
Gamma-Aminobutyric Acid Signaling in Damage Response, Metabolism, and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular Function of GABA in the Brain
3. Roles of GABA in Diverse Organ Systems
3.1. Liver
3.2. Liver Disease: Non-Alcoholic Fatty Liver Disease (NAFLD)
3.3. Liver Disease Symptom: Modulation of Fatty Acid Oxidation (FAO)
3.4. Liver Disease Symptom: Activation of Oxidative Stress
3.5. Liver Disease Symptom: Upregulation of Inflammation
3.6. Liver Disease Symptom: Misregulation of ER Stress and Insulin Signaling
3.7. Liver Disease Symptom: Ferroptosis
3.8. Liver Disease Symptom: Induction of DNA Damage
3.9. Liver Cancer, GABA, and DNA Damage
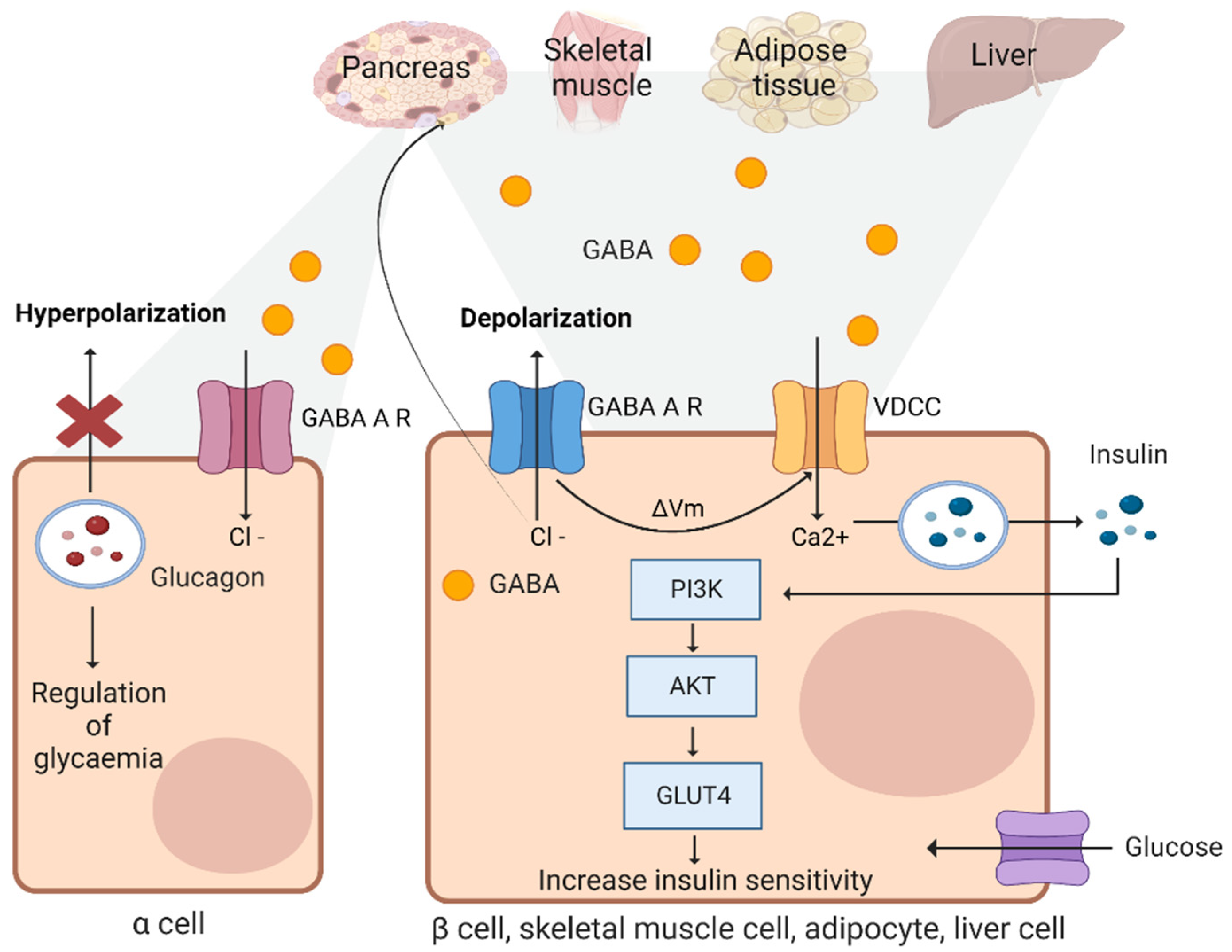
3.10. Beta Cells
3.11. Adipose Tissues and Skeletal Muscles
3.12. Clinical Perspective
4. GABA Metabolism
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lee, S.E.; Lee, Y.; Lee, G.H. The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Arch. Pharm. Res. 2019, 42, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yada, N.; Hagiwara, S.; Sakurai, T.; Kitano, M.; Kudo, M. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Mager, S.; Kleinberger-Doron, N.; Keshet, G.I.; Davidson, N.; Kanner, B.I.; Lester, H.A. Ion binding and permeation at the GABA transporter GAT1. J. Neurosci. 1996, 16, 5405–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltani, N.; Qiu, H.; Aleksic, M.; Glinka, Y.; Zhao, F.; Liu, R.; Li, Y.; Zhang, N.; Chakrabarti, R.; Ng, T.; et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci. USA 2011, 108, 11692–11697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petroff, O.A. GABA and glutamate in the human brain. Neuroscientist 2002, 8, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Chiodi, C.G.; Baptista-Hon, D.T.; Hunter, W.N.; Hales, T.G. Amino acid substitutions in the human homomeric β3 GABAA receptor that enable activation by GABA. J. Biol. Chem. 2019, 294, 2375–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mermer, F.; Poliquin, S.; Rigsby, K.; Rastogi, A.; Shen, W.; Romero-Morales, A.; Nwosu, G.; McGrath, P.; Demerast, S.; Aoto, J.; et al. Common molecular mechanisms of SLC6A1 variant-mediated neurodevelopmental disorders in astrocytes and neurons. Brain 2021, 144, 2499–2512. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Frøland, A.S.; Pettersen, M.N.; Storm-Mathisen, J.; Gundersen, V.; Rise, F.; Hassel, B. Propionate enters GABAergic neurons, inhibits GABA transaminase, causes GABA accumulation and lethargy in a model of propionic acidemia. Biochem. J. 2018, 475, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Porteous, R.; Herbison, A.E. Dynamics of GnRH Neuron Ionotropic GABA and Glutamate Synaptic Receptors Are Unchanged during Estrogen Positive and Negative Feedback in Female Mice. eNeuro 2017, 4, ENEURO.0259–17.2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoTurco, J.J.; Owens, D.F.; Heath, M.J.; Davis, M.B.; Kriegstein, A.R. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 1995, 15, 1287–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Han, W.; Tian, Q.; Li, Y.; Lu, W. Activity- and sleep-dependent regulation of tonic inhibition by Shisa7. Cell Rep. 2021, 34, 108899. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABAA and GABAB Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef] [PubMed]
- Kahanovitch, U.; Berlin, S.; Dascal, N. Collision coupling in the GABAB receptor-G protein-GIRK signaling cascade. FEBS Lett. 2017, 591, 2816–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunekawa, T.; Banno, R.; Yaginuma, H.; Taki, K.; Mizoguchi, A.; Sugiyama, M.; Onoue, T.; Takagi, H.; Hagiwara, D.; Ito, Y.; et al. GABAB Receptor Signaling in the Mesolimbic System Suppresses Binge-like Consumption of a High-Fat Diet. iScience 2019, 20, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Sun, L.; Tu, L. GABAB Receptor-Mediated PI3K/Akt Signaling Pathway Alleviates Oxidative Stress and Neuronal Cell Injury in a Rat Model of Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nan, S.; Zhang, Y.; Fan, J. Effects of GABAB receptor positive allosteric modulator BHF177 and IRS-1 on apoptosis of hippocampal neurons in rats with refractory epilepsy via the PI3K/Akt pathway. Cell Biol. Int. 2022, 46, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xiang, Y.Y.; Zhu, J.; Yi, F.; Li, J.; Liu, C.; Lu, W.Y. Protective roles of hepatic GABA signaling in acute liver injury of rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G208–G218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastgerdi, A.H.; Sharifi, M.; Soltani, N. GABA administration improves liver function and insulin resistance in offspring of type 2 diabetic rats. Sci. Rep. 2021, 11, 23155. [Google Scholar] [CrossRef] [PubMed]
- Sohrabipour, S.; Sharifi, M.R.; Talebi, A.; Sharifi, M.; Soltani, N. GABA dramatically improves glucose tolerance in streptozotocin-induced diabetic rats fed with high-fat diet. Eur. J. Pharmacol. 2018, 826, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Rehman, F.; Hori, T.; Nguyen, J.H. GABA, γ-Aminobutyric Acid, Protects Against Severe Liver Injury. J. Surg. Res. 2019, 236, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, J.; Hu, J.; He, H.; Wei, Y.; Ji, L.; Ma, X. Gama-aminobutyric acid (GABA) alleviates hepatic inflammation via GABA receptors/TLR4/NF-κB pathways in growing-finishing pigs generated by super-multiparous sows. Anim. Nutr. 2022, 9, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Galbo, T.; Shulman, G.I. Lipid-induced hepatic insulin resistance. Aging 2013, 5, 582–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, C.E.; Ghimire, S.; Hepler, C.; Miller, K.E.; Bruggink, S.M.; Kentch, K.P.; Higgins, M.R.; Banek, C.T.; Yoshino, J.; Klein, S.; et al. Hepatocyte membrane potential regulates serum insulin and insulin sensitivity by altering hepatic GABA release. Cell Rep. 2021, 35, 109298. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.E.; Ghimire, S.; Bruggink, S.M.; Miller, K.E.; Weninger, S.N.; Kronenfeld, J.M.; Yoshino, J.; Klein, S.; Duca, F.A.; Renquist, B.J. A critical role of hepatic GABA in the metabolic dysfunction and hyperphagia of obesity. Cell Rep. 2021, 35, 109301. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Funaba, M.; Matsui, T. Steatohepatitis is developed by a diet high in fat, sucrose, and cholesterol without increasing iron concentration in rat liver. Biol. Trace Elem. Res. 2016, 170, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Lanti, C.; Gatti, S.; Rametta, R.; Recalcati, S.; Maggioni, M.; Fracanzani, A.L.; Riso, P.; Cairo, G.; Fargion, S.; et al. High fat diet subverts hepatocellular iron uptake determining dysmetabolic iron overload. PLoS ONE 2015, 10, e0116855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Sun, W.; Qian, J.; Tang, Y. Fasting exacerbates hepatic growth differentiation factor 15 to promote fatty acid β-oxidation and ketogenesis via activating XBP1 signaling in liver. Redox Biol. 2018, 16, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Shi, Y.; Huang, C.; Huang, C.; Xu, P.; Zhou, C.; Liu, P.; Hu, R.; Zhuang, Y.; Li, G.; et al. Activation of AMP-activated protein kinase signaling pathway ameliorates steatosis in laying hen hepatocytes. Poult. Sci. 2021, 100, 100805. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Shimozono, R.; Asaoka, Y.; Yoshizawa, Y.; Aoki, T.; Noda, H.; Yamada, M.; Kaino, M.; Mochizuki, H. Nrf2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol. Pharmacol. 2013, 84, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kim, H.; Fu, Z.; Qiu, Y.; Yang, Z.; Wang, J.; Zhang, D.; Tong, X.; Yin, L.; Li, J.; et al. Deficiency of the Mitochondrial NAD Kinase Causes Stress-Induced Hepatic Steatosis in Mice. Gastroenterology 2018, 154, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Shi, Z.; Xie, C.; Gong, W.; Hu, Z.; Peng, Y. A novel mechanism of Gamma-aminobutyric acid (GABA) protecting human umbilical vein endothelial cells (HUVECs) against H2O2-induced oxidative injury. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2019, 217, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yu, R.; Zhou, Q.; Jiang, S.; Le, G. Protective effects of γ-aminobutyric acid against H2O2-induced oxidative stress in RIN-m5F pancreatic cells. Nutr. Metab. 2018, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, J.L.; Yoshimura, T.; Camporez, J.P.; Zhang, D.; Jornayvaz, F.R.; Kumashiro, N.; Guebre-Egziabher, F.; Jurczak, M.J.; Kahn, M.; Guigni, B.A.; et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 1869–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.H.; Bazuine, M.; Lumeng, C.N.; Geletka, L.M.; Mowers, J.; White, N.M.; Ma, J.T.; Zhou, J.; Qi, N.; Westcott, D.; et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009, 138, 961–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Han, Z.; Wu, Z.; Xia, Y.; Yang, G.; Yin, Y.; Ren, W. GABA regulates IL-1β production in macrophages. Cell Rep. 2022, 41, 111770. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Sun, S.-P.; Zhu, H.-S.; Jiao, X.-Q.; Zhong, K.; Guo, Y.-J.; Zha, G.-M.; Han, L.-Q.; Yang, G.-Y.; Li, H.-P. GABA regulates the proliferation and apoptosis of MAC-T cells through the LPS-induced TLR4 signaling pathway. Res. Vet. Sci. 2018, 118, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Jeong, S.-K.; Kim, H.-R.; Kim, D.-S.; Chae, S.-W.; Chae, H.J. Effects of triglyceride on ER stress and insulin resistance. Biochem. Biophys. Res. Commun. 2007, 363, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Sun, R.Q.; Zeng, X.Y.; Choong, Z.H.; Wang, H.; Watt, M.J.; Ye, J.M. Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes 2013, 62, 2095–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. AMPK exerts dual regulatory effects on the PI3K pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habegger, K.M.; Hoffman, N.J.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Kakino, S.; Ohki, T.; Nakayama, H.; Yuan, X.; Otabe, S.; Hashinaga, T.; Wada, N.; Kurita, Y.; Tanaka, K.; Hara, K.; et al. Pivotal Role of TNF-α in the Development and Progression of Nonalcoholic Fatty Liver Disease in a Murine Model. Horm. Metab. Res. 2018, 50, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.-P.; Denderen, B.J.V.; et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Invest. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochhead, P.A.; Salt, I.P.; Walker, K.S.; Hardie, D.G.; Sutherland, C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 2000, 49, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Moon, S.Y.; Kim, J.S.; Baek, C.H.; Kim, M.; Min, J.Y.; Lee, S.K. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am. J. Physiol. Renal Physiol. 2015, 308, F226–F236. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, E.; Hu, H. Role of Ferroptosis in Non-Alcoholic Fatty Liver Disease and Its Implications for Therapeutic Strategies. Biomedicines 2021, 9, 1660. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Jiang, Z.; Charloteaux, B.; Mayer, A.; Habraken, Y.; Tharun, L.; Klein, S.; Xu, X.; Duong, H.Q.; Vislovukh, A.; et al. The X-linked trichothiodystrophy-causing gene RNF113A links the spliceosome to cell survival upon DNA damage. Nat. Commun. 2020, 11, 1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Xia, Y.; Jin, S.; Xue, C.; Wang, Y.; Hu, R.; Jiang, H. Nrf2 attenuates ferroptosis-mediated IIR-ALI by modulating TERT and SLC7A11. Cell. Death. Dis. 2021, 12, 1027. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Saroul-Ainama, M.; Cordier, P.; Hammoutene, A.; Kabore, C.; Stadler, M.; Nemazanyy, I.; Galy-Fauroux, I.; Herrag, M.; Riedl, T.; et al. Replication stress triggered by nucleotide pool imbalance drives DNA damage and cGAS-STING pathway activation in NAFLD. Dev. Cell 2022, 57, 1728–1741. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Du, J.; Zhu, H.; Ling, Q. The role of cGAS-STING signalling in liver diseases. JHEP Rep. 2021, 3, 100324. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Hucke, F.; Sieghart, W.; Schöniger-Hekele, M.; Peck-Radosavljevic, M.; Müller, C. Clinical characteristics of patients with hepatocellular carcinoma in Austria—Is there a need for a structured screening program? Wien. Klin. Wochenschr. 2011, 123, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Peña, A.D.; Ilya, L.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, Y.; Tanaka, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Mohri, D.; Isomura, Y.; Seto, M.; Nakagawa, H.; Asaoka, Y.; et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J. Hepatol. 2011, 55, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Leavens, K.F.; Easton, R.M.; Shulman, G.I.; Previs, S.F.; Birnbaum, M.J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009, 10, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podsypanina, K.; Ellenson, L.H.; Nemes, A.; Gu, J.; Tamura, M.; Yamada, K.M.; Cordon-Cardo, C.; Catoretti, G.; Fisher, P.E.; Parsons, R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. USA 1999, 96, 1563–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teoh, N.C.; Dan, Y.Y.; Swisshelm, K.; Lehman, S.; Wright, J.H.; Haque, J.; Gu, Y.; Fausto, N. Defective DNA strand break repair causes chromosomal instability and accelerates liver carcinogenesis in mice. Hepatology 2008, 47, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- Hagan, D.W.; Ferreira, S.M.; Santos, G.J.; Phelps, E.A. The role of GABA in islet function. Front. Endocrinol. 2022, 13, 972115. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Ghani, M.W.; Ghani, H.; Jiang, W.; Birmani, M.W.; Ye, L.; Bin, L.; Cun, L.G.; Lilong, A.; Mei, X. Gimmicks of gamma-aminobutyric acid (GABA) in pancreatic β-cell regeneration through transdifferentiation of pancreatic α- to β-cells. Cell Biol. Int. 2020, 44, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lau, H.K.; Son, D.O.; Jin, T.; Yang, Y.; Zhang, Z.; Li, Y.; Prud’homme, G.J.; Wang, Q. Combined use of GABA and sitagliptin promotes human β-cell proliferation and reduces apoptosis. J. Endocrinol. 2021, 248, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Aikin, R.; Rosenberg, L.; Maysinger, D. Phosphatidylinositol 3-kinase signaling to Akt mediates survival in isolated canine islets of Langerhans. Biochem. Biophys. Res. Commun. 2000, 277, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Jo, K.; Shin, K.C.; Kim, J.I.; Ji, Y.; Park, Y.J.; Park, J.; Jeon, Y.G.; Ka, S.; Suk, S.; et al. GABA-stimulated adipose-derived stem cells suppress subcutaneous adipose inflammation in obesity. Proc. Natl. Acad. Sci. USA 2019, 116, 11936–11945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezazadeh, H.; Sharifi, M.R.; Soltani, N. Insulin resistance and the role of gamma-aminobutyric acid. J. Res. Med. Sci. 2021, 26, 39. [Google Scholar] [PubMed]
- Teixeira, A.L.; Fernandes, I.A.; Millar, P.J.; Vianna, L.C. GABAA receptor activation modulates the muscle sympathetic nerve activity responses at the onset of static exercise in humans. J. Appl. Physiol. 2021, 131, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Nakada, Y.; Sugimachi, K.; Yabuuchi, F.; Akai, T.; Mizuta, E.; Kuno, S.; Yamaguchi, M. Pharmacological characterization of the novel anxiolytic beta-carboline abecarnil in rodents and primates. Jpn. J. Pharmacol. 1994, 64, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, E.R.; Bojarski, J.T.; Yakatan, G.J. Kinetics of hydrolysis of barbituric acid derivatives. J. Pharm. Sci. 1971, 60, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Frølund, B.; Ebert, B.; Kristiansen, U.; Liljefors, T.; Krogsgaard-Larsen, P. GABA(A) receptor ligands and their therapeutic potentials. Curr. Top. Med. Chem. 2002, 2, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.R.; Burton, J.H. Clinical practice advisory: Emergency department procedural sedation with propofol. Ann. Emerg. Med. 2007, 50, 182–187.e1. [Google Scholar] [CrossRef] [PubMed]
- Assini, R.; Abercrombie, E.D. Zolpidem ameliorates motor impairments in the unilaterally 6-hydroxydopamine-lesioned rat. Eur. J. Neurosci. 2018, 48, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.M.; Rahman, A.; Husain, Z.; Mahmud, S.Z.; Ryan, W.G.; Feldman, J.M. Clozapine: A clinical review of adverse effects and management. Ann. Clin. Psychiatry 2003, 15, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A.R. Advantages of an antagonist: Bicuculline and other GABA antagonists. Br. J. Pharmacol. 2013, 169, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredenoord, A.J. Lesogaberan, a GABA(B) agonist for the potential treatment of gastroesophageal reflux disease. IDrugs 2009, 12, 576–584. [Google Scholar] [PubMed]
- Bhattacharya, D.; Becker, C.; Readhead, B.; Goossens, N.; Novik, J.; Fiel, M.I.; Cousens, L.P.; Magnusson, B.; Backmark, A.; Hicks, R.; et al. Repositioning of a novel GABA-B receptor agonist, AZD3355 (Lesogaberan), for the treatment of non-alcoholic steatohepatitis. Sci. Rep. 2021, 11, 20827. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, E.; Torgner, I.A.; Roberg, B. Kinetics and localization of brain phosphate activated glutaminase. J. Neurosci. Res. 2001, 66, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.; Frankel, S. gamma-Aminobutyric acid in brain: Its formation from glutamic acid. J. Biol. Chem. 1950, 187, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Sickmann, H.M.; Schousboe, A.; Waagepetersen, H.S. Activity of the lactate-alanine shuttle is independent of glutamate-glutamine cycle activity in cerebellar neuronal-astrocytic cultures. J. Neurosci. Res. 2005, 79, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Zaganas, I. Regulation of human glutamate dehydrogenases: Implications for glutamate, ammonia and energy metabolism in brain. J. Neurosci. Res. 2001, 66, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine-glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Yoon, H. Gamma-Aminobutyric Acid Signaling in Damage Response, Metabolism, and Disease. Int. J. Mol. Sci. 2023, 24, 4584. https://doi.org/10.3390/ijms24054584
Kim K, Yoon H. Gamma-Aminobutyric Acid Signaling in Damage Response, Metabolism, and Disease. International Journal of Molecular Sciences. 2023; 24(5):4584. https://doi.org/10.3390/ijms24054584
Chicago/Turabian StyleKim, Kimyeong, and Haejin Yoon. 2023. "Gamma-Aminobutyric Acid Signaling in Damage Response, Metabolism, and Disease" International Journal of Molecular Sciences 24, no. 5: 4584. https://doi.org/10.3390/ijms24054584
APA StyleKim, K., & Yoon, H. (2023). Gamma-Aminobutyric Acid Signaling in Damage Response, Metabolism, and Disease. International Journal of Molecular Sciences, 24(5), 4584. https://doi.org/10.3390/ijms24054584