S.; Makris, A.; Hennessy, A. TNF-alpha inhibits trophoblast integration into endothelial cellular networks. Placenta 2011, 32, 241–246. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Guo, C.; Myatt, L.; Maloyan, A. Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int. J. Obes. 2015, 39, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, Y.C.; Zhang, Z.; Barila, G.; Green-Brown, A.; Elovitz, M.A.; Simmons, R.A. Intrauterine Inflammation Alters the Transcriptome and Metabolome in Placenta. Front. Physiol. 2020, 11, 592689. [Google Scholar] [CrossRef]
- Batchuluun, B.; Al Rijjal, D.; Prentice, K.J.; Eversley, J.A.; Burdett, E.; Mohan, H.; Bhattacharjee, A.; Gunderson, E.P.; Liu, Y.; Wheeler, M.B. Elevated Medium-Chain Acylcarnitines Are Associated With Gestational Diabetes Mellitus and Early Progression to Type 2 Diabetes and Induce Pancreatic beta-Cell Dysfunction. Diabetes 2018, 67, 885–897. [Google Scholar] [CrossRef] [Green Version]
- Odegard, R.A.; Vatten, L.J.; Nilsen, S.T.; Salvesen, K.A.; Austgulen, R. Risk factors and clinical manifestations of pre-eclampsia. BJOG 2000, 107, 1410–1416. [Google Scholar] [CrossRef]
- Redman, C. Pre-eclampsia: A complex and variable disease. Pregnancy Hypertens. 2014, 4, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Smith, S.K.; He, Y.; Day, K.A.; Licence, D.R.; Corps, A.N.; Lammoglia, R.; Charnock-Jones, D.S. A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol. Reprod. 1998, 59, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and pregnancy: The role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Abu-Raya, B.; Michalski, C.; Sadarangani, M.; Lavoie, P.M. Maternal Immunological Adaptation During Normal Pregnancy. Front. Immunol. 2020, 11, 575197. [Google Scholar] [CrossRef]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef]
- Kirkman, D.L.; Robinson, A.T.; Rossman, M.J.; Seals, D.R.; Edwards, D.G. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2080–H2100. [Google Scholar] [CrossRef]
- Ricci, C.A.; Reid, D.M.; Sun, J.; Santillan, D.A.; Santillan, M.K.; Goulopoulou, S.; Phillips, N.R. Mitochondria-mediated Maternal-fetal Interactions and Consequences of Mitochondrial Dysregulation Indicate New Roles for Mitochondria in Hypertensive Pregnancies. medRxiv 2021. [Google Scholar] [CrossRef]
- Sanchez-Aranguren, L.C.; Espinosa-Gonzalez, C.T.; Gonzalez-Ortiz, L.M.; Sanabria-Barrera, S.M.; Riano-Medina, C.E.; Nunez, A.F.; Ahmed, A.; Vasquez-Vivar, J.; Lopez, M. Soluble Fms-Like Tyrosine Kinase-1 Alters Cellular Metabolism and Mitochondrial Bioenergetics in Preeclampsia. Front. Physiol. 2018, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.; Vaka, V.R.; McMaster, K.M.; Wallace, K.; Cornelius, D.C.; Amaral, L.M.; Cunningham, M.W.; LaMarca, B. Vascular endothelial mitochondrial oxidative stress in response to preeclampsia: A role for angiotension II type 1 autoantibodies. Am. J. Obstet. Gynecol. MFM 2021, 3, 100275. [Google Scholar] [CrossRef] [PubMed]
- Duley, L. Pre-eclampsia and the hypertensive disorders of pregnancy. Br. Med. Bull. 2003, 67, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Uriho, A.; Tang, X.; Le, G.; Yang, S.; Harimana, Y.; Ishimwe, S.P.; Yiping, L.; Zhang, K.; Ma, S.; Muhoza, B. Effects of resveratrol on mitochondrial biogenesis and physiological diseases. Adv. Tradit. Med. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D. Resveratrol and Cardiovascular Diseases. Nutrients 2016, 8, 250. [Google Scholar] [CrossRef]
- Poudel, R.; Stanley, J.L.; Rueda-Clausen, C.F.; Andersson, I.J.; Sibley, C.P.; Davidge, S.T.; Baker, P.N. Effects of resveratrol in pregnancy using murine models with reduced blood supply to the uterus. PLoS ONE 2013, 8, e64401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cudmore, M.J.; Ramma, W.; Cai, M.; Fujisawa, T.; Ahmad, S.; Al-Ani, B.; Ahmed, A. Resveratrol inhibits the release of soluble fms-like tyrosine kinase (sFlt-1) from human placenta. Am. J. Obstet. Gynecol. 2012, 206, 253.e10–253.e15. [Google Scholar] [CrossRef]
- Ding, J.; Kang, Y.; Fan, Y.; Chen, Q. Efficacy of resveratrol to supplement oral nifedipine treatment in pregnancy-induced preeclampsia. Endocr. Connect. 2017, 6, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Giusti, L.; Tesi, M.; Ciregia, F.; Marselli, L.; Zallocco, L.; Suleiman, M.; De Luca, C.; Del Guerra, S.; Zuccarini, M.; Trerotola, M.; et al. The Protective Action of Metformin against Pro-Inflammatory Cytokine-Induced Human Islet Cell Damage and the Mechanisms Involved. Cells 2022, 11, 2465. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, S.G.; Kang, Y.J.; Choi, H.C. Metformin mitigates stress-induced premature senescence by upregulating AMPKalpha at Ser485 phosphorylation induced SIRT3 expression and inactivating mitochondrial oxidants. Mech. Ageing Dev. 2022, 206, 111708. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef]
- Feng, J.; Wang, X.; Ye, X.; Ares, I.; Lopez-Torres, B.; Martinez, M.; Martinez-Larranaga, M.R.; Wang, X.; Anadon, A.; Martinez, M.A. Mitochondria as an important target of metformin: The mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol. Res. 2022, 177, 106114. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Tuohey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1–356.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Cao, G.; Yi, W.; Li, L.; Cao, X. Effect of Metformin on a Preeclampsia-Like Mouse Model Induced by High-Fat Diet. Biomed Res. Int. 2019, 2019, 6547019. [Google Scholar] [CrossRef]
- Alqudah, A.; McKinley, M.C.; McNally, R.; Graham, U.; Watson, C.J.; Lyons, T.J.; McClements, L. Risk of pre-eclampsia in women taking metformin: A systematic review and meta-analysis. Diabet. Med. 2018, 35, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glueck, C.J.; Goldenberg, N.; Pranikoff, J.; Loftspring, M.; Sieve, L.; Wang, P. Height, weight, and motor-social development during the first 18 months of life in 126 infants born to 109 mothers with polycystic ovary syndrome who conceived on and continued metformin through pregnancy. Hum. Reprod. 2004, 19, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glueck, C.J.; Goldenberg, N.; Pranikoff, J.; Khan, Z.; Padda, J.; Wang, P. Effects of metformin-diet intervention before and throughout pregnancy on obstetric and neonatal outcomes in patients with polycystic ovary syndrome. Curr. Med. Res. Opin. 2013, 29, 55–62. [Google Scholar] [CrossRef]
- De Leo, V.; Musacchio, M.C.; Piomboni, P.; Di Sabatino, A.; Morgante, G. The administration of metformin during pregnancy reduces polycystic ovary syndrome related gestational complications. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 63–66. [Google Scholar] [CrossRef]
- Cluver, C.A.; Hiscock, R.; Decloedt, E.H.; Hall, D.R.; Schell, S.; Mol, B.W.; Brownfoot, F.; Kaitu’u-Lino, T.J.; Walker, S.P.; Tong, S. Use of metformin to prolong gestation in preterm pre-eclampsia: Randomised, double blind, placebo controlled trial. BMJ 2021, 374, n2103. [Google Scholar] [CrossRef]
- Thompson, J.D.; Diers, D. Management of nursing intensity. Nurs. Clin. N. Am. 1988, 23, 473–492. [Google Scholar] [CrossRef]
- Long, J.; Huang, Y.; Tang, Z.; Shan, Y.; Feng, D.; Wang, W.; Liu, J.; Huang, Y.; Gu, H.; Guo, D.; et al. Mitochondria Targeted Antioxidant Significantly Alleviates Preeclampsia Caused by 11beta-HSD2 Dysfunction via OPA1 and MtDNA Maintenance. Antioxidants 2022, 11, 1505. [Google Scholar] [CrossRef]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study, G. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Kirkman, D.L.; Muth, B.J.; Ramick, M.G.; Townsend, R.R.; Edwards, D.G. Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2018, 314, F423–F429. [Google Scholar] [CrossRef]
- Mason, S.A.; Wadley, G.D.; Keske, M.A.; Parker, L. Effect of mitochondrial-targeted antioxidants on glycaemic control, cardiovascular health, and oxidative stress in humans: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2022, 24, 1047–1060. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, P.; Zhu, F.; Liao, J.; Wu, Y.; Hu, M.; Fu, H.; Qiao, J.; Lin, L.; Huang, B.; et al. The Potent Antioxidant MitoQ Protects Against Preeclampsia During Late Gestation but Increases the Risk of Preeclampsia When Administered in Early Pregnancy. Antioxid. Redox Signal. 2021, 34, 118–136. [Google Scholar] [CrossRef]
- Ayla, S.; Seckin, I.; Tanriverdi, G.; Cengiz, M.; Eser, M.; Soner, B.C.; Oktem, G. Doxorubicin induced nephrotoxicity: Protective effect of nicotinamide. Int. J. Cell Biol. 2011, 2011, 390238. [Google Scholar] [CrossRef] [Green Version]
- Wahlberg, G.; Carlson, L.A.; Wasserman, J.; Ljungqvist, A. Protective effect of nicotinamide against nephropathy in diabetic rats. Diabetes Res. 1985, 2, 307–312. [Google Scholar]
- Rongvaux, A.; Andris, F.; Van Gool, F.; Leo, O. Reconstructing eukaryotic NAD metabolism. Bioessays 2003, 25, 683–690. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell. Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Fushima, T.; Oyanagi, G.; Townley-Tilson, H.W.; Sato, E.; Nakada, H.; Oe, Y.; Hagaman, J.R.; Wilder, J.; Li, M.; et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc. Natl. Acad. Sci. USA 2016, 113, 13450–13455. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Tian, Y.J.; Guo, J.; Sun, W.P.; Lun, Y.Z.; Guo, M.; Luo, N.; Cao, Y.; Cao, J.M.; Gong, X.J.; et al. Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br. J. Nutr. 2013, 110, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, Y.I.; Mahmoud, A.A. Role of nicotinamide (vitamin B3) in acetaminophen-induced changes in rat liver: Nicotinamide effect in acetaminophen-damged liver. Exp. Toxicol. Pathol. 2016, 68, 345–354. [Google Scholar] [CrossRef]
- Tian, Y.J.; Luo, N.; Chen, N.N.; Lun, Y.Z.; Gu, X.Y.; Li, Z.; Ma, Q.; Zhou, S.S. Maternal nicotinamide supplementation causes global DNA hypomethylation, uracil hypo-incorporation and gene expression changes in fetal rats. Br. J. Nutr. 2014, 111, 1594–1601. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.P.; Zhai, M.Z.; Li, D.; Zhou, Y.; Chen, N.N.; Guo, M.; Zhou, S.S. Comparison of the effects of nicotinic acid and nicotinamide degradation on plasma betaine and choline levels. Clin. Nutr. 2017, 36, 1136–1142. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Healthy Pregnancy | PE | ||||||
|---|---|---|---|---|---|---|---|
| Early | Late | Early-Onset/Severe | Late-Onset/Mild | ||||
| Content | ⇓ |
 | ⇑ | ⇑⇓ | ⇑= | ||
| Structure and dynamics |
|
|
|
| |||
| Function | Respiration and ATP generation | ⇓ |
| ⇑ | ⇑⇓ | ⇑= | |
| ROS | ⇓ |
| ⇑ | ⇑ | ⇑= | ||
| Antioxidant capacity | ⇓ |
| ⇑ | ⇑⇓ | ⇑ | ||
| Canonical PE | Immunological PE | Gestational Parent Driven PE | |
|---|---|---|---|
| Onset and severity of disease |
|
|
|
| Pathogenesis |
|
|
|
| Genes expressed in placenta |
|
|
|
| Placenta pathology |
|
|
|
| Potential risk factors |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahan, F.; Vasam, G.; Green, A.E.; Bainbridge, S.A.; Menzies, K.J. Placental Mitochondrial Function and Dysfunction in Preeclampsia. Int. J. Mol. Sci. 2023, 24, 4177. https://doi.org/10.3390/ijms24044177
Jahan F, Vasam G, Green AE, Bainbridge SA, Menzies KJ. Placental Mitochondrial Function and Dysfunction in Preeclampsia. International Journal of Molecular Sciences. 2023; 24(4):4177. https://doi.org/10.3390/ijms24044177
Chicago/Turabian StyleJahan, Fahmida, Goutham Vasam, Alex E. Green, Shannon A. Bainbridge, and Keir J. Menzies. 2023. "Placental Mitochondrial Function and Dysfunction in Preeclampsia" International Journal of Molecular Sciences 24, no. 4: 4177. https://doi.org/10.3390/ijms24044177
APA StyleJahan, F., Vasam, G., Green, A. E., Bainbridge, S. A., & Menzies, K. J. (2023). Placental Mitochondrial Function and Dysfunction in Preeclampsia. International Journal of Molecular Sciences, 24(4), 4177. https://doi.org/10.3390/ijms24044177