
Aggregation Limiting Cell-Penetrating Peptides Derived from Protein Signal Sequences

 , , , and
, , , and
Abstract
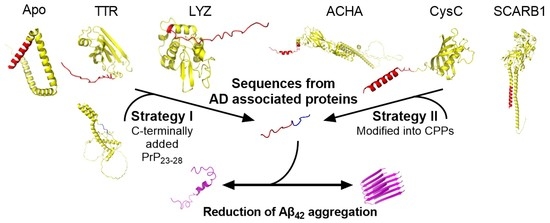
:
1. Introduction
2. Results and Discussion
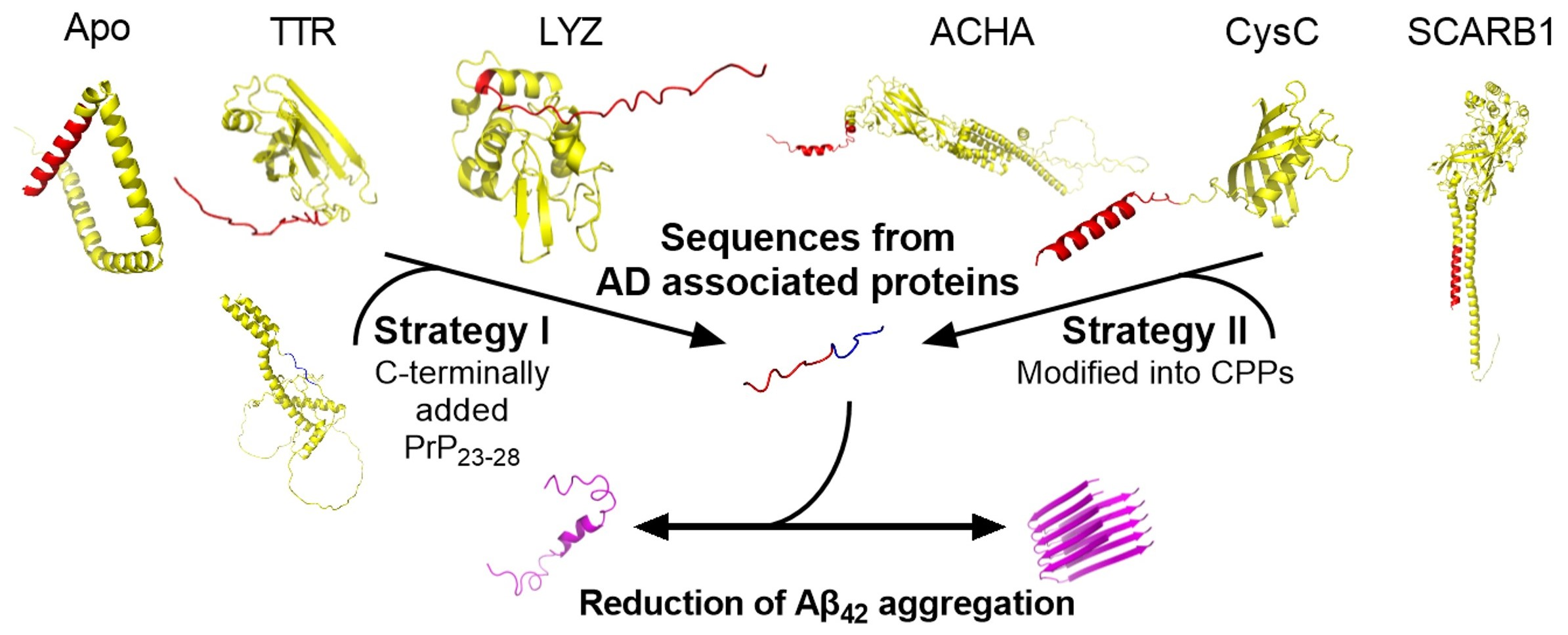
2.1. Peptide Design and Characterisation
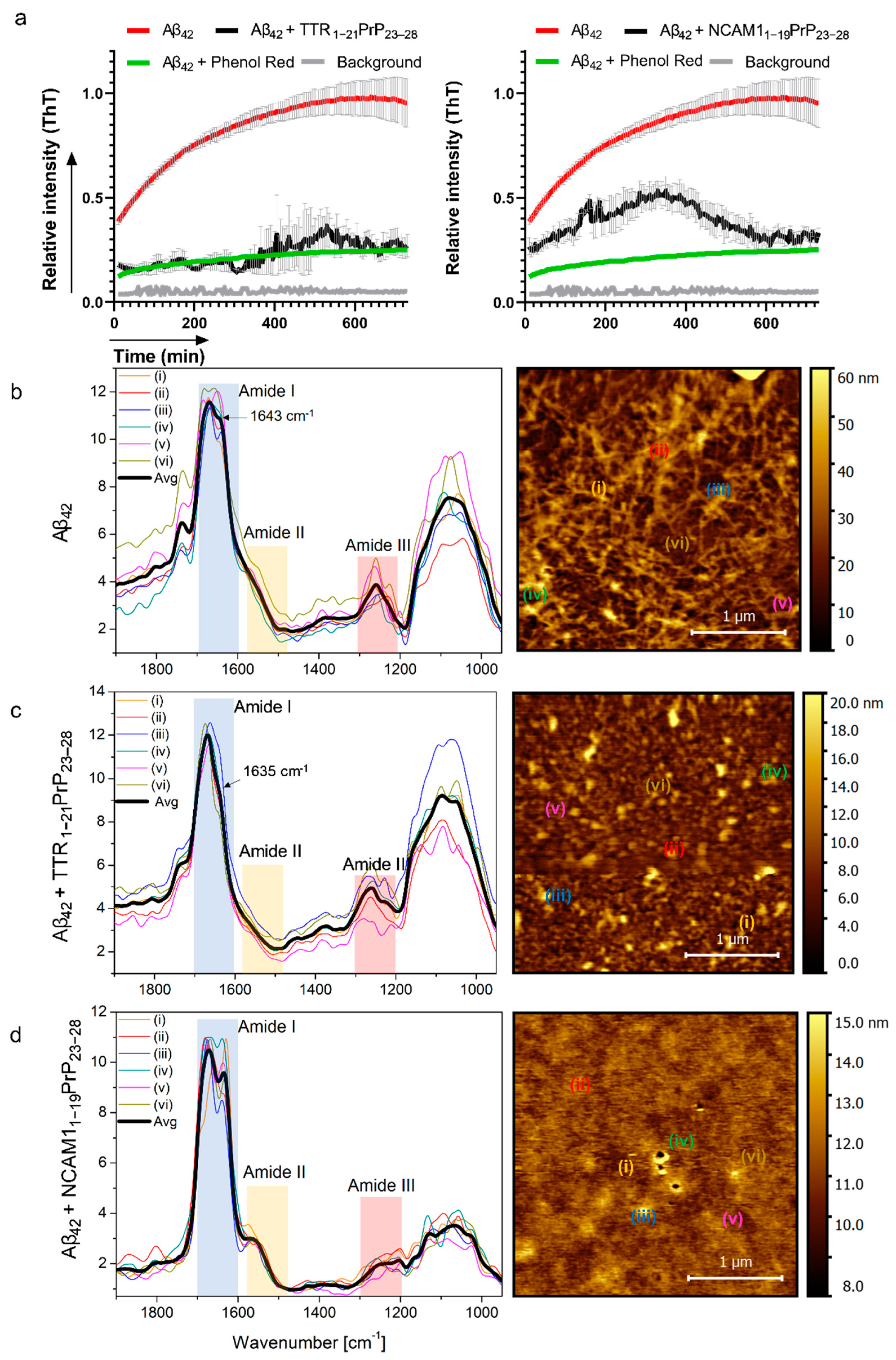
2.2. Interaction with Aβ and Reduction in Amyloid Beta Aggregation In Vitro
2.3. Reduction in Amyloid Aggregates in Cell Culture
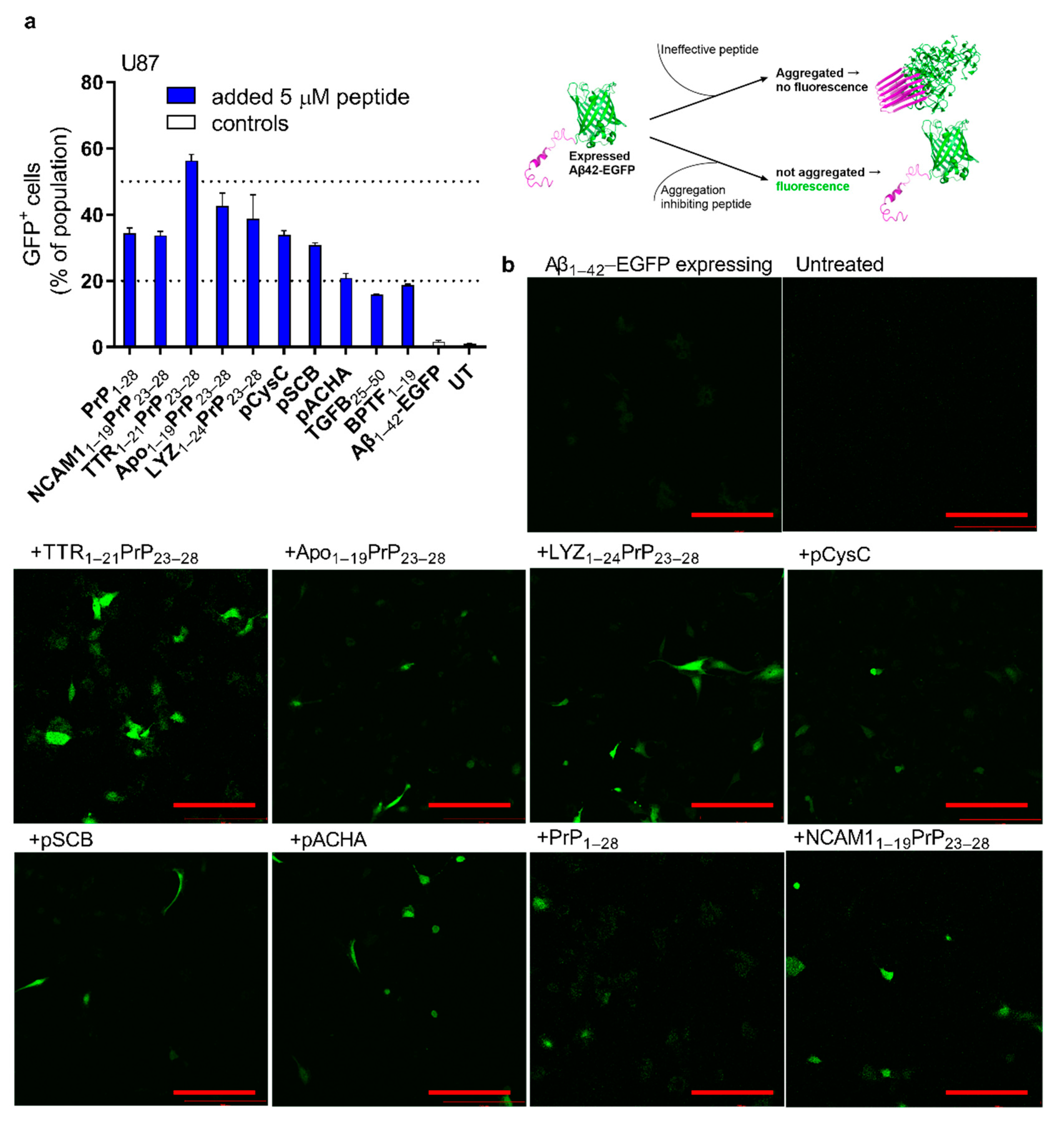
2.4. Reduction in Expressed Aβ Aggregation in Cells
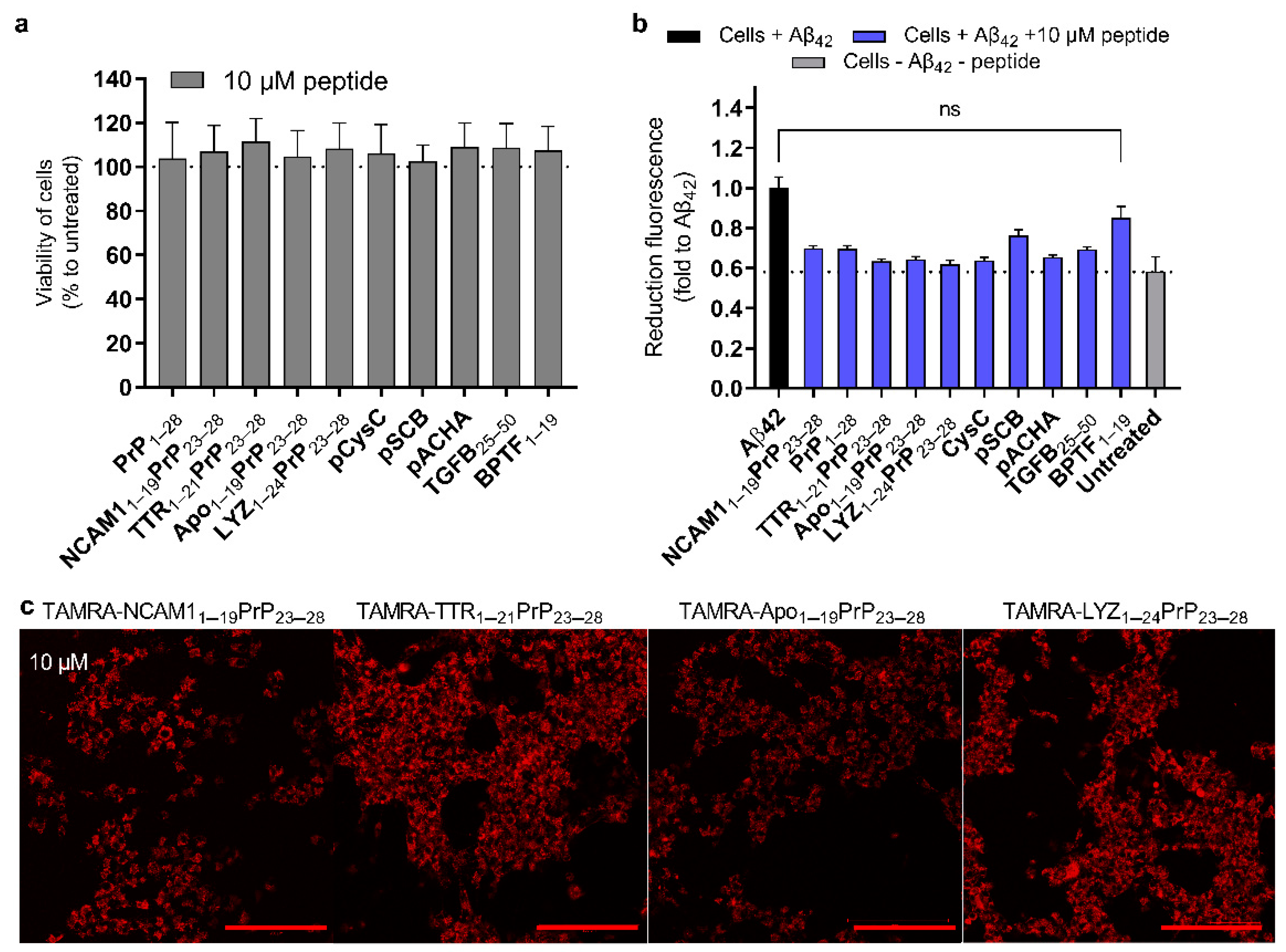
2.5. Reduction in Amyloid Toxicity and the Safety of Peptides to the Cells
2.6. The Chimeric Peptides Efficient in Mammalian Cell Test System Have CPP Properties
3. Materials and Methods
3.1. Vizualisation and Analysis
3.2. Predictions and Calculations
3.3. Peptide Synthesis
3.4. Secondary Structures of Peptides-Circular Dichroism (CD) Spectra
3.5. Nanoscale Characterization of Interactions and Reduction in Aβ42 Aggregation-AFM-IR, SAXS, and DLS
3.6. Reducing Aggregation of Aβ42-Thioflavin T Assay
3.7. Cell Culture Maintenance
3.8. Plasmids
3.9. Complex Formation for Transfection and Transfection
3.10. Detection of Aβ42 Aggregates on Cells-IHC
3.11. The Internalization of the Labelled Peptides and the Reduction in Aβ42 Aggregation Visualized on Live Cells Using Confocal Microscopy
3.12. Reduction in the Aβ42 Aggregation Assessed with Flow Cytometry
3.13. Cell Viability Detected with MTS
3.14. Proliferation of Cells Detected with CellTox Green Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.A.; Bartnik, D.; Glück, J.M.; Piorkowska, K.; Wiesehan, K.; Weber, U.; Gulyas, B.; Halldin, C.; Pfeifer, A.; Spenger, C.; et al. Development of a Small D-Enantiomeric Alzheimer’s Amyloid-β Binding Peptide Ligand for Future In Vivo Imaging Applications. PLoS ONE 2012, 7, e41457. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, M.H.; Ahmad, K.; Saeed, M.; Alharbi, A.M.; Barreto, G.E.; Ashraf, G.M.; Choi, I. Peptide based therapeutics and their use for the treatment of neurodegenerative and other diseases. Biomed. Pharmacother. 2018, 103, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Brinkmalm, G.; Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Sun, X.; Frosch, M.P.; Selkoe, D.J.; Portelius, E.; Zetterberg, H.; et al. Identification of neurotoxic cross-linked amyloid-β dimers in the Alzheimer’s brain. Brain 2019, 142, 1441–1457. [Google Scholar] [CrossRef]
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef] [Green Version]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Funke, S.A. Peptides for Therapy and Diagnosis of Alzheimer’s Disease. Curr. Pharm. Des. 2012, 18, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Król, S.; Österlund, N.; Vosough, F.; Jarvet, J.; Wärmländer, S.; Barth, A.; Ilag, L.L.; Magzoub, M.; Gräslund, A.; Mörman, C. The amyloid-inhibiting NCAM-PrP peptide targets Aβ peptide aggregation in membrane-mimetic environments. Iscience 2021, 24, 102852. [Google Scholar] [CrossRef]
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedrós, I.; Marin, M.; Olloquequi, J.; Camins, A. Una revisión de los avances en la terapéutica de la enfermedad de Alzheimer: Estrategia frente a la proteína β-amiloide. Neurología 2018, 33, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-β Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020, 21, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Anderson, D.H.; Liang, W.Y.; Chou, J.A.; Saelices, L. The inhibition of cellular toxicity of amyloid-β by dissociated transthyretin. J. Biol. Chem. 2020, 295, 14015–14024. [Google Scholar] [CrossRef] [PubMed]
- Cho, P.Y.; Joshi, G.; Johnson, J.A.; Murphy, R.M. Transthyretin-Derived Peptides as β-Amyloid Inhibitors. ACS Chem. Neurosci. 2014, 5, 542–551. [Google Scholar] [CrossRef]
- Pate, K.M.; Kim, B.J.; Shusta, E.V.; Murphy, R.M. Transthyretin Mimetics as Anti-β-Amyloid Agents: A Comparison of Peptide and Protein Approaches. Chemmedchem 2018, 13, 968–979. [Google Scholar] [CrossRef]
- Aono, M.; Bennett, E.; Kim, K.; Lynch, J.; Myers, J.; Pearlstein, R.; Warner, D.; Laskowitz, D. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-d-aspartate excitotoxicity in primary rat neuronal–glial cell cultures. Neuroscience 2003, 116, 437–445. [Google Scholar] [CrossRef]
- Li, F.-Q.; Sempowski, G.D.; McKenna, S.E.; Laskowitz, D.T.; Colton, C.A.; Vitek, M.P. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. Experiment 2006, 318, 956–965. [Google Scholar] [CrossRef]
- Yamauchi, K.; Tozuka, M.; Hidaka, H.; Nakabayashi, T.; Sugano, M.; Kondo, Y.; Nakagawara, A.; Katsuyama, T. Effect of apolipoprotein AII on the interaction of apolipoprotein E with beta-amyloid: Some apo(E-AII) complexes inhibit the internalization of beta-amyloid in cultures of neuroblastoma cells. J. Neurosci. Res. 2000, 62, 608–614. [Google Scholar] [CrossRef]
- Sarkar, G.; Curran, G.L.; Mahlum, E.; Decklever, T.; Wengenack, T.M.; Blahnik, A.; Hoesley, B.; Lowe, V.J.; Poduslo, J.F.; Jenkins, R.B. A carrier for non-covalent delivery of functional beta-galactosidase and antibodies against amyloid plaques and igm to the brain. PLoS ONE 2011, 6, e28881. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Shen, Q.; Pang, Y.; Li, X.; Chen, Y.; Wang, X.; Luo, X.; Wu, Z.; Bao, Z.; Zhang, J.; et al. The synthesized transporter K16APoE enabled the therapeutic HAYED peptide to cross the blood-brain barrier and remove excess iron and radicals in the brain, thus easing Alzheimer’s disease. Drug Deliv. Transl. Res. 2018, 9, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Shi, C.; Di Lorenzo, D.; Van Baelen, A.-C.; Tonali, N. The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules 2020, 25, 2439. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.T.S.; Gräslund, A.; Abrahams, J.P. Human lysozyme inhibits the in vitro aggregation of Aβ peptides, which in vivo are associated with Alzheimer’s disease. Chem. Commun. 2013, 49, 6507–6509. [Google Scholar] [CrossRef]
- Sandin, L.; Bergkvist, L.; Nath, S.; Kielkopf, C.; Janefjord, C.; Helmfors, L.; Zetterberg, H.; Blennow, K.; Li, H.; Nilsberth, C.; et al. Beneficial effects of increased lysozyme levels in Alzheimer’s disease modelled in Drosophila melanogaster. FEBS J. 2016, 283, 3508–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmfors, L.; Boman, A.; Civitelli, L.; Nath, S.; Sandin, L.; Janefjord, C.; McCann, H.; Zetterberg, H.; Blennow, K.; Halliday, G.; et al. Protective properties of lysozyme on β-amyloid pathology: Implications for Alzheimer disease. Neurobiol. Dis. 2015, 83, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, M.; Mehrnejad, F. Molecular Insight into Human Lysozyme and Its Ability to Form Amyloid Fibrils in High Concentrations of Sodium Dodecyl Sulfate: A View from Molecular Dynamics Simulations. PLoS ONE 2016, 11, e0165213. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Levy, E. Cystatin C in Alzheimer’s disease. Front. Mol. Neurosci. 2012, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Deng, A.; Irizarry, M.C.; Nitsch, R.M.; Growdon, J.H.; Rebeck, G.W. Elevation of cystatin c in susceptible neurons in alzheimer’s disease. Am. J. Pathol. 2001, 159, 1061–1068. [Google Scholar] [CrossRef]
- Wilkinson, K.; El Khoury, J. Microglial scavenger receptors and their roles in the pathogenesis of alzheimer’s disease. Int. J. Alzheimer’s Dis. 2012, 2012, 489456. [Google Scholar] [CrossRef] [Green Version]
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Chiba, T.; Yamada, M.; Nawa, M.; Kanekura, K.; Suzuki, H.; Terashita, K.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Transforming growth factor β2 is a neuronal death-inducing ligand for amyloid-β precursor protein. Mol. Cell. Biol. 2005, 25, 9304–9317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, M.R.; Nagele, R.G.; Wang, H.-Y.; Peterson, P.A.; Lee, D.H.S. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 2001, 38, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Conejero-Goldberg, C.; Davies, P.; Ulloa, L. Alpha7 nicotinic acetylcholine receptor: A link between inflammation and neurodegeneration. Neurosci. Biobehav. Rev. 2008, 32, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The cellular prion protein traps Alzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, K.L.; Guterstam, P.; Langel, Ü.; Gräslund, A. Targeting prion propagation using peptide constructs with signal sequence motifs. Arch. Biochem. Biophys. 2014, 564, 254–261. [Google Scholar] [CrossRef]
- Oglęcka, K.; Lundberg, P.; Magzoub, M.; Eriksson, L.G.; Langel, Ü.; Gräslund, A. Relevance of the N-terminal NLS-like sequence of the prion protein for membrane perturbation effects. Biochim. Biophys. Acta (BBA)—Biomembr. 2008, 1778, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Löfgren, K.; Wahlström, A.; Lundberg, P.; Langel, Ö.; Gräslund, A.; Bedecs, K. Antiprion properties of prion protein-derived cell-penetrating peptides. FASEB J. 2008, 22, 2177–2184. [Google Scholar] [CrossRef]
- Henning-Knechtel, A.; Kumar, S.; Wallin, C.; Król, S.; Wärmländer, S.K.T.S.; Jarvet, J.; Esposito, G.; Kirmizialtin, S.; Gräslund, A.; Hamilton, A.D. Designed cell-penetrating peptide inhibitors of amyloid-beta aggregation and cytotoxicity. Cell Rep. Phys. Sci. 2020, 1, 100014. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Yu, S. Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, F.S.; Longo, G.; Faggiano, S.; Lipiec, E.; Pastore, A.; Dietler, G. Infrared nanospectroscopy characterization of oligomeric and fibrillar aggregates during amyloid formation. Nat. Commun. 2015, 6, 7831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mello, L.; Hamley, I.W.; Castelletto, V.; Garcia, B.B.M.; Han, S.W.; De Oliveira, C.L.P.; Da Silva, E.R. Nanoscopic Structure of Complexes Formed between DNA and the Cell-Penetrating Peptide Penetratin. J. Phys. Chem. B 2019, 123, 8861–8871. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Mie, M.; Andou, T.; Mihara, H.; Kobatake, E. Fluorescent and luminescent fusion proteins for analyses of amyloid beta peptide aggregation. J. Pept. Sci. 2017, 23, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Hussein, R.M.; Hashem, R.M.; Rashed, L.A. Evaluation of the amyloid beta-GFP fusion protein as a model of amyloid beta peptides-mediated aggregation: A study of DNAJB6 chaperone. Front. Mol. Neurosci. 2015, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikkanen, R.; Herzog, V.; Schmitz, A. Cytosolic and nuclear aggregation of the amyloid beta-peptide following its expression in the endoplasmic reticulum. Histochem Cell Biol. 2002, 118, 353–360. [Google Scholar] [CrossRef]
- Ochiishi, T.; Doi, M.; Yamasaki, K.; Hirose, K.; Kitamura, A.; Urabe, T.; Hattori, N.; Kinjo, M.; Ebihara, T.; Shimura, H. Development of new fusion proteins for visualizing amyloid-β oligomers in vivo. Sci. Rep. 2016, 6, 22712. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Hällbrink, M.; Kilk, K.; Elmquist, A.; Lundberg, P.; Lindgren, M.; Jiang, Y.; Pooga, M.; Soomets, U.; Langel, Ü. Prediction of Cell-Penetrating Peptides. Int. J. Pept. Res. Ther. 2005, 11, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A.; Open Source Drug Discovery Consortium; Raghava, G.P.S. In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Patra, M.C. MLCPP 2.0: An Updated Cell-penetrating Peptides and Their Uptake Efficiency Predictor. J. Mol. Biol. 2022, 434, 167604. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. The camsol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015, 427, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Amery, L.; Ekizoglou, S.; Vendruscolo, M.; Popovic, B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci. Rep. 2017, 7, 8200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, P., Jr.; Gerald, D.F. Chapter 9: Chou-Fasman Prediction of the Secondary Structure of Proteins: The Chou-Fasman-Prevelige Algorithm. In Prediction of Protein Structure and the Principles of Protein Conformation; Gerald, D.F., Ed.; Plenum: New York, NY, USA, 1989; pp. 391–416. ISBN 0306431319. [Google Scholar]
- Garnier, J.; Osguthorpe, D.; Robson, B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978, 120, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Qian, N.; Sejnowski, T.J. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol. 1988, 202, 865–884. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Garg, A.; Keizer, D.; Pires, D.E.V.; Ascher, D.B. CSM-peptides: A computational approach to rapid identification of therapeutic peptides. Protein Sci. 2022, 31, e4442. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. Peptide Toxicity Prediction. In Computational Peptidology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 1268, pp. 143–157. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, K.; Kumar, R.; Singh, S.; Tuknait, A.; Gautam, A.; Mathur, D.; Anand, P.; Varshney, G.C.; Raghava, G.P.S. A Web Server and Mobile App for Computing Hemolytic Potency of Peptides. Sci. Rep. 2016, 6, 22843. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Q.; Li, B.; Lu, C.; Yang, S.; Long, J.; He, B.; Chen, H.; Huang, J. BBPpredict: A Web Service for Identifying Blood-Brain Barrier Penetrating Peptides. Front. Genet. 2022, 13, 845747. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mello, L.R.; Porosk, L.; Lourenço, T.C.; Garcia, B.B.M.; Costa, C.A.R.; Han, S.W.; de Souza, J.S.; Langel, U.; da Silva, E.R. Amyloid-like Self-Assembly of a Hydrophobic Cell-Penetrating Peptide and Its Use as a Carrier for Nucleic Acids. ACS Appl. Bio. Mater. 2021, 4, 6404–6416. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Sun, W.; Shao, Y.; Wei, F.; Zhang, X.; Wang, W.; Li, P. Recent development of PeakForce Tapping mode atomic force microscopy and its applications on nanoscience. Nanotechnol. Rev. 2018, 7, 605–621. [Google Scholar] [CrossRef]
- Arukuusk, P.; Pärnaste, L.; Oskolkov, N.; Copolovici, D.-M.; Margus, H.; Padari, K.; Möll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New generation of efficient peptide-based vectors, NickFects, for the delivery of nucleic acids. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Peptide Sequence 1,2 | Net Charge 3 | Mw 4 |
|---|---|---|---|
| PrP1–28 | MANLGYWLLALFVTMWTDVGLCKKRPKP | 3.9 | 3253 |
| NCAM11–19PrP23–28 | MLRTKDLIWTLFFLGTAVSKKRPKP | 6 | 2946 |
| TTR1–21PrP23–28 | MASLRLFLLCLAGLVFVSEAGKKRPKP | 4.9 | 2944 |
| Apo1–19PrP23–28 | MKLLAMVALLVTICSLEGAKKRPKP | 4.9 | 2712 |
| LYZ1–24PrP23–28 | MKALIVLGLVLLSVTVQGKVFERCKKRPKP | 6.9 | 3352 |
| pCysC | MASPLRSLLFLLAVLAVAWAATPKQGPRKK | 6 | 3235 |
| pSCB | MGGSSRARWVALGLGALGLLFAAKKRA | 6 | 2759 |
| pACHA | MCGRRGGIWLALAAALLHVSLQRRPK | 6 | 2875 |
| TGFB25–50 | STLDMDQFMRKRIEAIRGQILSKLKL | 4 | 3089 |
| BPTF1–19 | MRGRRGRPPKQPAAPAAER | 6 | 2085 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porosk, L.; Härk, H.H.; Bicev, R.N.; Gaidutšik, I.; Nebogatova, J.; Armolik, E.-J.; Arukuusk, P.; da Silva, E.R.; Langel, Ü. Aggregation Limiting Cell-Penetrating Peptides Derived from Protein Signal Sequences. Int. J. Mol. Sci. 2023, 24, 4277. https://doi.org/10.3390/ijms24054277
Porosk L, Härk HH, Bicev RN, Gaidutšik I, Nebogatova J, Armolik E-J, Arukuusk P, da Silva ER, Langel Ü. Aggregation Limiting Cell-Penetrating Peptides Derived from Protein Signal Sequences. International Journal of Molecular Sciences. 2023; 24(5):4277. https://doi.org/10.3390/ijms24054277
Chicago/Turabian StylePorosk, Ly, Heleri Heike Härk, Renata Naporano Bicev, Ilja Gaidutšik, Jekaterina Nebogatova, Eger-Jasper Armolik, Piret Arukuusk, Emerson Rodrigo da Silva, and Ülo Langel. 2023. "Aggregation Limiting Cell-Penetrating Peptides Derived from Protein Signal Sequences" International Journal of Molecular Sciences 24, no. 5: 4277. https://doi.org/10.3390/ijms24054277
APA StylePorosk, L., Härk, H. H., Bicev, R. N., Gaidutšik, I., Nebogatova, J., Armolik, E.-J., Arukuusk, P., da Silva, E. R., & Langel, Ü. (2023). Aggregation Limiting Cell-Penetrating Peptides Derived from Protein Signal Sequences. International Journal of Molecular Sciences, 24(5), 4277. https://doi.org/10.3390/ijms24054277