
Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
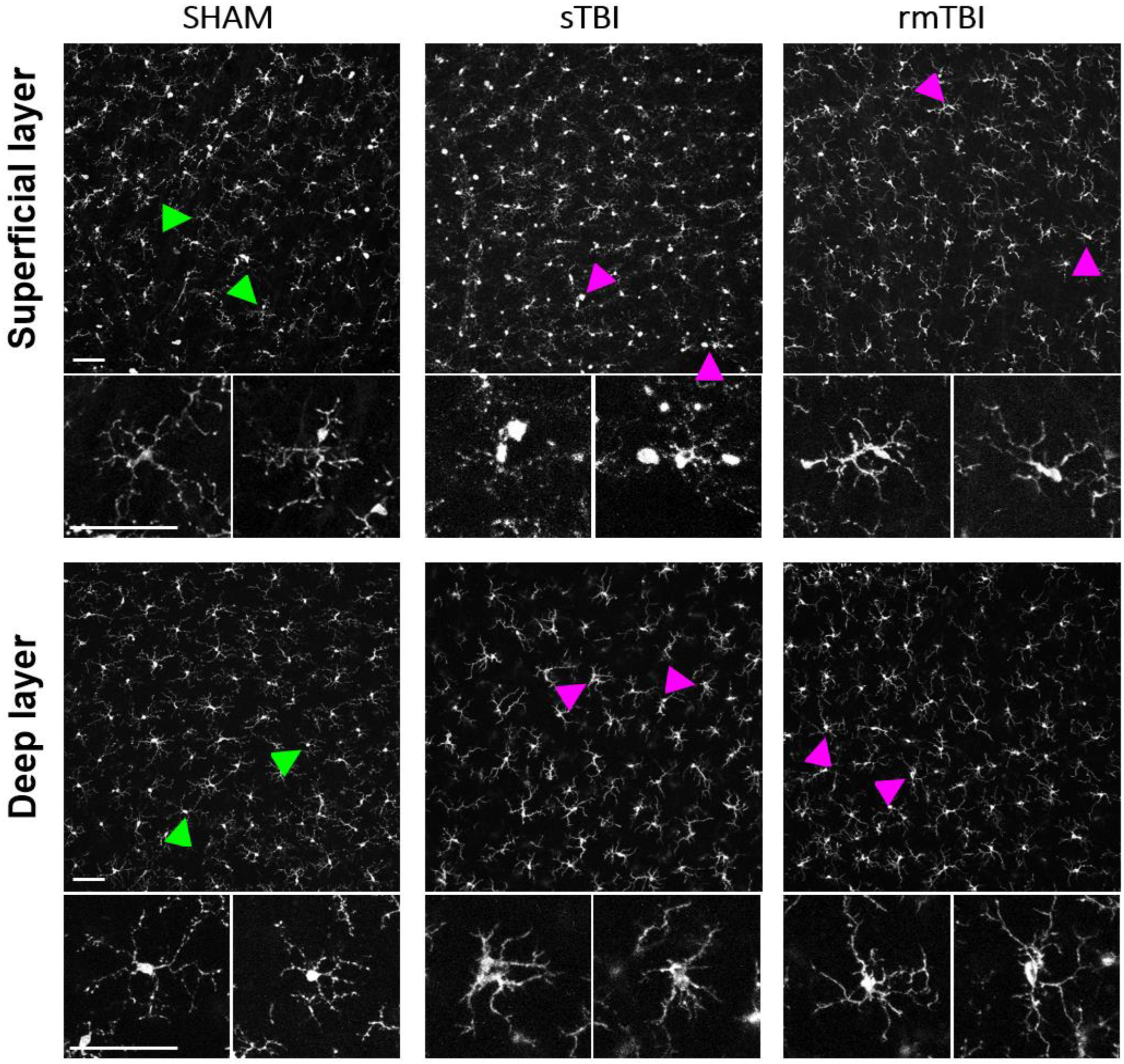
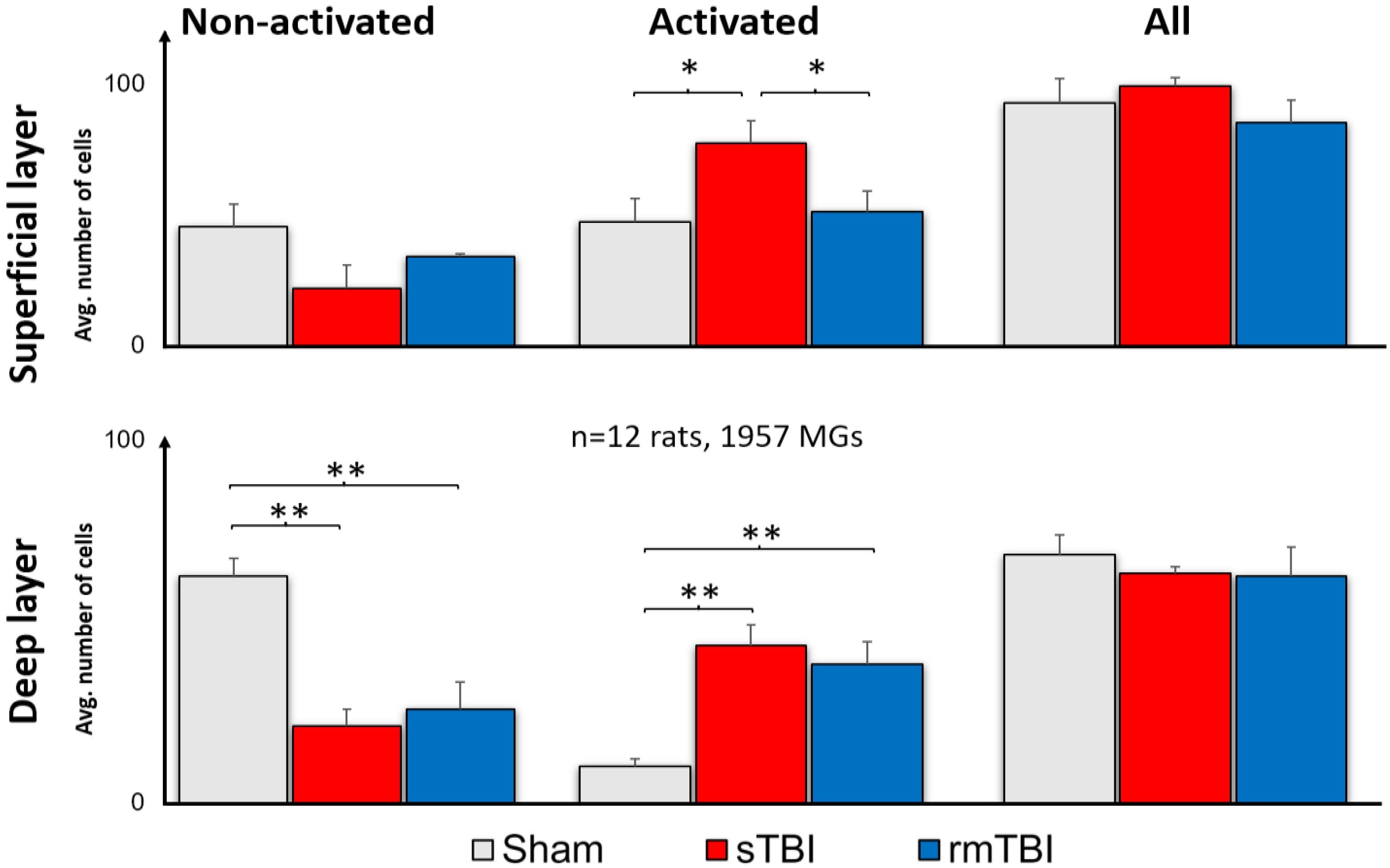
2.1. Effects of TBI on Retinal Microglia
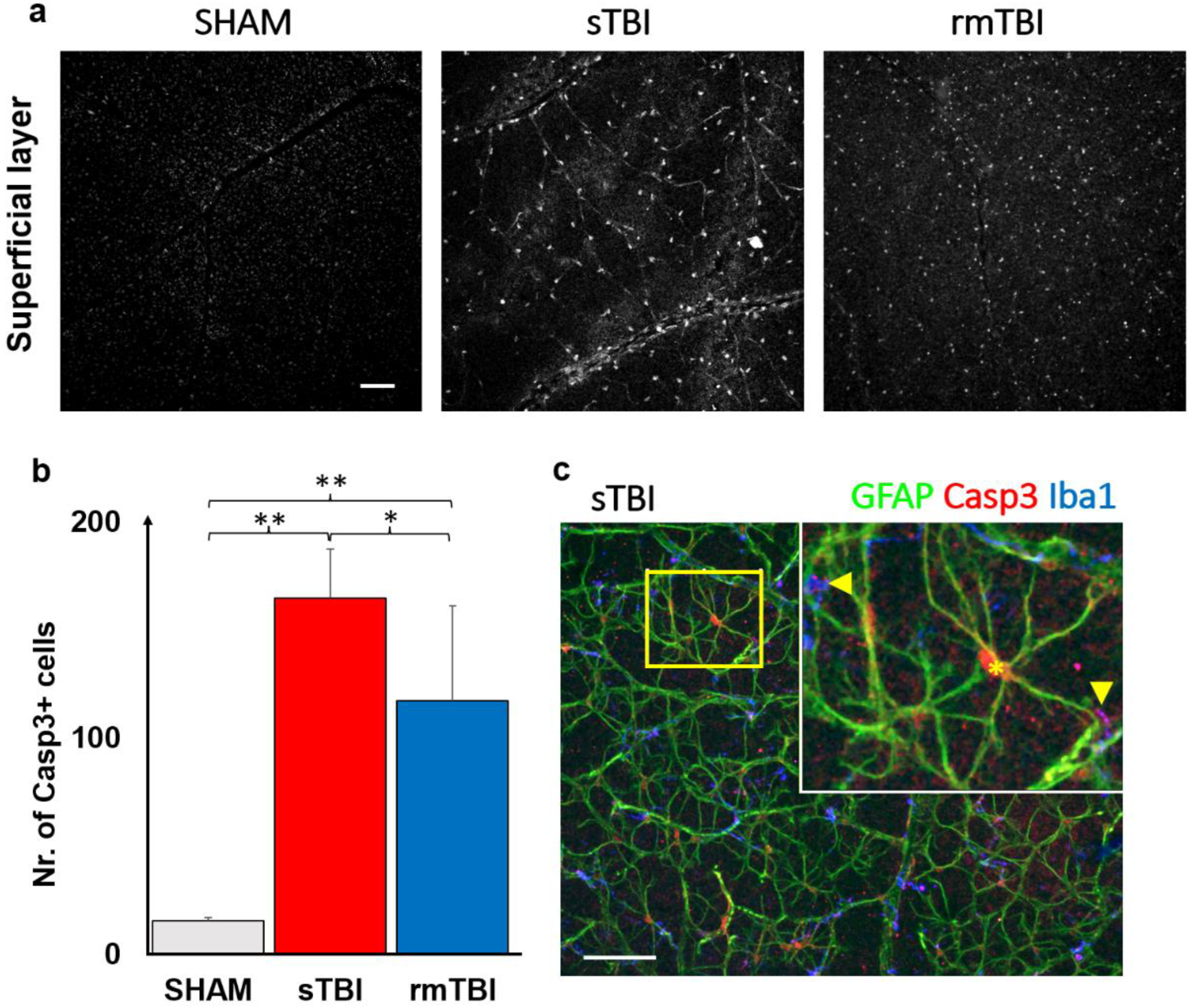
2.2. Specific Caspase3 Activation due to Traumatic Brain Injury in the Superficial Layer of the Retina
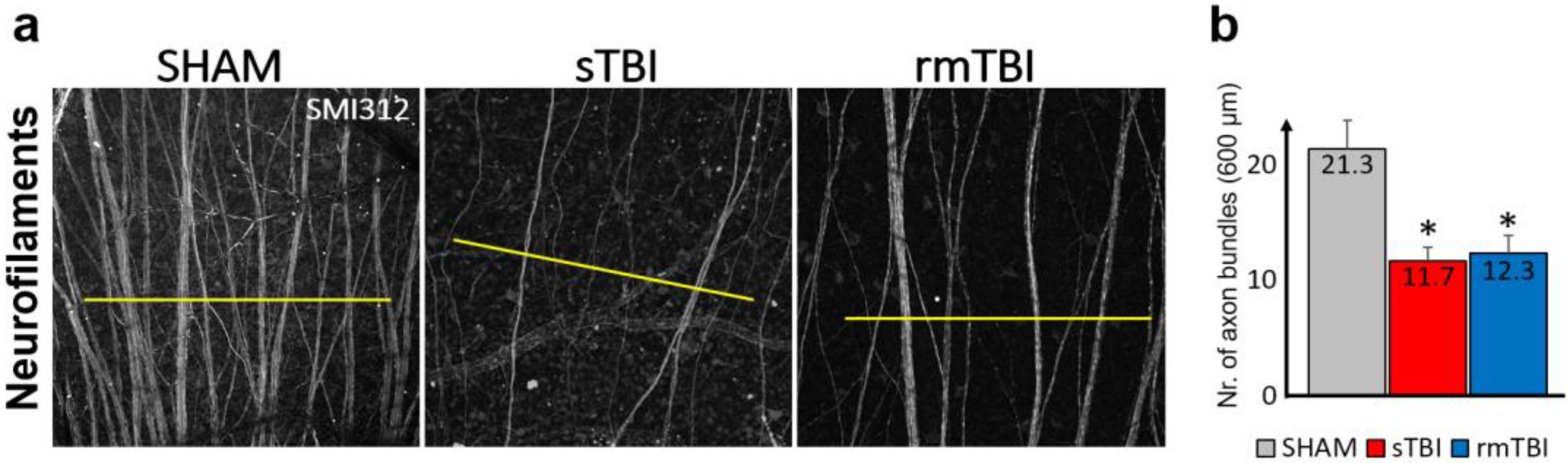
2.3. Loss of Axonal Connections due to Traumatic Brain Injury in the Neurofilament Layer of the Retina
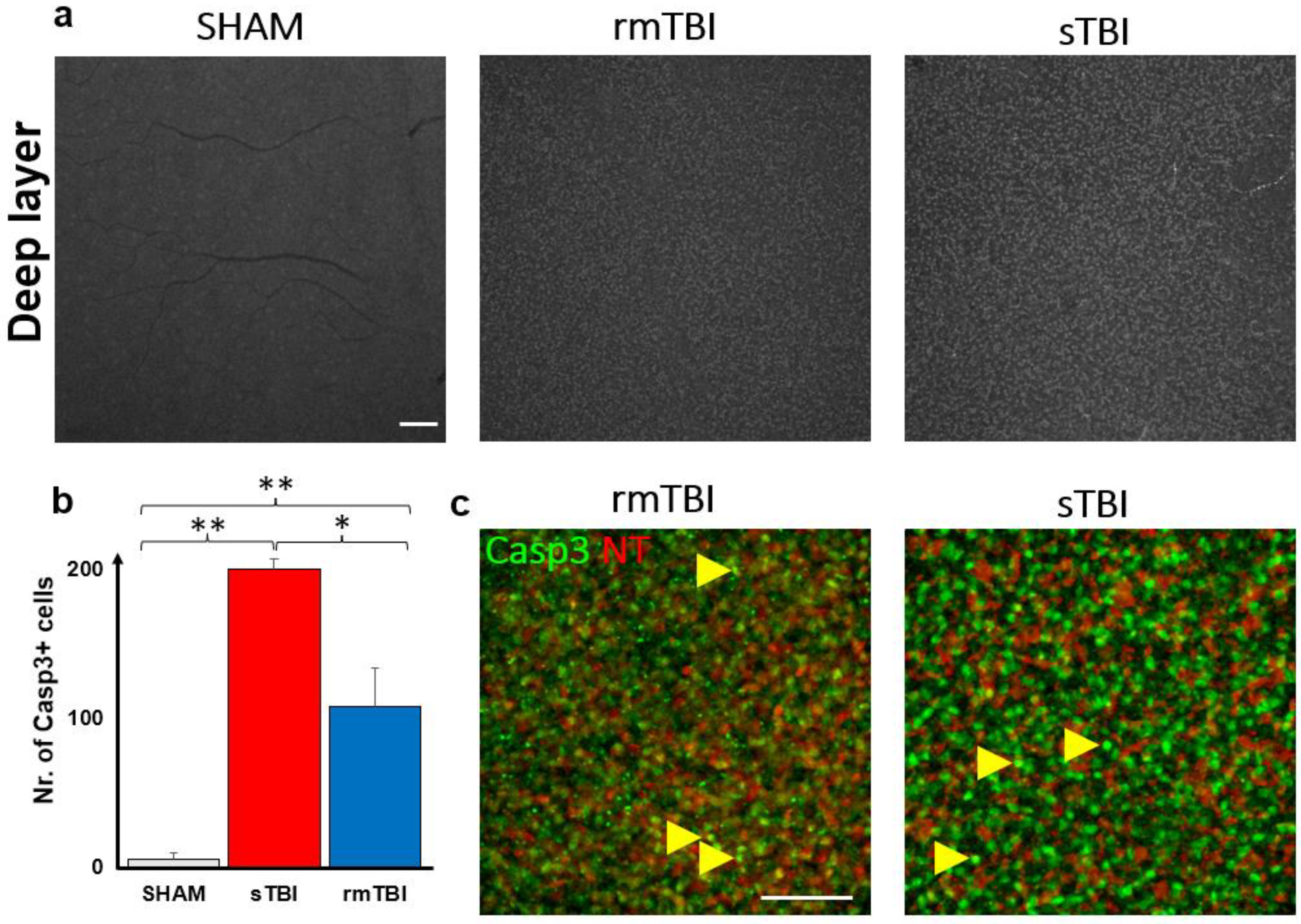
2.4. Caspase3 Is Activated due to Traumatic Brain Injury in the DL of the Retina
3. Discussion
3.1. Microglial Activation due to Traumatic Brain Injury
3.2. Caspase3 Activation, Cell Death Marker
3.3. Fate of Different Cell Types under the Influence of TBI
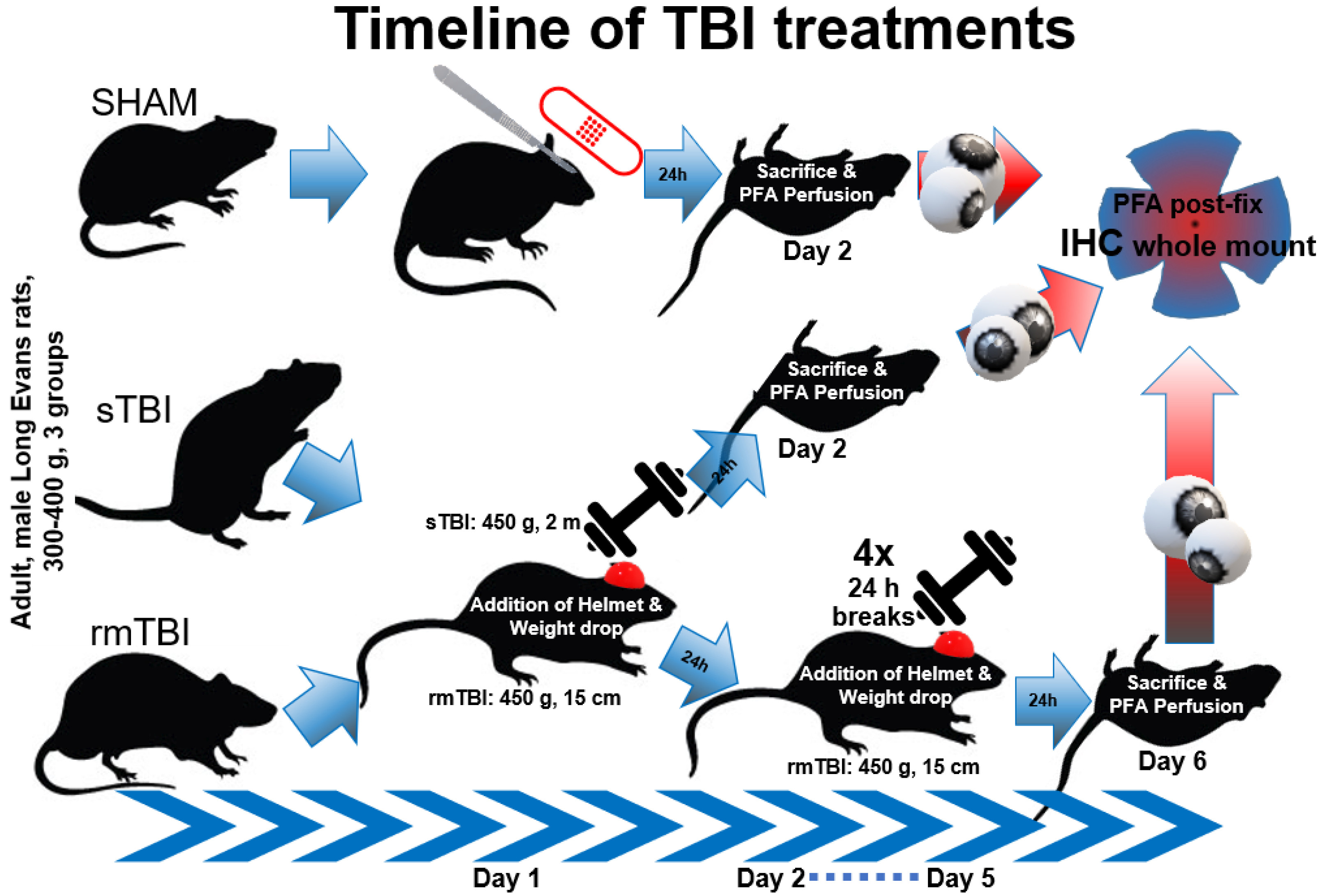
4. Materials and Methods
4.1. Animals and Preparation
4.2. Immunohistochemistry
4.3. Microscopy
4.4. Measurement of Microglial and Casp3 Activation
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orff, H.J.; Ayalon, L.; Drummond, S.P.A. Traumatic Brain Injury and Sleep Disturbance: A Review of Current Research. J. Head Trauma Rehabil. 2009, 24, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Jennekens, N.; de Casterlé, B.D.; Dobbels, F. A Systematic Review of Care Needs of People with Traumatic Brain Injury (TBI) on a Cognitive, Emotional and Behavioural Level. J. Clin. Nurs. 2010, 19, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.A.; Close, J.C.T. Traumatic Brain Injury in Older Adults: Characteristics, Causes and Consequences. Injury 2012, 43, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Hardy, J.; Zetterberg, H. Neurological Consequences of Traumatic Brain Injuries in Sports. Mol. Cell. Neurosci. 2015, 66, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.N.; Smoliga, J.M. Physical Activity Intolerance and Cardiorespiratory Dysfunction in Patients with Moderate-to-Severe Traumatic Brain Injury. Sports Med. 2019, 49, 1183–1198. [Google Scholar] [CrossRef]
- de Souza, R.L.; Thais, M.E.; Cavallazzi, G.; Diaz, A.P.; Schwarzbold, M.L.; Nau, A.L.; Rodrigues, G.M.; Souza, D.S.; Hohl, A.; Walz, R. Side of Pupillary Mydriasis Predicts the Cognitive Prognosis in Patients with Severe Traumatic Brain Injury. Acta Anaesthesiol. Scand. 2015, 59, 392–405. [Google Scholar] [CrossRef]
- van Dijck, J.T.J.M.; Bartels, R.H.M.A.; Lavrijsen, J.C.M.; Ribbers, G.M.; Kompanje, E.J.O.; Peul, W.C. The Patient with Severe Traumatic Brain Injury: Clinical Decision-Making: The First 60 min and Beyond. Curr. Opin. Crit. Care 2019, 25, 622–629. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, X.; Kleiven, S. Biomechanics of Periventricular Injury. J. Neurotrauma 2020, 37, 1074–1090. [Google Scholar] [CrossRef]
- Mckee, A.C.; Daneshvar, D.H. The Neuropathology of Traumatic Brain Injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [CrossRef]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Das, M.; Tang, X.; Mohapatra, S.S.; Mohapatra, S. Vision Impairment after Traumatic Brain Injury: Present Knowledge and Future Directions. Rev. Neurosci. 2019, 30, 305–315. [Google Scholar] [CrossRef]
- Ciuffreda, K.J.; Kapoor, N.; Rutner, D.; Suchoff, I.B.; Han, M.E.; Craig, S. Occurrence of Oculomotor Dysfunctions in Acquired Brain Injury: A Retrospective Analysis. Optometry 2007, 78, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Choi, S.; Bikkannavar, P.; Cordeiro, M.F. Microglia: Key Players in Retinal Ageing and Neurodegeneration. Front. Cell. Neurosci. 2022, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, S.; Heinrich, A.; Biber, K. The Brain’s Best Friend: Microglial Neurotoxicity Revisited. Front. Cell. Neurosci. 2013, 7, 71. [Google Scholar] [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia Drive APOE-Dependent Neurodegeneration in a Tauopathy Mouse Model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Clark, R.S.B.; Kochanek, P.M.; Watkins, S.C.; Chen, M.; Dixon, C.E.; Seidberg, N.A.; Melick, J.; Loeffert, J.E.; Nathaniel, P.D.; Jin, K.L.; et al. Caspase-3 Mediated Neuronal Death After Traumatic Brain Injury in Rats. J. Neurochem. 2000, 74, 740–753. [Google Scholar] [CrossRef]
- Balogh, B.; Szarka, G.; Tengölics, Á.J.; Hoffmann, G.; Völgyi, B.; Kovács-öller, T. Led-induced Microglial Activation and Rise in Caspase3 Suggest a Reorganization in the Retina. Int. J. Mol. Sci. 2021, 22, 10418. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase Signalling Controls Microglia Activation and Neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Eyolfson, E.; Khan, A.; Mychasiuk, R.; Lohman, A.W. Microglia Dynamics in Adolescent Traumatic Brain Injury. J. Neuroinflamm. 2020, 17, 1–19. [Google Scholar] [CrossRef]
- Mcilwain, D.R.; Berger, T.; Mak, T.W.; Baehrecke, E.H.; Green, D.R.; Kornbluth, S.; Salvesen, G.S. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Bruera, M.G.; Benedetto, M.M.; Guido, M.E.; Degano, A.L.; Contin, M.A. Glial Cell Response to Constant Low Light Exposure in Rat Retina. Vis. Neurosci. 2022, 39, E005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, G.; Ishimoto, S.-I.; Pararajasegaram, G.; Rao, N.A. Expression of Major Histocompatibility Complex Molecules in Rodent Retina. Immunohistochemical Study. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1848–1857. [Google Scholar]
- Tsukamoto, Y.; Omi, N. Classification of Mouse Retinal Bipolar Cells: Type-Specific Connectivity with Special Reference to Rod-Driven AII Amacrine Pathways. Front. Neuroanat. 2017, 11, 92. [Google Scholar] [CrossRef]
- Liu, J.H.; Singh, J.B.; Veruki, M.L.; Hartveit, E. Morphological Properties of the Axon Initial Segment-like Process of AII Amacrine Cells in the Rat Retina. J. Comp. Neurol. 2021, 529, 3593–3620. [Google Scholar] [CrossRef]
- Seabrook, T.A.; Burbridge, T.J.; Crair, M.C.; Huberman, A.D. Architecture, Function, and Assembly of the Mouse Visual System. Annu. Rev. Neurosci. 2017, 40, 499–538. [Google Scholar] [CrossRef]
- Lukas, T.J.; Wang, A.L.; Yuan, M.; Neufeld, A.H. Early Cellular Signaling Responses to Axonal Injury. Cell Commun. Signal. 2009, 7, 1–16. [Google Scholar] [CrossRef]
- Mannix, R.; Monuteaux, M.C.; Schutzman, S.A.; Meehan, W.P.; Nigrovic, L.E.; Neuman, M.I. Isolated Skull Fractures: Trends in Management in US Pediatric Emergency Departments. Ann. Emerg. Med. 2013, 62, 327–331. [Google Scholar] [CrossRef]
- Rasiah, P.K.; Geier, B.; Jha, K.A.; Gangaraju, R. Visual Deficits after Traumatic Brain Injury. Histol. Histopathol. 2021, 36, 711–724. [Google Scholar] [CrossRef]
- Fehily, B.; Fitzgerald, M. Repeated Mild Traumatic Brain Injury. Cell Transplant. 2017, 26, 1131–1155. [Google Scholar] [CrossRef]
- Singaravelu, J.; Zhao, L.; Fariss, R.N.; Nork, T.M.; Wong, W.T. Microglia in the Primate Macula: Specializations in Microglial Distribution and Morphology with Retinal Position and with Aging. Brain Struct. Funct. 2017, 222, 2759–2771. [Google Scholar] [CrossRef]
- Honig, M.G.; del Mar, N.A.; Henderson, D.L.; O’Neal, D.; Doty, J.B.; Cox, R.; Li, C.; Perry, A.M.; Moore, B.M.; Reiner, A. Raloxifene Modulates Microglia and Rescues Visual Deficits and Pathology After Impact Traumatic Brain Injury. Front. Neurosci. 2021, 15, 1274. [Google Scholar] [CrossRef] [PubMed]
- Childs, C.; Barker, L.A.; Gage, A.; Loosemore, M. Investigating Possible Retinal Biomarkers of Head Trauma in Olympic Boxers Using Optical Coherence Tomography. Eye Brain 2018, 10, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Oller, T.; Ivanova, E.; Bianchimano, P.; Sagdullaev, B.T. The Pericyte Connectome: Spatial Precision of Neurovascular Coupling Is Driven by Selective Connectivity Maps of Pericytes and Endothelial Cells and Is Disrupted in Diabetes. Cell Discov. 2020, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Hammer, D.X.; Agrawal, A.; Villanueva, R.; Saeedi, O.; Liu, Z. Label-Free Adaptive Optics Imaging of Human Retinal Macrophage Distribution and Dynamics. Proc. Natl. Acad. Sci. USA 2020, 117, 30661–30669. [Google Scholar] [CrossRef]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia Activation as a Biomarker for Traumatic Brain Injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Gao, L.; Fan, X.; Xu, H. Friend or Foe? Resident Microglia vs Bone Marrow-Derived Microglia and Their Roles in the Retinal Degeneration. Mol. Neurobiol. 2016, 54, 4094–4112. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Mechanisms Mediating Caspase Activation in Cell Death. Cell Death Differ. 1999, 6, 1060–1066. [Google Scholar] [CrossRef]
- Hengartner, M.O. The Biochemistry of Apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Knoblach, S.M.; Nikolaeva, M.; Huang, X.; Fan, L.; Krajewski, S.; Reed, J.C.; Faden, A.I. Multiple Caspases Are Activated after Traumatic Brain Injury: Evidence for Involvement in Functional Outcome. J. Neurotrauma 2004, 19, 1155–1170. [Google Scholar] [CrossRef]
- Glushakov, A.O.; Glushakova, O.Y.; Korol, T.Y.; Acosta, S.A.; Borlongan, C.V.; Valadka, A.B.; Hayes, R.L.; Glushakov, A.V. Chronic Upregulation of Cleaved-Caspase-3 Associated with Chronic Myelin Pathology and Microvascular Reorganization in the Thalamus after Traumatic Brain Injury in Rats. Int. J. Mol. Sci. 2018, 19, 3151. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Salvesen, G.S. Caspase Activation. Biochem. Soc. Symp. 2003, 70, 233–242. [Google Scholar] [CrossRef]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of Caspase Activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Sun, H.; Guo, W.Y. Astrocyte Polarization in Glaucoma: A New Opportunity. Neural. Regen. Res. 2022, 17, 2582. [Google Scholar] [CrossRef] [PubMed]
- Szabo, E.; Patko, E.; Vaczy, A.; Molitor, D.; Csutak, A.; Toth, G.; Reglodi, D.; Atlasz, T. Retinoprotective Effects of PACAP Eye Drops in Microbead-Induced Glaucoma Model in Rats. Int. J. Mol. Sci. 2021, 22, 8825. [Google Scholar] [CrossRef]
- Fusz, K.; Kovács-öller, T.; Kóbor, P.; Szabó-Meleg, E.; Völgyi, B.; Buzás, P.; Telkes, I. Regional Variation of Gap Junctional Connections in the Mammalian Inner Retina. Cells 2021, 10, 2396. [Google Scholar] [CrossRef]
- Cardona, S.M.; Mendiola, A.S.; Yang, Y.C.; Adkins, S.L.; Torres, V.; Cardona, A.E. Disruption of Fractalkine Signaling Leads to Microglial Activation and Neuronal Damage in the Diabetic Retina. ASN Neuro. 2015, 7, 1759091415608204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fox, M.A.; Povlishock, J.T. Diffuse Traumatic Axonal Injury in the Optic Nerve Does Not Elicit Retinal Ganglion Cell Loss. J. Neuropathol. Exp. Neurol. 2013, 72, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, K.; Wang, Z.; Chen, G. Progress of Research on Diffuse Axonal Injury after Traumatic Brain Injury. Neural Plast. 2016, 2016, 9746313. [Google Scholar] [CrossRef]
- Klimo, K.R.; Stern-Green, E.A.; Shelton, E.; Day, E.; Jordan, L.; Robich, M.; Racine, J.; McDaniel, C.E.; VanNasdale, D.A.; Yuhas, P.T. Structure and Function of Retinal Ganglion Cells in Subjects with a History of Repeated Traumatic Brain Injury. Front. Neurol. 2022, 13, 1779. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–Neuron Interactions in the Mammalian Retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Kavanagh, E.; Rodhe, J.; Burguillos, M.A.; Venero, J.L.; Joseph, B. Regulation of Caspase-3 Processing by CIAP2 Controls the Switch between pro-Inflammatory Activation and Cell Death in Microglia. Cell Death Dis. 2014, 5, e1565. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Hoek, R.M.; Vos, R.M.; Aartsen, W.M.; Klimczak, R.R.; Hoyng, S.A.; Flannery, J.G.; Wijnholds, J. Specific Tools for Targeting and Expression in Müller Glial Cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14009. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, Y.; Slater, B.J.; Miller, N.R.; Bernstein, S.L. Axonal Degeneration, Regeneration and Ganglion Cell Death in a Rodent Model of Anterior Ischemic Optic Neuropathy (RAION). Exp. Eye Res. 2010, 91, 286–292. [Google Scholar] [CrossRef]
- Tan, H.; Li, X.; Huang, K.; Luo, M.; Wang, L. Morphological and Distributional Properties of SMI-32 Immunoreactive Ganglion Cells in the Rat Retina. J. Comp. Neurol. 2022, 530, 1276–1287. [Google Scholar] [CrossRef]
- Tadepalli, S.A.; Bali, Z.K.; Bruszt, N.; Nagy, L.V.; Amrein, K.; Fazekas, B.; Büki, A.; Czeiter, E.; Hernádi, I. Long-Term Cognitive Impairment without Diffuse Axonal Injury Following Repetitive Mild Traumatic Brain Injury in Rats. Behav. Brain Res. 2020, 378, 112268. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A.; Abd-Elfattah Foda, M.A.; van den Brink, W.; Campbell, J.; Kita, H.; Demetriadou, K. A New Model of Diffuse Brain Injury in Rats: Part I: Pathophysiology and Biomechanics. J. Neurosurg. 1994, 80, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, N.; Hammamieh, R.; Gautam, A.; Miller, S.A.; Condlin, M.L.; Jett, M.; Scrimgeour, A.G. TBI Weight-Drop Model with Variable Impact Heights Differentially Perturbs Hippocampus-Cerebellum Specific Transcriptomic Profile. Exp. Neurol. 2021, 335, 113516. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Meaney, D.F.; Cullen, D.K.; Smith, D.H. Animal Models of Traumatic Brain Injury. Handb. Clin. Neurol. 2015, 127, 115–128. [Google Scholar] [CrossRef]
- Büchele, F.; Morawska, M.M.; Schreglmann, S.R.; Penner, M.; Muser, M.; Baumann, C.R.; Noain, D. Novel Rat Model of Weight Drop-Induced Closed Diffuse Traumatic Brain Injury Compatible with Electrophysiological Recordings of Vigilance States. J. Neurotrauma 2016, 33, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Kovács-Öller, T.; Szarka, G.; Tengölics, Á.J.; Ganczer, A.; Balogh, B.; Szabó-Meleg, E.; Nyitrai, M.; Völgyi, B. Spatial Expression Pattern of the Major Ca2+-Buffer Proteins in Mouse Retinal Ganglion Cells. Cells 2020, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive Microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Arshadi, C.; Günther, U.; Eddison, M.; Harrington, K.I.S.; Ferreira, T.A. SNT: A Unifying Toolbox for Quantification of Neuronal Anatomy. Nat. Methods 2021, 18, 374–377. [Google Scholar] [CrossRef]
- Green, T.R.F.; Murphy, S.M.; Rowe, R.K. Comparisons of Quantitative Approaches for Assessing Microglial Morphology Reveal Inconsistencies, Ecological Fallacy, and a Need for Standardization. Sci. Rep. 2022, 12, 18196. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; De Groef, L. Characterizing Microglia Activation: A Spatial Statistics Approach to Maximize Information Extraction. Sci. Rep. 2017, 7, 1576. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Secondary Antibodies, Dyes | ||||||
|---|---|---|---|---|---|---|---|
| Name | Dilution | Source | Code | Name | Dilution | Source | Code |
| ms-SMI312 | 1:1000 | Calbiochem | NE1022/NE1023 | anti-ms-Alexa488 | 1:1000 | Invitrogen | A11017 |
| gp-Iba1 | 1:2000 | SySy | 234004 | anti-ms-Alexa647 | 1:1000 | Invitrogen | A21237 |
| rb-Caspase-3 | 1:1000 | NovusBio | AF835 | anti-gp-Alexa647 | 1:1000 | Invitrogen | A21450 |
| anti-rb-Cy3 | 1:500 | Jackson | 715-165-150 | ||||
| Non-Activated (Naïve) Morphology | Activated Morphology |
|---|---|
| Small, round soma | Enlarged, disorganized soma |
| Low soma to surroundings ratio | Soma to surroundings ratio increases |
| No amoeboid or leaf-like appendages are found | Amoeboid and leaf-like structures |
| Sporadic occurrence of act-MGs | Aggregate occurrence of act-MGs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács-Öller, T.; Zempléni, R.; Balogh, B.; Szarka, G.; Fazekas, B.; Tengölics, Á.J.; Amrein, K.; Czeiter, E.; Hernádi, I.; Büki, A.; et al. Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina. Int. J. Mol. Sci. 2023, 24, 4451. https://doi.org/10.3390/ijms24054451
Kovács-Öller T, Zempléni R, Balogh B, Szarka G, Fazekas B, Tengölics ÁJ, Amrein K, Czeiter E, Hernádi I, Büki A, et al. Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina. International Journal of Molecular Sciences. 2023; 24(5):4451. https://doi.org/10.3390/ijms24054451
Chicago/Turabian StyleKovács-Öller, Tamás, Renáta Zempléni, Boglárka Balogh, Gergely Szarka, Bálint Fazekas, Ádám J. Tengölics, Krisztina Amrein, Endre Czeiter, István Hernádi, András Büki, and et al. 2023. "Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina" International Journal of Molecular Sciences 24, no. 5: 4451. https://doi.org/10.3390/ijms24054451
APA StyleKovács-Öller, T., Zempléni, R., Balogh, B., Szarka, G., Fazekas, B., Tengölics, Á. J., Amrein, K., Czeiter, E., Hernádi, I., Büki, A., & Völgyi, B. (2023). Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina. International Journal of Molecular Sciences, 24(5), 4451. https://doi.org/10.3390/ijms24054451