

Manipulation of Oxidative Stress Responses by Non-Thermal Plasma to Treat Herpes Simplex Virus Type 1 Infection and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
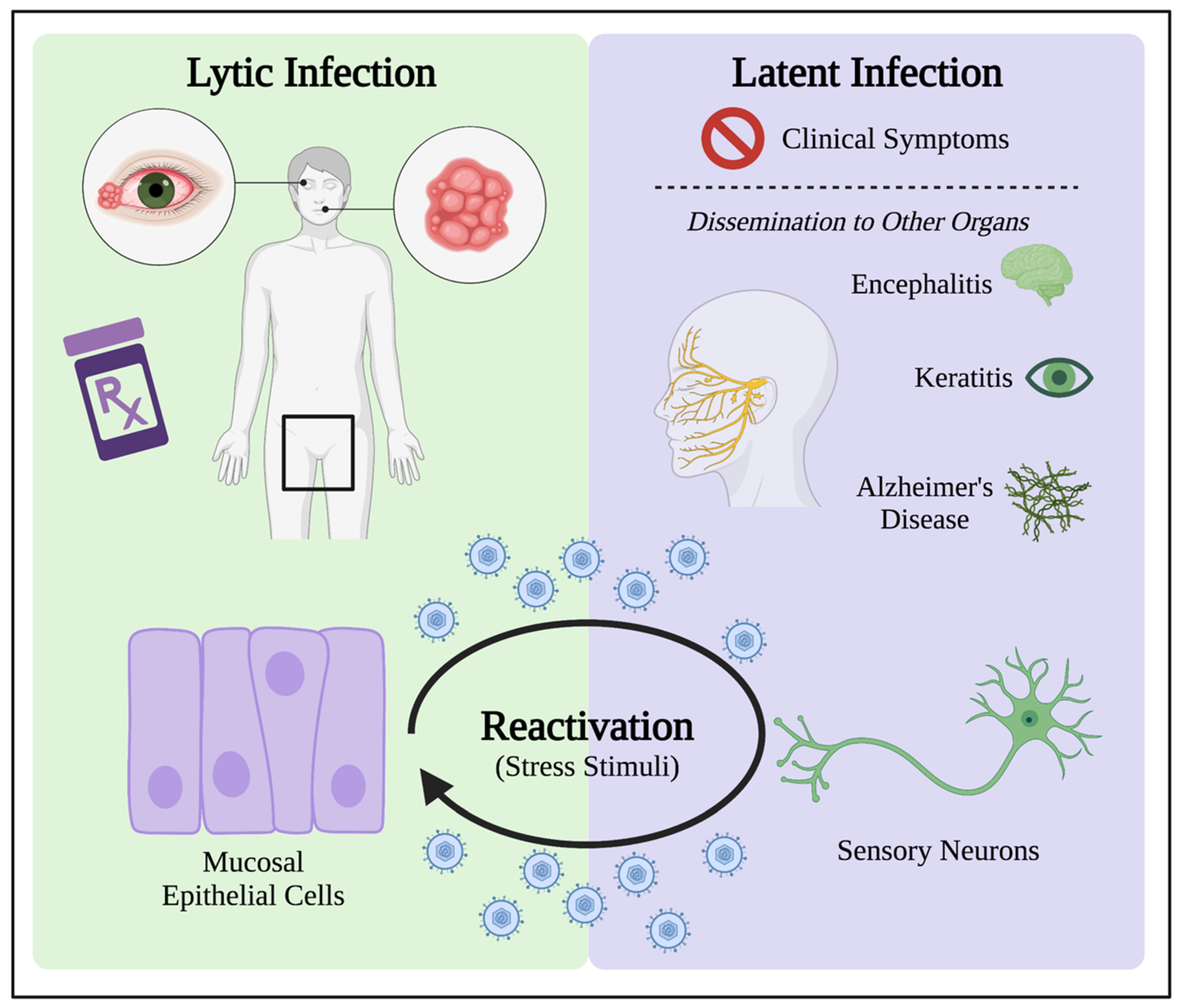
:1. Introduction to HSV-1 Infection
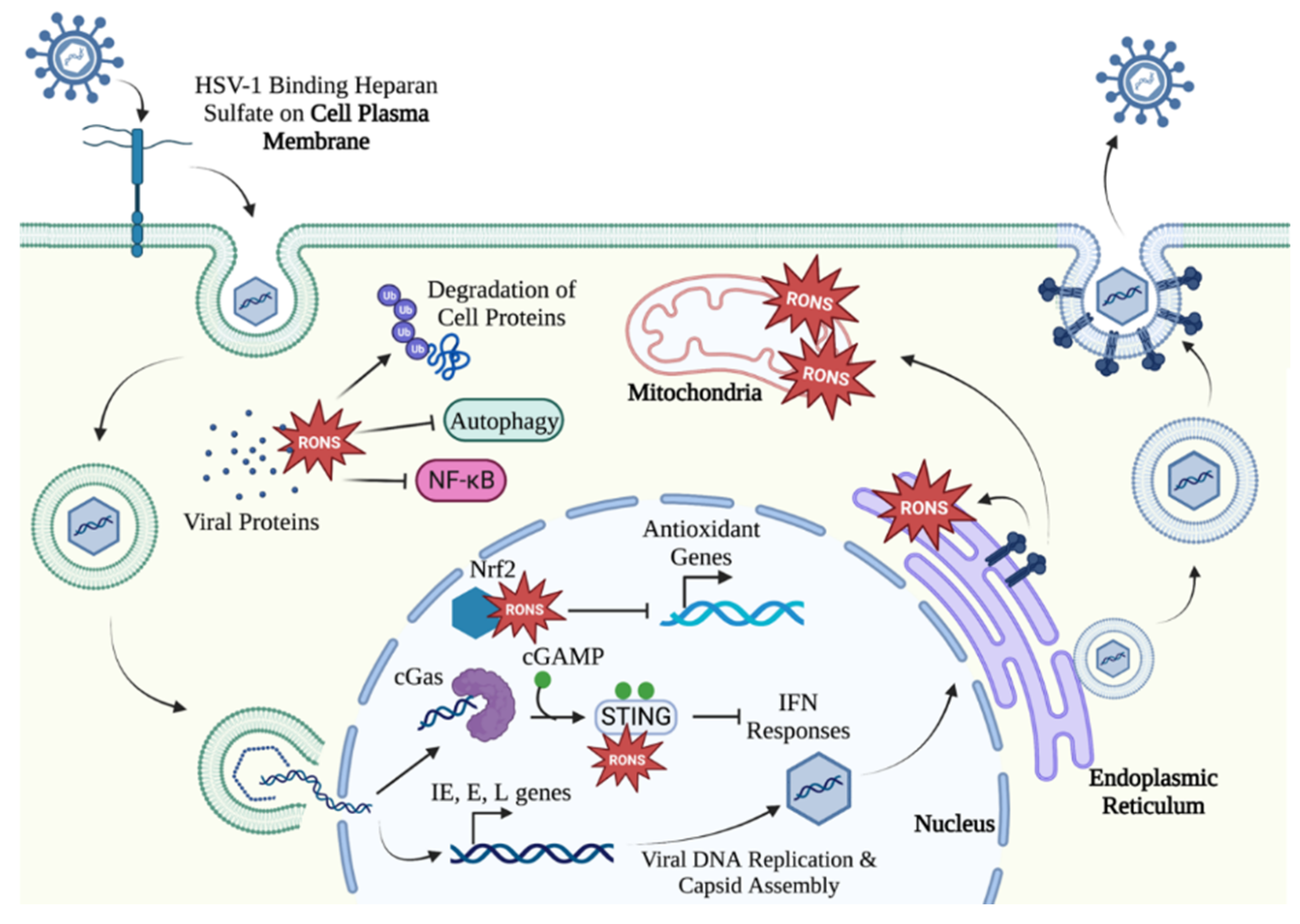
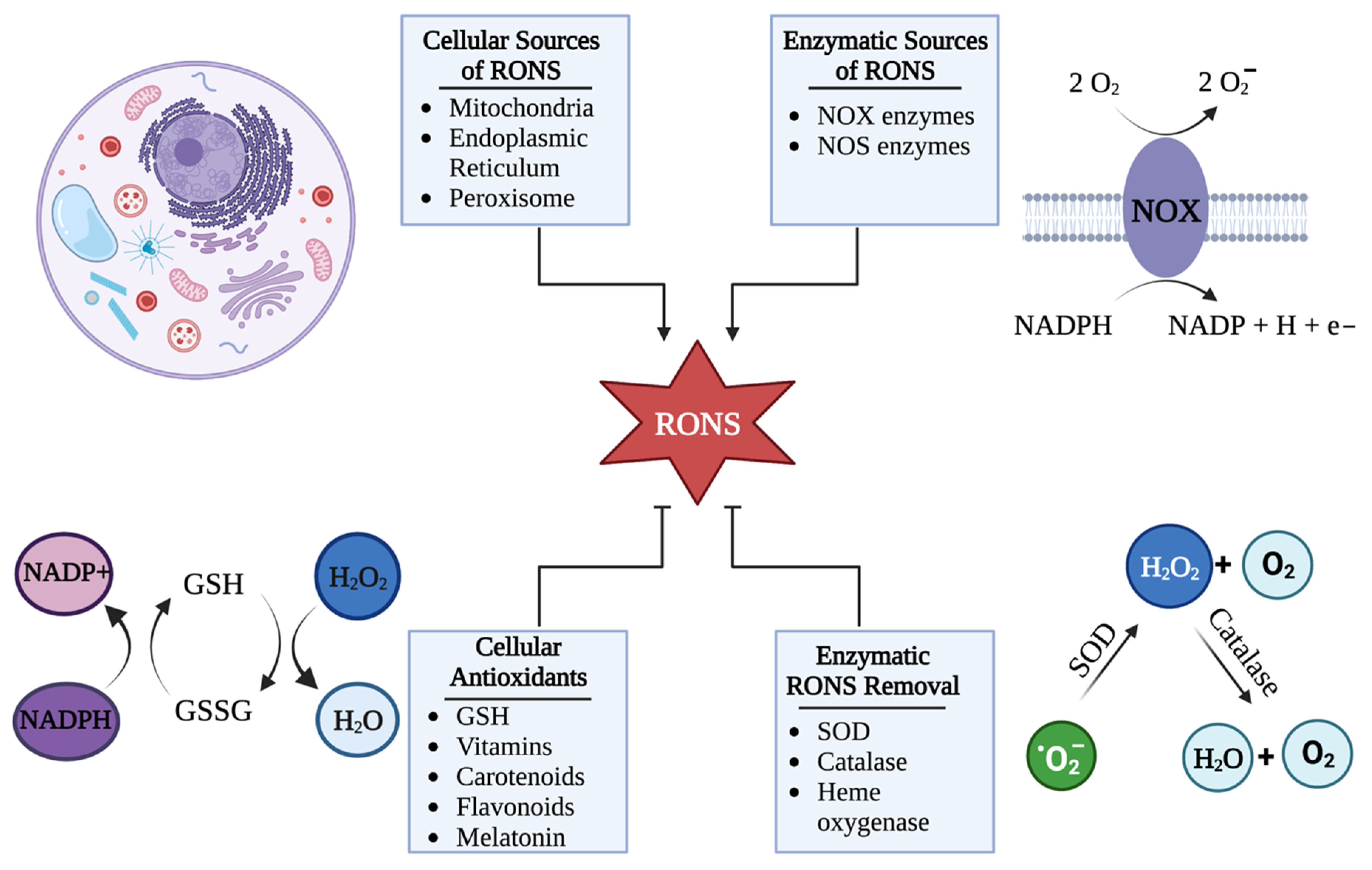
2. HSV-1 Disrupts Cellular Redox Homeostasis during Infection
HSV-1 Can Hijack Cellular RONS Generation to Cause Oxidative Stress
3. HSV-1 Lytic Infection Manipulates Oxidative Stress in the Cell
3.1. HSV-1 Alters Cellular Redox Homeostasis Early in Infection
3.2. HSV-1 Maniplates the Cell Environment to Facilitate Viral Gene Expression and Assembly
4. Latent Infection Induces Oxidative Stress for HSV-1 Persistence in Patients
4.1. HSV-1 Latency Results in Long-Term Oxidative Damage in Neurons
4.2. Oxidative Stress in the Reactivation from Latency
Oxidative Stress as Stimulus in Models for HSV-1 Reactivation
5. Cellular Mechanisms That Control RONS Concentrations
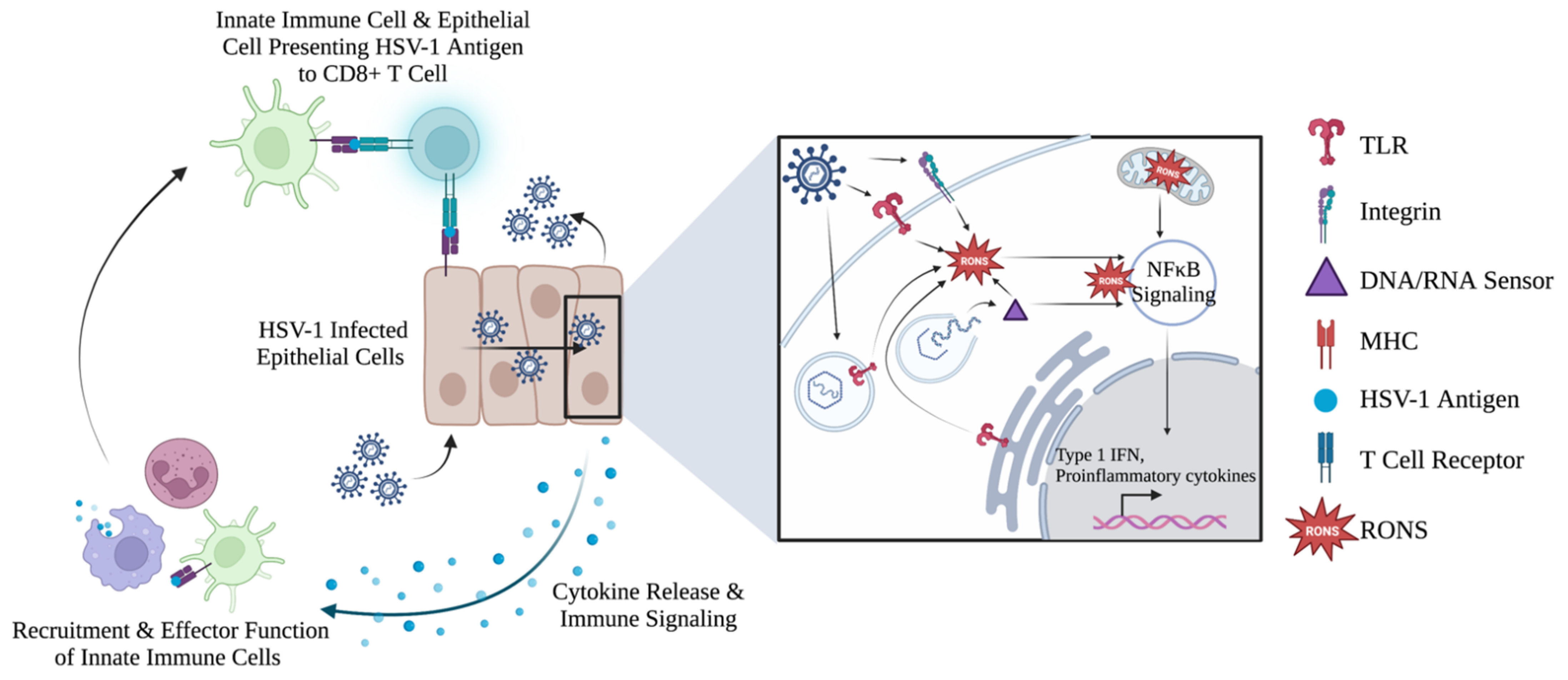
5.1. Cells Control the Upregulation of RONS in Response to HSV-1
5.2. Cellular RONS Mediate Innate Immune Responses against HSV-1
5.3. Evidence of RONS Involvement in Adaptive Immune Response against HSV-1
6. Examination of RONS as Antiviral Agents
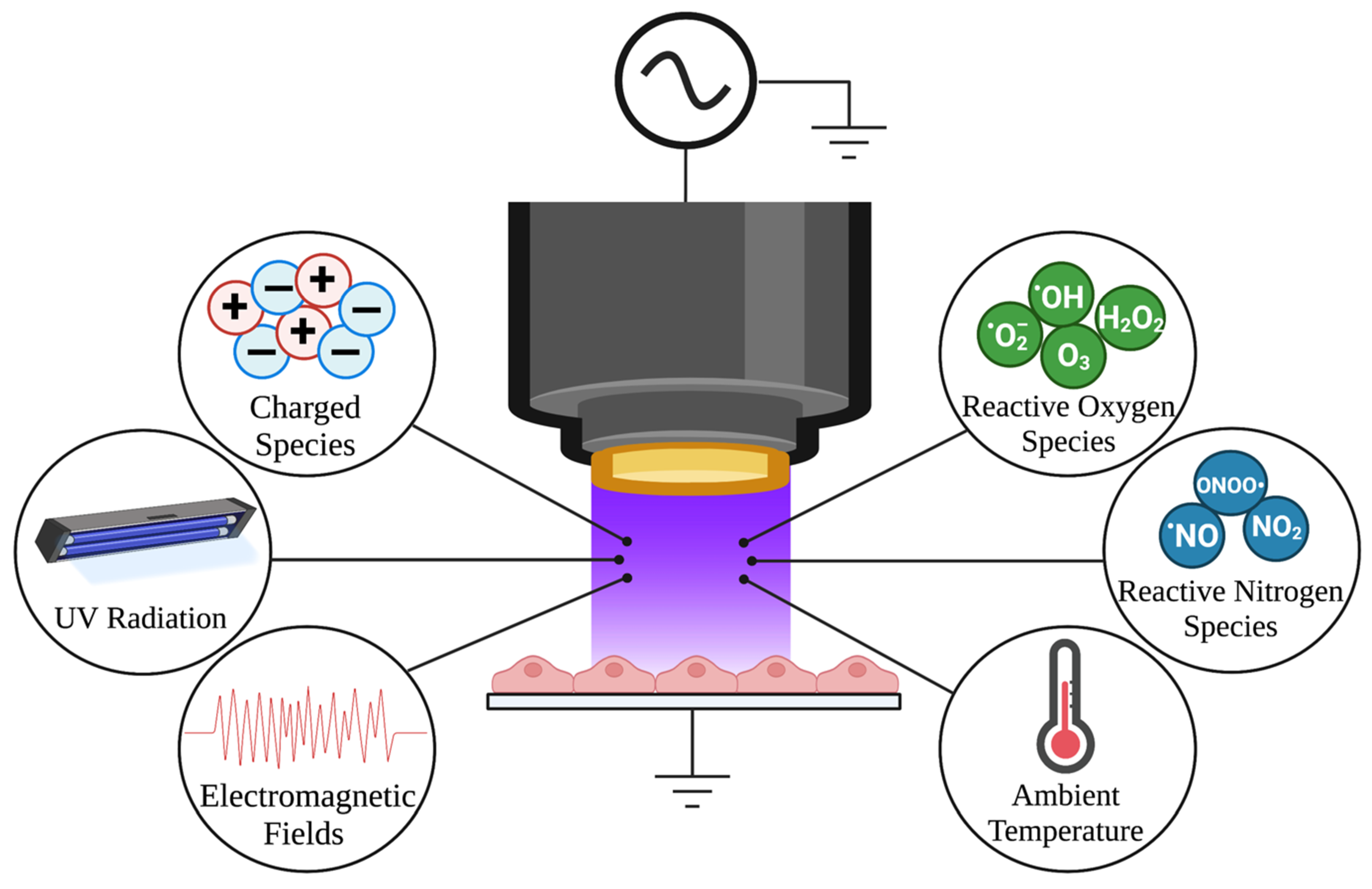
7. NTP as a Method for Controlled RONS Delivery
7.1. NTP Adds an Additional Layer of Oxidative Stress to Control HSV-1 Infection
7.2. Specialized Devices Allow Controllable Delivery of RONS
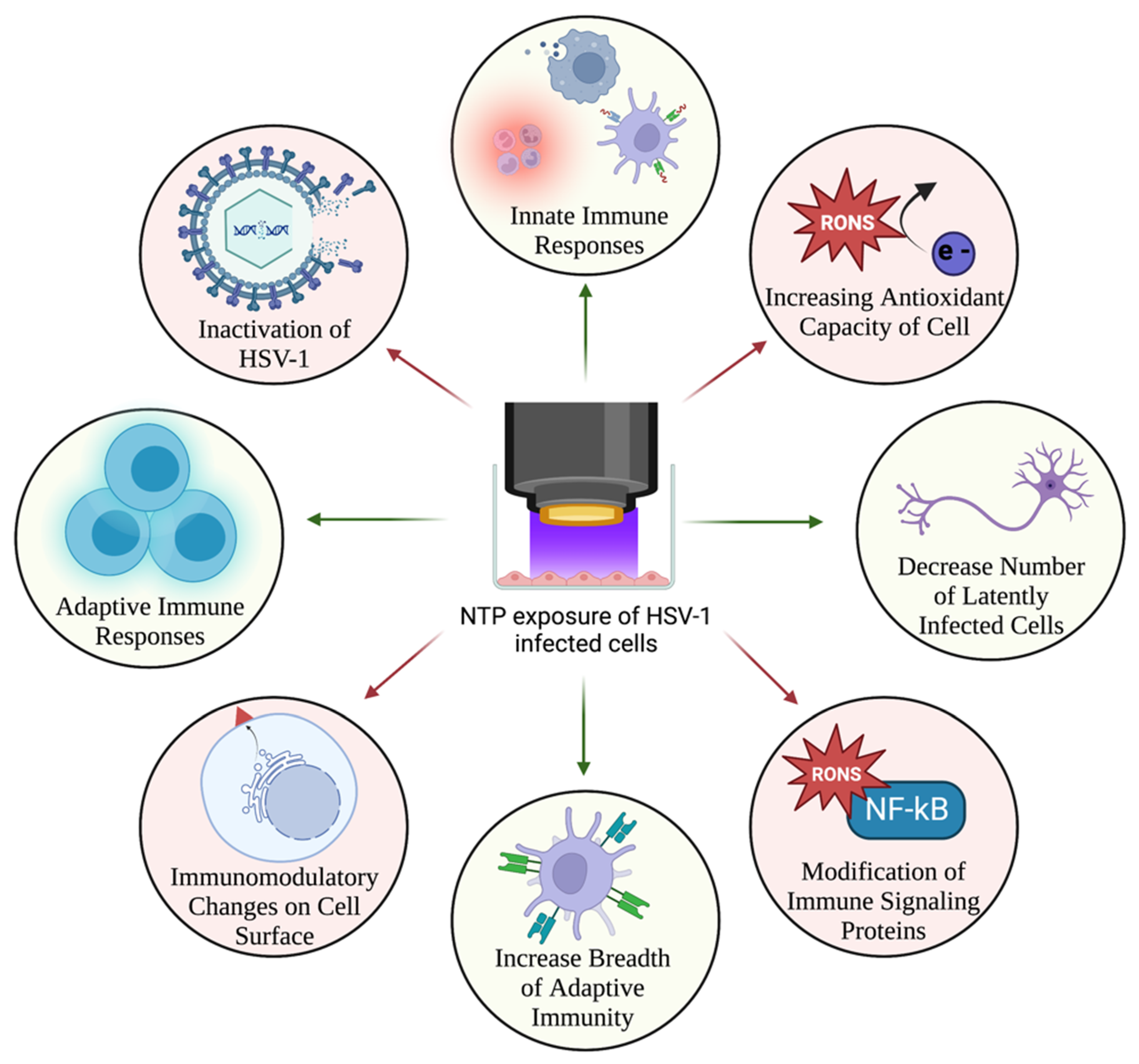
8. NTP as a Virucidal and Antiviral Agent
9. NTP as a Therapeutic Alternative for HSV-1 Infection
9.1. Direct Effects of NTP-Generated RONS on HSV-1 Infection
9.2. Effects of NTP-Induced Oxidative Stress on HSV-1 Replication
10. NTP as an Immunomodulatory Agent for Treatment of HSV-1 Infection
Enhancement of Anti-HSV-1 Host Immune Responses by NTP
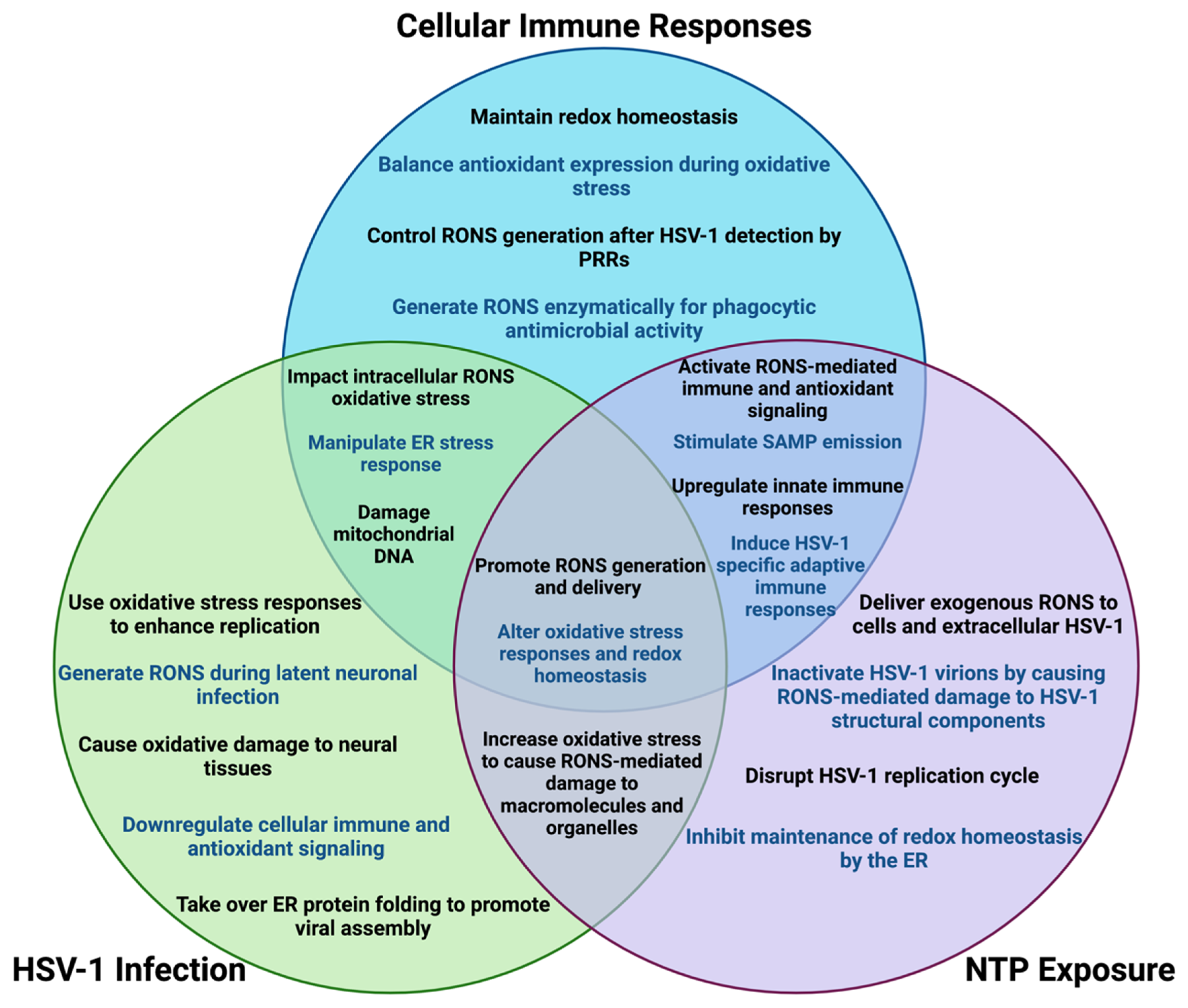
11. Overlapping Roles for Oxidative Stress in Treating HSV-1 Infection with NTP
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herpes Simplex Virus. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 16 December 2022).
- Egan, K.P.; Allen, A.G.; Wigdahl, B.; Jennings, S.R. Modeling the pathology, immune responses, and kinetics of HSV-1 replication in the lip scarification model. Virology 2018, 514, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Research 2018, 7, 1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. NeuroTherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Farooq, A.V.; Tiwari, V.; Kim, M.-J.; Shukla, D. HSV-1 infection of human corneal epithelial cells: Receptor-mediated entry and trends of re-infection. Mol. Vis. 2010, 16, 2476–2486. [Google Scholar] [PubMed]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D human brain–like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 2014, 6, 202. [Google Scholar] [CrossRef]
- Santana, S.; Sastre, I.; Recuero, M.; Bullido, M.J.; Aldudo, J. Oxidative Stress Enhances Neurodegeneration Markers Induced by Herpes Simplex Virus Type 1 Infection in Human Neuroblastoma Cells. PLoS ONE 2013, 8, e75842. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, F.J.; Farías, M.A.; Gonzalez-Troncoso, M.P.; Corrales, N.; Duarte, L.F.; Retamal-Díaz, A.; Gonzalez, P.A. Experimental Dissection of the Lytic Replication Cycles of Herpes Simplex Viruses in vitro. Front. Microbiol. 2018, 9, 2406. [Google Scholar] [CrossRef]
- Cernik, C.; Gallina, K.; Brodell, R.T. The Treatment of Herpes Simplex InfectionsAn Evidence-Based Review. Arch. Intern. Med. 2008, 168, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes Simplex Virus Resistance to Acyclovir and Penciclovir after Two Decades of Antiviral Therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.-C.; Feng, H.; Lin, Y.-C.; Guo, X.-R. New strategies against drug resistance to herpes simplex virus. Int. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef]
- Su, C.; Zhan, G.; Zheng, C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. 2016, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Laurent, A.; Nicco, C.; Chéreau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Lévy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling Tumor Growth by Modulating Endogenous Production of Reactive Oxygen Species. Cancer Res. 2005, 65, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [Green Version]
- Goldkorn, T.; Balaban, N.; Matsukuma, K.; Chea, V.; Gould, R.; Last, J.; Chan, C.; Chavez, C. EGF-Receptor Phosphorylation and Signaling Are Targeted by H2O2 Redox Stress. Am. J. Respir. Cell Mol. Biol. 1998, 19, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, A.; Saegusa, K.; Noguchi, T.; Sadamitsu, C.; Nishitoh, H.; Nagai, S.; Koyasu, S.; Matsumoto, K.; Takeda, K.; Ichijo, H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 2005, 6, 587–592. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharm. Pharm. 2014, 5, 1000141. [Google Scholar]
- Jha, N.; Ryu, J.J.; Choi, E.H.; Kaushik, N.K. Generation and Role of Reactive Oxygen and Nitrogen Species Induced by Plasma, Lasers, Chemical Agents, and Other Systems in Dentistry. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A.Y. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation–reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates Than Complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. An Update on Mitochondrial Reactive Oxygen Species Production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [Green Version]
- Ozgur, R.; Uzilday, B.; Iwata, Y.; Koizumi, N.; Turkan, I. Interplay between the unfolded protein response and reactive oxygen species: A dynamic duo. J. Exp. Bot. 2018, 69, 3333–3345. [Google Scholar] [CrossRef] [Green Version]
- Dansen, T.B.; Wirtz, K.W.A. The Peroxisome in Oxidative Stress. IUBMB Life 2001, 51, 223–230. [Google Scholar]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta (BBA) -Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Croen, K.D. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J. Clin. Investig. 1993, 91, 2446–2452. [Google Scholar] [CrossRef]
- Feng, C.; Li, J.; Zheng, H. Deciphering mechanism of conformationally controlled electron transfer in nitric oxide synthases. Front. Biosci. 2018, 23, 1803–1821. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Dumas, A.; Knaus, U.G. Raising the ’Good’ Oxidants for Immune Protection. Front. Immunol. 2021, 12, 698042. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory Syncytial Virus Infection: Mechanisms of Redox Control and Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshi, M.L.; Su, Y.C.; Hong, J.-R. RNA Viruses: ROS-Mediated Cell Death. Int. J. Cell Biol. 2014, 2014, 467452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebrat, Y.; Amogne, W.; Mekasha, A.; Gleason, R.L., Jr.; Seifu, D. Lipid Peroxidation and Altered Antioxidant Profiles with Pediatric HIV Infection and Antiretroviral Therapy in Addis Ababa, Ethiopia. J. Trop. Pediatr. 2017, 63, 196–202. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Meng, C.; Sun, Y.; Safdar, A.; Pasha, R.H.; Munir, M.; Ding, C. Oxidative Stress in Poultry: Lessons from the Viral Infections. Oxidative Med. Cell. Longev. 2018, 2018, 5123147. [Google Scholar] [CrossRef] [Green Version]
- Kavouras, J.H.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovács, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. NeuroVirology 2007, 13, 416–425. [Google Scholar] [CrossRef]
- Sebastiano, M.; Chastel, O.; de Thoisy, B.; Eens, M.; Costantini, D. Oxidative stress favours herpes virus infection in vertebrates: A meta-analysis. Curr. Zool. 2016, 62, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Laine, R.F.; Albecka, A.; van de Linde, S.; Rees, E.J.; Crump, C.M.; Kaminski, C.F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 2015, 6, 5980. [Google Scholar] [CrossRef] [Green Version]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S.N. Human herpes simplex virus: Life cycle and development of inhibitors. Biochem. (Moscow) 2014, 79, 1635–1652. [Google Scholar] [CrossRef]
- Hilterbrand, A.T.; Daly, R.E.; Heldwein, E.E. Contributions of the Four Essential Entry Glycoproteins to HSV-1 Tropism and the Selection of Entry Routes. Mbio 2021, 12, e00143-21. [Google Scholar] [CrossRef]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2021, 10, 617578. [Google Scholar] [CrossRef]
- Choudhary, S.; Marquez, M.; Alencastro, F.; Spors, F.; Zhao, Y.; Tiwari, V. Herpes Simplex Virus Type-1 (HSV-1) Entry into Human Mesenchymal Stem Cells Is Heavily Dependent on Heparan Sulfate. J. Biomed. Biotechnol. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [Green Version]
- Danastas, K.; Miranda-Saksena, M.; Cunningham, A.L. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int. J. Mol. Sci. 2020, 21, 5150. [Google Scholar] [CrossRef]
- Mulvey, M.; Arias, C.; Mohr, I. Maintenance of Endoplasmic Reticulum (ER) Homeostasis in Herpes Simplex Virus Type 1-Infected Cells through the Association of a Viral Glycoprotein with PERK, a Cellular ER Stress Sensor. J. Virol. 2007, 81, 3377–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molteni, C.G.; Principi, N.; Esposito, S. Reactive oxygen and nitrogen species during viral infections. Free. Radic. Res. 2014, 48, 1163–1169. [Google Scholar] [CrossRef]
- Aubert, M.; Chen, Z.; Lang, R.; Chung, H.D.; Fowler, C.; Derek, D.S.; Keith, R.J. The Antiapoptotic Herpes Simplex Virus Glycoprotein J Localizes to Multiple Cellular Organelles and Induces Reactive Oxygen Species Formation. J. Virol. 2008, 82, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Tebaldi, G.; Pritchard, S.M.; Nicola, A.V. Herpes Simplex Virus Entry by a Nonconventional Endocytic Pathway. J. Virol. 2020, 94, e01910-20. [Google Scholar] [CrossRef]
- Fan, D.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Zhao, X.; Chen, S.; Liu, M.; et al. The Role of VP16 in the Life Cycle of Alphaherpesviruses. Front. Microbiol. 2020, 11, 1910. [Google Scholar] [CrossRef]
- Bearer, E.L.; Breakefield, X.O.; Schuback, D.; Reese, T.S.; LaVail, J.H. Retrograde axonal transport of herpes simplex virus: Evidence for a single mechanism and a role for tegument. Proc. Natl. Acad. Sci. USA 2000, 97, 8146–8150. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Che, Y.; Li, Q. HSV-1 tegument protein and the development of its genome editing technology. Virol. J. 2016, 13, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt-Jones, E.A.; Orzalli, M.H.; Knipe, D.M. Innate Immune Mechanisms and Herpes Simplex Virus Infection and Disease. In Cell Biology of Herpes Viruses; Springer: Berlin/Heidelberg, Germany, 2017; Volume 223, pp. 49–75. [Google Scholar] [CrossRef]
- Weller, S.K. Herpes Simplex Virus Reorganizes the Cellular DNA Repair and Protein Quality Control Machinery. PLoS Pathog. 2010, 6, e1001105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkness, J.M.; Kader, M.; DeLuca, N.A. Transcription of the Herpes Simplex Virus 1 Genome during Productive and Quiescent Infection of Neuronal and Nonneuronal Cells. J. Virol. 2014, 88, 6847–6861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2021, 11, 613799. [Google Scholar] [CrossRef] [PubMed]
- Copeland, A.M.; Newcomb, W.W.; Brown, J.C. Herpes Simplex Virus Replication: Roles of Viral Proteins and Nucleoporins in Capsid-Nucleus Attachment. J. Virol. 2009, 83, 1660–1668. [Google Scholar] [CrossRef] [Green Version]
- Shahin, V.; Hafezi, W.; Oberleithner, H.; Ludwig, Y.; Windoffer, B.; Schillers, H.; Kühn, J.E. The genome of HSV-1 translocates through the nuclear pore as a condensed rod-like structure. J. Cell Sci. 2006, 119, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Lemoff, A.; Wang, G.; Zarek, C.; Lowe, A.; Yan, N.; Reese, T.A. Reactive oxygen species oxidize STING and suppress interferon production. Elife 2020, 9, e57837. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Wang, Z.; Li, Y.; Wang, H.; Liu, H. Upregulation of nuclear factor E2-related factor 2 (Nrf2) represses the replication of herpes simplex virus type 1. Virol. J. 2022, 19, 1–8. [Google Scholar] [CrossRef]
- Honess, R.W.; Roizman, B. Regulation of Herpesvirus Macromolecular Synthesis I. Cascade Regulation of the Synthesis of Three Groups of Viral Proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Packard, J.E.; Dembowski, J.A. HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors. Viruses 2021, 13, 2015. [Google Scholar] [CrossRef]
- Rodríguez, M.C.; Dybas, J.M.; Hughes, J.; Weitzman, M.D.; Boutell, C. The HSV-1 ubiquitin ligase ICP0: Modifying the cellular proteome to promote infection. Virus Res. 2020, 285, 198015. [Google Scholar] [CrossRef]
- Tang, S.; Patel, A.; Krause, P.R. Hidden regulation of herpes simplex virus 1 pre-mRNA splicing and polyadenylation by virally encoded immediate early gene ICP27. PLoS Pathog. 2019, 15, e1007884. [Google Scholar] [CrossRef] [Green Version]
- Fierer, D.S.; Challberg, M.D. Purification and characterization of UL9, the herpes simplex virus type 1 origin-binding protein. J. Virol. 1992, 66, 3986–3995. [Google Scholar] [CrossRef] [Green Version]
- Muylaert, I.; Tang, K.-W.; Elias, P. Replication and Recombination of Herpes Simplex Virus DNA. J. Biol. Chem. 2011, 286, 15619–15624. [Google Scholar] [CrossRef] [Green Version]
- Weller, S.K.; Coen, D.M. Herpes Simplex Viruses: Mechanisms of DNA Replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 Exonuclease, UL12, Stimulates Recombination by a Single Strand Annealing Mechanism. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef]
- Cavignac, Y.; Esclatine, A. Herpesviruses and autophagy: Catch me if you can! Viruses 2010, 2, 314–333. [Google Scholar] [CrossRef]
- Marino-Merlo, F.; Papaianni, E.; Frezza, C.; Pedatella, S.; De Nisco, M.; Macchi, B.; Grelli, S.; Mastino, A. NF-κB-Dependent Production of ROS and Restriction of HSV-1 Infection in U937 Monocytic Cells. Viruses 2019, 11, 428. [Google Scholar] [CrossRef] [Green Version]
- Valyi-Nagy, T.; Dermody, T.S. Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol. Histopathol. 2005, 20, 957–967. [Google Scholar] [CrossRef]
- Castro-Acosta, R.M.; Rodríguez-Limas, W.A.; Valderrama, B.; Ramírez, O.T.; Palomares, L.A. Effect of metal catalyzed oxidation in recombinant viral protein assemblies. Microb. Cell Factories 2014, 13, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- England, K.; Odriscoll, C.; Cotter, T.G. Carbonylation of glycolytic proteins is a key response to drug-induced oxidative stress and apoptosis. Cell Death Differ. 2003, 11, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, S.S.; Bryant, P.W.; Burch, A.D. Accumulation of oxidized proteins in Herpesvirus infected cells. Free. Radic. Biol. Med. 2010, 49, 383–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus Capsid Assembly and DNA Packaging. In Cell Biology of Herpes Viruses; Springer: Berlin/Heidelberg, Germany, 2017; Volume 223, pp. 119–142. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.H.; Radtke, K.; Kniesmeijer, E.; Geertsema, H.; Sodeik, B.; Wuite, G.J.L. Scaffold expulsion and genome packaging trigger stabilization of herpes simplex virus capsids. Proc. Natl. Acad. Sci. USA 2009, 106, 9673–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albright, B.S.; Nellissery, J.; Szczepaniak, R.; Weller, S.K. Disulfide Bond Formation in the Herpes Simplex Virus 1 UL6 Protein Is Required for Portal Ring Formation and Genome Encapsidation. J. Virol. 2011, 85, 8616–8624. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Baines, J. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. USA 2011, 108, 14276. [Google Scholar] [CrossRef] [Green Version]
- Leuzinger, H.; Ziegler, U.; Schraner, E.M.; Fraefel, C.; Glauser, D.L.; Heid, I.; Ackermann, M.; Mueller, M.; Wild, P. Herpes Simplex Virus 1 Envelopment Follows Two Diverse Pathways. J. Virol. 2005, 79, 13047–13059. [Google Scholar] [CrossRef] [Green Version]
- Sandbaumhüter, M.; Döhner, K.; Schipke, J.; Binz, A.; Pohlmann, A.; Sodeik, B.; Bauerfeind, R. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 2012, 15, 248–269. [Google Scholar] [CrossRef]
- Ahmad, I.; Wilson, D.W. HSV-1 Cytoplasmic Envelopment and Egress. Int. J. Mol. Sci. 2020, 21, 5969. [Google Scholar] [CrossRef]
- Pasdeloup, D.; McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J. Herpesvirus Tegument Protein pUL37 Interacts with Dystonin/BPAG1 To Promote Capsid Transport on Microtubules during Egress. J. Virol. 2013, 87, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Millhouse, S.; Wigdahl, B. Molecular circuitry regulating herpes simplex virus type 1 latency in neurons. J. Neurovirology 2000, 6, 6–24. [Google Scholar] [CrossRef]
- Kenny, J.J.; Millhouse, S.; Wotring, M.; Wigdahl, B. Upstream Stimulatory Factor Family Binds to the Herpes Simplex Virus Type 1 Latency-Associated Transcript Promoter. Virology 1997, 230, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Millhouse, S.; Kenny, J.J.; Quinn, P.G.; Lee, V.; Wigdahl, B. ATF/CREB elements in the herpes simplex virus type 1 latency-associated transcript promoter interact with members of the ATF/CREB and AP-1 transcription factor families. J. Biomed. Sci. 1998, 5, 451–464. [Google Scholar] [CrossRef]
- Kenny, J.J.; Krebs, F.C.; Hartle, H.T.; Gartner, A.E.; Chatton, B.; Leiden, J.M.; Hoeffler, J.P.; Weber, P.C.; Wigdahl, B. Identification of a Second ATF/CREB-like Element in the Herpes Simplex Virus Type 1 (HSV-1) Latency-Associated Transcript (LAT) Promoter. Virology 1994, 200, 220–235. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.R.; Connor, V.; Coleman, H.M.; Proença, J.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef]
- Knipe, D.M.; Raja, P.; Lee, J.S. Clues to mechanisms of herpesviral latent infection and potential cures. Proc. Natl. Acad. Sci. USA 2015, 112, 11993–11994. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free. Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Dosal, R.; Horan, K.A.; Rahbek, S.H.; Ichijo, H.; Chen, Z.; Mieyal, J.J.; Hartmann, R.; Paludan, S.R. HSV Infection Induces Production of ROS, which Potentiate Signaling from Pattern Recognition Receptors: Role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 2011, 7, e1002250. [Google Scholar] [CrossRef] [PubMed]
- Grinde, B. Herpesviruses: Latency and reactivation—viral strategies and host response. J. Oral Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Sheng, W.S.; Schachtele, S.J.; Lokensgard, J.R. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflammation 2011, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Schachtele, S.J.; Hu, S.; Little, M.R.; Lokensgard, J.R. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflammation 2010, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Marques, C.P.; Cheeran, M.C.-J.; Palmquist, J.M.; Hu, S.; Lokensgard, J.R. Microglia are the major cellular source of inducible nitric oxide synthase during experimental herpes encephalitis. J. NeuroVirology 2008, 14, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Doll, J.R.; Hoebe, K.; Thompson, R.L.; Sawtell, N.M. Resolution of herpes simplex virus reactivation in vivo results in neuronal destruction. PLoS Pathog. 2020, 16, e1008296. [Google Scholar] [CrossRef] [Green Version]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and Mouse Models of HSV-1 Latency, Reactivation, and Recurrent Eye Diseases. J. Biomed. Biotechnol. 2012, 2012, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Heck, D.E.; Vetrano, A.M.; Mariano, T.M.; Laskin, J.D. UVB Light Stimulates Production of Reactive Oxygen Species: Unexpected Role for Catalase. J. Biol. Chem. 2003, 278, 22432–22436. [Google Scholar] [CrossRef] [Green Version]
- Hogk, I.; Kaufmann, M.; Finkelmeier, D.; Rupp, S.; Burger-Kentischer, A. An In Vitro HSV-1 Reactivation Model Containing Quiescently Infected PC12 Cells. BioResearch Open Access 2013, 2, 250–257. [Google Scholar] [CrossRef]
- Shimeld, C.; Easty, D.L.; Hill, T.J. Reactivation of Herpes Simplex Virus Type 1 in the Mouse Trigeminal Ganglion: An In Vivo Study of Virus Antigen and Cytokines. J. Virol. 1999, 73, 1767–1773. [Google Scholar] [CrossRef] [Green Version]
- Brem, R.; Macpherson, P.; Guven, M.; Karran, P. Oxidative stress induced by UVA photoactivation of the tryptophan UVB photoproduct 6-formylindolo[3,2-b]carbazole (FICZ) inhibits nucleotide excision repair in human cells. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.P.; Richa, U.; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Ruzza, P.; Honisch, C.; Hussain, R.; Siligardi, G. Free Radicals and ROS Induce Protein Denaturation by UV Photostability Assay. Int. J. Mol. Sci. 2021, 22, 6512. [Google Scholar] [CrossRef]
- Preston, C.M.; Nicholl, M.J. Induction of Cellular Stress Overcomes the Requirement of Herpes Simplex Virus Type 1 for Immediate-Early Protein ICP0 and Reactivates Expression from Quiescent Viral Genomes. J. Virol. 2008, 82, 11775–11783. [Google Scholar] [CrossRef] [Green Version]
- Stoeger, T.; Adler, H. “Novel” Triggers of Herpesvirus Reactivation and Their Potential Health Relevance. Front. Microbiol. 2019, 9, 3207. [Google Scholar] [CrossRef]
- Wigdahl, B.; Scheck, A.C.; Ziegler, R.J.; De Clercq, E.; Rapp, F. Analysis of the herpes simplex virus genome during in vitro latency in human diploid fibroblasts and rat sensory neurons. J. Virol. 1984, 49, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Wigdahl, B.L.; Isom, H.C.; Rapp, F. Repression and activation of the genome of herpes simplex viruses in human cells. Proc. Natl. Acad. Sci. USA 1981, 78, 6522–6526. [Google Scholar] [CrossRef] [Green Version]
- Wigdahl, B.L.; Scheck, A.C.; De Clercq, E.; Rapp, F. High Efficiency Latency and Activation of Herpes Simplex Virus in Human Cells. Science 1982, 217, 1145–1146. [Google Scholar] [CrossRef]
- Wigdahl, B.L.; Ziegler, R.J.; Sneve, M.; Rapp, F. Herpes simplex virus latency and reactivation in isolated rat sensory neurons. Virology 1983, 127, 159–167. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.-C.; Nakanishi, I.; Matsumoto, K.-I.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nencioni, L.; Sgarbanti, R.; Amatore, D.; Checconi, P.; Celestino, I.; Limongi, D.; Anticoli, S.; Palamara, A.T.; Garaci, E. Intracellular Redox Signaling as Therapeutic Target for Novel Antiviral Strategy. Curr. Pharm. Des. 2011, 17, 3898–3904. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Leoni, V.; Campadelli-Fiume, G. Type I interferon and NF-κB activation elicited by herpes simplex virus gH/gL via αvβ3 integrin in epithelial and neuronal cell lines. J. Virol. 2013, 87, 13911–13916. [Google Scholar] [CrossRef] [Green Version]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Jensen, S.B.; Nielsen, C.; Quartin, E.; Kato, H.; Chen, Z.; Silverman, R.H.; Akira, S.; Paludan, S.R. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene- like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 2009, 90, 74–78. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, H.; Iles, K.E.; Rinna, A.; Merrill, G.; Yodoi, J.; Torres, M.; Forman, H.J. The ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophages. Free. Radic. Res. 2006, 40, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, H.; Min, S.H.; Rhee, S.G.; Jeong, W. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J. Biol. Chem. 2008, 283, 23863–23871. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Trzyna, W.; McClintock, D.; Schumacker, P. Role of Oxidants in NF-κB Activation and TNF-α Gene Transcription Induced by Hypoxia and Endotoxin. J. Immunol. 2000, 165, 1013. [Google Scholar] [CrossRef] [Green Version]
- Muri, J.; Thut, H.; Feng, Q.; Kopf, M. Thioredoxin-1 distinctly promotes NF-κB target DNA binding and NLRP3 inflammasome activation independently of Txnip. ELife 2020, 9, e53627. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta (BBA) -Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription Factors NRF2 and NF-κB Are Coordinated Effectors of the Rho Family, GTP-binding Protein RAC1 during Inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M.; Takeuchi, C.; Ruedi, J.; Gutierrez, A.; Wentworth, P., Jr. Investigating antibody-catalyzed ozone generation by human neutrophils. Proc. Natl. Acad. Sci. USA 2003, 100, 3031–3034. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C.; Hampton, M.B.; Livesey, J.H.; Kettle, A.J. Modeling the Reactions of Superoxide and Myeloperoxidase in the Neutrophil Phagosome. J. Biol. Chem. 2006, 281, 39860–39869. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Lu, M.; Zhang, C.; Wu, X.; Chen, J.; Lv, W.; Sun, T.; Qiu, H.; Huang, S. Oxidative stress modulates the expression of toll-like receptor 3 during respiratory syncytial virus infection in human lung epithelial A549 cells. Mol. Med. Rep. 2018, 18, 1867–1877. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Wong, P.K.; Engelbrecht, R.S.; Chian, E.S. Mechanism of enteroviral inactivation by ozone. Appl. Environ. Microbiol. 1981, 41, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Petry, G.; Rossato, L.G.; Nespolo, J.; Kreutz, L.C.; Bertol, C.D. In Vitro Inactivation of Herpes Virus by Ozone. Ozone Sci. Eng. 2014, 36, 249–252. [Google Scholar] [CrossRef]
- Moldgy, A.; Nayak, G.; Aboubakr, H.A.; Goyal, S.M.; Bruggeman, P.J. Inactivation of virus and bacteria using cold atmospheric pressure air plasmas and the role of reactive nitrogen species. J. Phys. D Appl. Phys. 2020, 53, 434004. [Google Scholar] [CrossRef]
- Tsukidate, D.; Takashima, K.; Sasaki, S.; Miyashita, S.; Kaneko, T.; Takahashi, H.; Ando, S. Activation of plant immunity by exposure to dinitrogen pentoxide gas generated from air using plasma technology. PLoS ONE 2022, 17, e0269863. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.M.; Chander, Y.; Yezli, S.; Otter, J.A. Evaluating the virucidal efficacy of hydrogen peroxide vapour. J. Hosp. Infect. 2014, 86, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Mileto, D.; Mancon, A.; Staurenghi, F.; Rizzo, A.; Econdi, S.; Gismondo, M.; Guidotti, M. Inactivation of SARS-CoV-2 in the Liquid Phase: Are Aqueous Hydrogen Peroxide and Sodium Percarbonate Efficient Decontamination Agents? ACS Chem. Health Saf. 2021, 28, 260–267. [Google Scholar] [CrossRef]
- Rudnick, S.N.; McDevitt, J.J.; First, M.W.; Spengler, J.D. Inactivating influenza viruses on surfaces using hydrogen peroxide or triethylene glycol at low vapor concentrations. Am. J. Infect. Control. 2009, 37, 813–819. [Google Scholar] [CrossRef]
- Aboubakr, H.A.; Gangal, U.; Youssef, M.M.; Goyal, S.M.; Bruggeman, P.J. Inactivation of virus in solution by cold atmospheric pressure plasma: Identification of chemical inactivation pathways. J. Phys. D: Appl. Phys. 2016, 49, 204001. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Hypochlorous Acid Chemistry in Mammalian Cells—Influence on Infection and Role in Various Pathologies. Int. J. Mol. Sci. 2022, 23, 10735. [Google Scholar] [CrossRef]
- Block, M.S.; Rowan, B.G. Hypochlorous Acid: A Review. J. Oral Maxillofac. Surg. 2020, 78, 1461–1466. [Google Scholar] [CrossRef]
- Pinto, A.K.; Richner, J.M.; Poore, E.A.; Patil, P.P.; Amanna, I.J.; Slifka, M.K.; Diamond, M.S. A Hydrogen Peroxide-Inactivated Virus Vaccine Elicits Humoral and Cellular Immunity and Protects against Lethal West Nile Virus Infection in Aged Mice. J. Virol. 2013, 87, 1926–1936. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Wang, M.-D.; Ming, S.-L.; Li, G.-L.; Yu, P.-W.; Qi, Y.-L.; Jiang, D.-W.; Yang, G.-Y.; Wang, J.; Chu, B.-B. An effective inactivant based on singlet oxygen-mediated lipid oxidation implicates a new paradigm for broad-spectrum antivirals. Redox Biol. 2020, 36, 101601. [Google Scholar] [CrossRef]
- Kipshidze, N.; Yeo, N.; Kipshidze, N. Photodynamic therapy for COVID-19. Nat. Photonics 2020, 14, 651–652. [Google Scholar] [CrossRef]
- Garren, M.R.; Ashcraft, M.; Qian, Y.; Douglass, M.; Brisbois, E.J.; Handa, H. Nitric oxide and viral infection: Recent developments in antiviral therapies and platforms. Appl. Mater. Today 2020, 22, 100887. [Google Scholar] [CrossRef]
- Gamba, G.; Cavalieri, H.; Courreges, M.C.; Massouh, E.J.; Benencia, F. Early inhibition of nitric oxide production increases HSV-1 intranasal infection. J. Med. Virol. 2004, 73, 313–322. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Bhartiya, P.; Kaushik, N.; Shin, Y.; Nguyen, L.N.; Park, J.S.; Kim, D.; Choi, E.H. Nitric-oxide enriched plasma-activated water inactivates 229E coronavirus and alters antiviral response genes in human lung host cells. Bioact. Mater. 2023, 19, 569–580. [Google Scholar] [CrossRef]
- Von Woedtke, T.; Emmert, S.; Metelmann, H.-R.; Rupf, S.; Weltmann, K.-D. Perspectives on cold atmospheric plasma (CAP) applications in medicine. Phys. Plasmas 2020, 27, 070601. [Google Scholar] [CrossRef]
- Von Woedtke, T.; Schmidt, A.; Bekeschus, S.; Wende, K.; Weltmann, K.-D. Plasma Medicine: A Field of Applied Redox Biology. In Vivo 2019, 33, 1011–1026. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Chernets, N.; Han, J.; Alicea, Y.; Dobrynin, D.; Fridman, G.; Freeman, T.A.; Fridman, A.; Miller, V. Non-Equilibrium Dielectric Barrier Discharge Treatment of Mesenchymal Stem Cells: Charges and Reactive Oxygen Species Play the Major Role in Cell Death. Plasma Process. Polym. 2015, 12, 1117–1127. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Eom, S.; Yoon, S.-Y.; Ryu, S.; Kim, S.B.; Yi, J.M.; Hyun, J.W. Non-thermal dielectric-barrier discharge plasma induces reactive oxygen species by epigenetically modifying the expression of NADPH oxidase family genes in keratinocytes. Redox Biol. 2020, 37, 101698. [Google Scholar] [CrossRef]
- Graves, D.B. The emerging role of reactive oxygen and nitrogen species in redox biology and some implications for plasma applications to medicine and biology. J. Phys. D Appl. Phys. 2012, 45, 263001. [Google Scholar] [CrossRef]
- Mohamed, H.; Nayak, G.; Rendine, N.; Wigdahl, B.; Krebs, F.C.; Bruggeman, P.J.; Miller, V. Non-Thermal Plasma as a Novel Strategy for Treating or Preventing Viral Infection and Associated Disease. Front. Phys. 2021, 9, 683118. [Google Scholar] [CrossRef]
- Grahl, T.; Märkl, H. Killing of microorganisms by pulsed electric fields. Appl. Microbiol. Biotechnol. 1996, 45, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Biasin, M.; Bianco, A.; Pareschi, G.; Cavalleri, A.; Cavatorta, C.; Fenizia, C.; Galli, P.; Lessio, L.; Lualdi, M.; Tombetti, E.; et al. UV-C irradiation is highly effective in inactivating SARS-CoV-2 replication. Sci. Rep. 2021, 11, 6260. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-C.; Li, C.-S. Inactivation of Viruses on Surfaces by Ultraviolet Germicidal Irradiation. J. Occup. Environ. Hyg. 2007, 4, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Fernández, A.R.; Rosemblatt, M.; Perez-Acle, T. Nanosecond pulsed electric field (nsPEF) and vaccines: A novel technique for the inactivation of SARS-CoV-2 and other viruses? Ann. Med. 2022, 54, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Kulms, D.; Zeise, E.; Pöppelmann, B.; Schwarz, T. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene 2002, 21, 5844–5851. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.A.; Silva-Júnior, A.C.T.; Oliveira, E.B.; Asad, L.M.B.O.; Reis, N.C.S.C.; Felzenszwalb, I.; Kovary, K.; Asad, N.R. Reactive oxygen species mediate lethality induced by far-UV in Escherichia coli cells. Redox Rep. 2005, 10, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Oshin, E.A.; Guo, S.; Scott, M.; Li, X.; Mangiamele, C.; Heller, R. Synergistic Effects of an Atmospheric-Pressure Plasma Jet and Pulsed Electric Field on Cells and Skin. IEEE Trans. Plasma Sci. 2021, 49, 3317–3324. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Antinore, M.J.; Huang, Y.; Fornace, A.J. Ultraviolet-irradiation-induced apoptosis is mediated via ligand independent activation of tumor necrosis factor receptor 1. Oncogene 1998, 17, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Pakhomova, O.N.; Khorokhorina, V.A.; Bowman, A.M.; Rodaitė-Riševičienė, R.; Saulis, G.; Xiao, S.; Pakhomov, A.G. Oxidative effects of nanosecond pulsed electric field exposure in cells and cell-free media. Arch. Biochem. Biophys. 2012, 527, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Aboubakr, H.A.; Williams, P.; Gangal, U.; Youssef, M.M.; El-Sohaimy, S.A.A.; Bruggeman, P.J.; Goyal, S.M. Virucidal Effect of Cold Atmospheric Gaseous Plasma on Feline Calicivirus, a Surrogate for Human Norovirus. Appl. Environ. Microbiol. 2015, 81, 3612–3622. [Google Scholar] [CrossRef] [Green Version]
- Kalghatgi, S.; Kelly, C.M.; Cerchar, E.; Torabi, B.; Alekseev, O.; Fridman, A.; Friedman, G.; Azizkhan-Clifford, J. Effects of Non-Thermal Plasma on Mammalian Cells. PLoS ONE 2011, 6, e16270. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, R. Dielectric barrier discharges: Progress on plasma sources and on the understanding of regimes and single filaments. Plasma Sources Sci. Technol. 2017, 26, 053001. [Google Scholar] [CrossRef]
- Winter, J.; Brandenburg, R.; Weltmann, K.-D. Atmospheric pressure plasma jets: An overview of devices and new directions. Plasma Sources Sci. Technol. 2015, 24, 064001. [Google Scholar] [CrossRef]
- Lietz, A.M.; Kushner, M.J. Air plasma treatment of liquid covered tissue: Long timescale chemistry. J. Phys. D Appl. Phys. 2016, 49, 425204. [Google Scholar] [CrossRef]
- Filipić, A.; Primc, G.; Zaplotnik, R.; Mehle, N.; Gutierrez-Aguirre, I.; Ravnikar, M.; Mozetič, M.; Žel, J.; Dobnik, D. Cold Atmospheric Plasma as a Novel Method for Inactivation of Potato Virus Y in Water Samples. Food Environ. Virol. 2019, 11, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Ahlfeld, B.; Li, Y.; Boulaaba, A.; Binder, A.; Schotte, U.; Zimmermann, J.L.; Morfill, G.; Klein, G. Inactivation of a Foodborne Norovirus Outbreak Strain with Nonthermal Atmospheric Pressure Plasma. Mbio 2015, 6, e02300-14. [Google Scholar] [CrossRef] [Green Version]
- Nayak, G.; Aboubakr, H.A.; Goyal, S.M.; Bruggeman, P.J. Reactive species responsible for the inactivation of feline calicivirus by a two-dimensional array of integrated coaxial microhollow dielectric barrier discharges in air. Plasma Process. Polym. 2017, 15, 1700119. [Google Scholar] [CrossRef]
- Yamashiro, R.; Misawa, T.; Sakudo, A. Key role of singlet oxygen and peroxynitrite in viral RNA damage during virucidal effect of plasma torch on feline calicivirus. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.-M.; Zhang, G.-J.; Wu, X.-L.; Peng, Z.-Y.; Zhang, Z.-H.; Shao, X.-J.; Chang, Z.-S. Effect of Low-Temperature Plasma on Deactivation of Hepatitis B Virus. IEEE Trans. Plasma Sci. 2012, 40, 2711–2716. [Google Scholar] [CrossRef]
- Volotskova, O.; Dubrovsky, L.; Keidar, M.; Bukrinsky, M. Cold Atmospheric Plasma Inhibits HIV-1 Replication in Macrophages by Targeting Both the Virus and the Cells. PLoS ONE 2016, 11, e0165322. [Google Scholar] [CrossRef]
- Jin, T.; Xu, Y.; Dai, C.; Zhou, X.; Xu, Q.; Wu, Z. Cold atmospheric plasma: A non-negligible strategy for viral RNA inactivation to prevent SARS-CoV-2 environmental transmission. AIP Adv. 2021, 11, 085019. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, O.; Donovan, K.; Limonnik, V.; Azizkhan-Clifford, J. Nonthermal Dielectric Barrier Discharge (DBD) Plasma Suppresses Herpes Simplex Virus Type 1 (HSV-1) Replication in Corneal Epithelium. Transl. Vis. Sci. Technol. 2014, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, P.; Vono, M.; Venier, P.; Tarricone, E.; Deligianni, V.; Martines, E.; Zuin, M.; Spagnolo, S.; Cavazzana, R.; Cardin, R.; et al. Disinfection of Ocular Cells and Tissues by Atmospheric-Pressure Cold Plasma. PLoS ONE 2012, 7, e33245. [Google Scholar] [CrossRef] [PubMed]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by Nitrosative Agents and H2O2 Involves KEAP1 Disulfide Formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Laporte, A.; Lortz, S.; Schaal, C.; Lenzen, S.; Elsner, M. Hydrogen peroxide permeability of cellular membranes in insulin-producing cells. Biochim. Biophys. Acta (BBA) -Biomembr. 2019, 1862, 183096. [Google Scholar] [CrossRef]
- Kang, S.U.; Kim, H.J.; Kim, D.H.; Han, C.H.; Lee, Y.S.; Kim, C.-H. Nonthermal plasma treated solution inhibits adipocyte differentiation and lipogenesis in 3T3-L1 preadipocytes via ER stress signal suppression. Sci. Rep. 2018, 8, 2277. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Dietrich, S.; Steuer, A.; Weltmann, K.-D.; von Woedtke, T.; Masur, K.; Wende, K. Non-thermal Plasma Activates Human Keratinocytes by Stimulation of Antioxidant and Phase II Pathways. J. Biol. Chem. 2015, 290, 6731–6750. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Choi, E.H.; Han, I. Regulation of Redox Homeostasis by Nonthermal Biocompatible Plasma Discharge in Stem Cell Differentiation. Oxidative Med. Cell. Longev. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Bekeschus, S.; von Woedtke, T.; Kramer, A.; Weltmann, K.-D.; Masur, K. Cold Physical Plasma Treatment Alters Redox Balance in Human Immune Cells. Plasma Med. 2013, 3, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-thermal plasma induces immunogenic cell death in vivo in murine CT26 colorectal tumors. OncoImmunology 2018, 7, e1484978. [Google Scholar] [CrossRef] [Green Version]
- Freund, E.; Liedtke, K.R.; van der Linde, J.; Metelmann, H.-R.; Heidecke, C.-D.; Partecke, L.-I.; Bekeschus, S. Physical plasma-treated saline promotes an immunogenic phenotype in CT26 colon cancer cells in vitro and in vivo. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Clemen, R.; Nießner, F.; Sagwal, S.K.; Freund, E.; Schmidt, A. Medical Gas Plasma Jet Technology Targets Murine Melanoma in an Immunogenic Fashion. Adv. Sci. 2020, 7, 1903438. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Lippert, M.; Diepold, K.; Chiosis, G.; Seufferlein, T.; Azoitei, N. Physical plasma-triggered ROS induces tumor cell death upon cleavage of HSP90 chaperone. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Rödder, K.; Fregin, B.; Otto, O.; Lippert, M.; Weltmann, K.-D.; Wende, K.; Schmidt, A.; Gandhirajan, R.K. Toxicity and Immunogenicity in Murine Melanoma following Exposure to Physical Plasma-Derived Oxidants. Oxid. Med. Cell. Longev. 2017, 2017, 4396467. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Bekeschus, S.; Rödder, K.; Schmidt, A.; Stope, M.B.; von Woedtke, T.; Miller, V.; Fridman, A.; Weltmann, K.-D.; Masur, K.; Metelmann, H.-R.; et al. Cold physical plasma selects for specific T helper cell subsets with distinct cells surface markers in a caspase-dependent and NF-κB-independent manner. Plasma Process. Polym. 2016, 13, 1144–1150. [Google Scholar] [CrossRef]
- Mohamed, H.; Clemen, R.; Freund, E.; Lackmann, J.-W.; Wende, K.; Connors, J.; Haddad, E.K.; Dampier, W.; Wigdahl, B.; Miller, V.; et al. Non-thermal plasma modulates cellular markers associated with immunogenicity in a model of latent HIV-1 infection. PLoS ONE 2021, 16, e0247125. [Google Scholar] [CrossRef]
- Reske, A.; Pollara, G.; Krummenacher, C.; Katz, D.R.; Chain, B.M. Glycoprotein-Dependent and TLR2-Independent Innate Immune Recognition of Herpes Simplex Virus-1 by Dendritic Cells. J. Immunol. 2008, 180, 7525–7536. [Google Scholar] [CrossRef] [Green Version]
- Bedoui, S.; Greyer, M. The role of dendritic cells in immunity against primary herpes simplex virus infections. Front. Microbiol. 2014, 5, 533. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Chen, S.H.; Oakes, J.E.; Lausch, R.N. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J. Virol. 1996, 70, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, H.; Bin Wei, B. Immune response of T cells during herpes simplex virus type 1 (HSV-1) infection. J. Zhejiang Univ. B 2017, 18, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upasani, V.; Rodenhuis-Zybert, I.; Cantaert, T. Antibody-independent functions of B cells during viral infections. PLoS Pathog. 2021, 17, e1009708. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutter, J.; Bruggeman, P.J.; Wigdahl, B.; Krebs, F.C.; Miller, V. Manipulation of Oxidative Stress Responses by Non-Thermal Plasma to Treat Herpes Simplex Virus Type 1 Infection and Disease. Int. J. Mol. Sci. 2023, 24, 4673. https://doi.org/10.3390/ijms24054673
Sutter J, Bruggeman PJ, Wigdahl B, Krebs FC, Miller V. Manipulation of Oxidative Stress Responses by Non-Thermal Plasma to Treat Herpes Simplex Virus Type 1 Infection and Disease. International Journal of Molecular Sciences. 2023; 24(5):4673. https://doi.org/10.3390/ijms24054673
Chicago/Turabian StyleSutter, Julia, Peter J. Bruggeman, Brian Wigdahl, Fred C. Krebs, and Vandana Miller. 2023. "Manipulation of Oxidative Stress Responses by Non-Thermal Plasma to Treat Herpes Simplex Virus Type 1 Infection and Disease" International Journal of Molecular Sciences 24, no. 5: 4673. https://doi.org/10.3390/ijms24054673
APA StyleSutter, J., Bruggeman, P. J., Wigdahl, B., Krebs, F. C., & Miller, V. (2023). Manipulation of Oxidative Stress Responses by Non-Thermal Plasma to Treat Herpes Simplex Virus Type 1 Infection and Disease. International Journal of Molecular Sciences, 24(5), 4673. https://doi.org/10.3390/ijms24054673