


The Role of BCL-2 and PD-1/PD-L1 Pathway in Pathogenesis of Myelodysplastic Syndromes


Abstract
:1. Introduction
2. Function of the BCL-2 Family of Proteins
3. Genetic Background, Structure and Polymorphism
4. The Importance of the BCL-2 Family in the Course of MDS
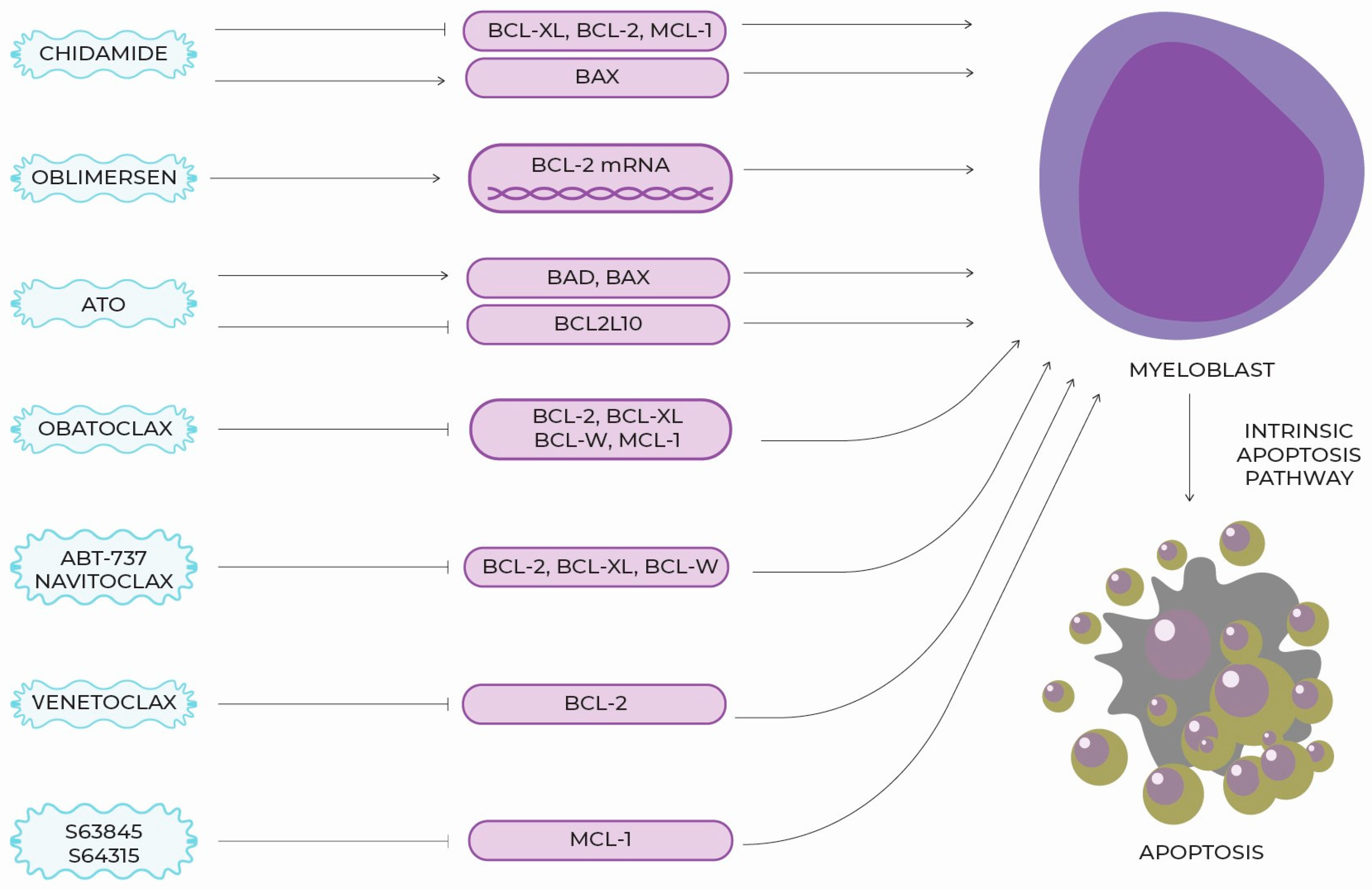
5. New Targeted Molecules, Therapeutic Perspectives and Clinical Use
6. BCL-2 Section Summary
7. PD-1—Structure and Function
8. PD-L1 and PD-L-2 Structure and Function
9. The Role of the Pro-Inflammatory Microenvironment in the Pathogenesis of MDSs
10. The Importance of PD-1 and PD-L1 Signaling Pathway in the Course of MDSs
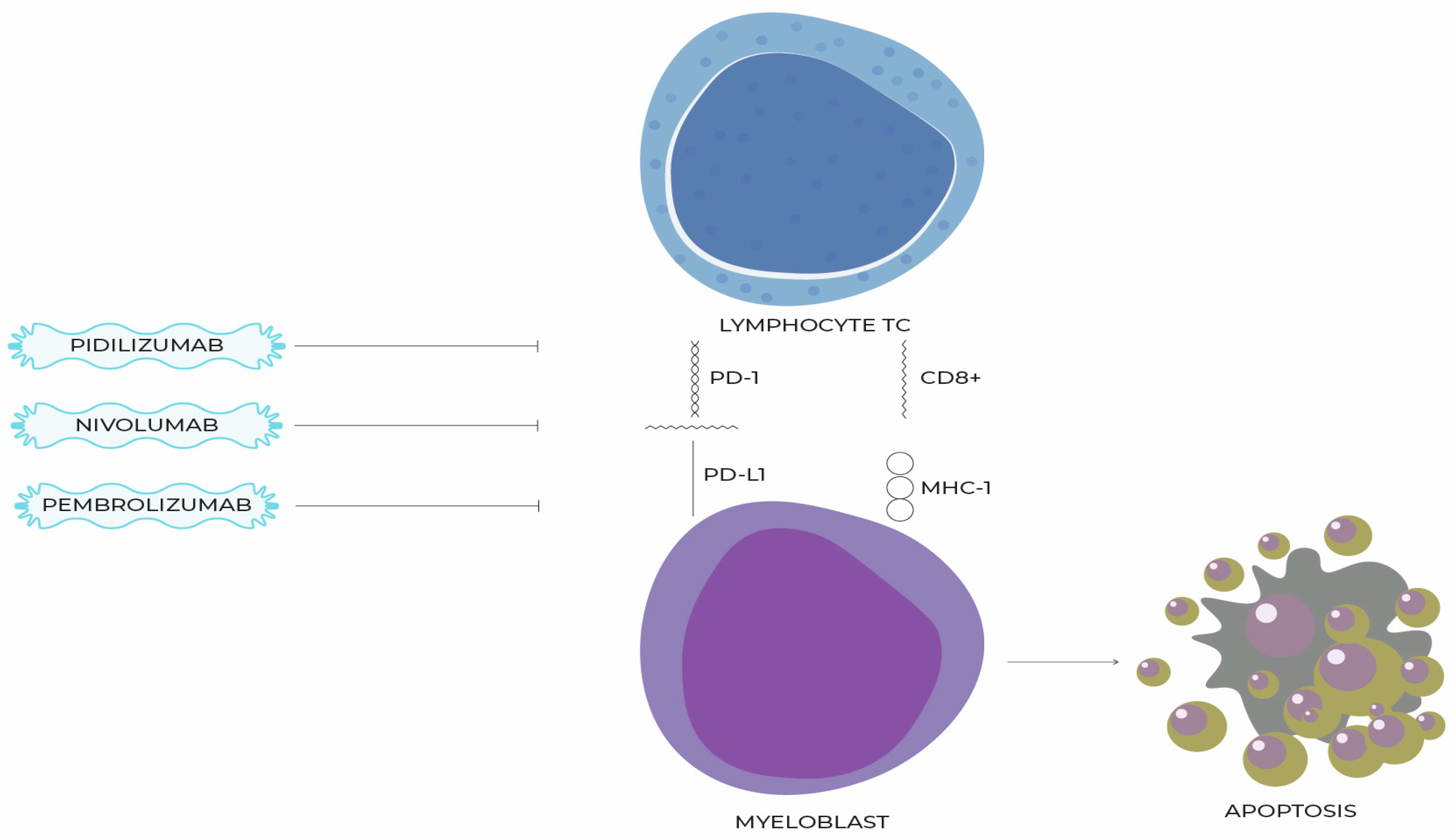
11. Molecules That Inhibit the PD-1/PD-L1 Pathway and Their Use in the Treatment of MDSs
12. Preclinical Data on the Association of BCL-2 Protein and the PD-1/PD-1L Pathway
13. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Mądry, K.; Machowicz, R.; Waszczuk-Gajda, A.; Drozd-Sokołowska, J.; Hołowiecka, B.S.; Wiater, E.; Mital, A.; Obara, A.; Szmigielska-Kapłon, A.; Kołkowska-Lesniak, A.; et al. Demographic, Hematologic, and Clinical Features of Myelodysplastic Syndrome Patients: Results from the First Polish Myelodysplastic Syndrome Registry. Acta Haematol. 2015, 134, 125–134. [Google Scholar] [CrossRef]
- Rollison, D.E.; Howlader, N.; Smith, M.T.; Strom, S.S.; Merritt, W.D.; Ries, L.A.; Edwards, B.K.; List, A.F. Epidemiology of Myelodysplastic Syndromes and Chronic Myeloproliferative Disorders in the United States, 2001–2004, Using Data from the NAACCR and SEER Programs. Blood 2008, 112, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, A.M.; Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X. Epidemiology of Myelodysplastic Syndromes: Why Characterizing the Beast Is a Prerequisite to Taming It. Blood Rev. 2019, 34, 1–15. [Google Scholar] [CrossRef]
- Bhatia, S. Therapy-Related Myelodysplasia and Acute Myeloid Leukemia. Semin. Oncol. 2013, 40, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Visconte, V.; Tiu, R.V.; Rogers, H.J. Pathogenesis of Myelodysplastic Syndromes: An Overview of Molecular and Non-Molecular Aspects of the Disease. Blood Res. 2014, 49, 216–227. [Google Scholar] [CrossRef]
- Komrokji, R.S. Treatment of Higher-Risk Myelodysplastic Syndromes After Failure of Hypomethylating Agents. Clin. Lymphoma Myeloma Leuk. 2015, 15, S56–S59. [Google Scholar] [CrossRef]
- Liu, L.; Jia, M.; Sun, L.; Tian, W.; Tang, P.; Jiang, Z. Meta-Analysis of the Benefit of Hypomethylating Agents before Allogeneic Hematopoietic Stem Cell Transplantation in Myelodysplastic Syndromes. Clin. Exp. Med. 2021, 21, 537–543. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [Green Version]
- Calvi, L.M.; Li, A.J.; Becker, M.W. What Is the Role of the Microenvironment in MDS? Best Pract. Res. Clin. Haematol. 2019, 32, 101113. [Google Scholar] [CrossRef]
- Ganan-Gomez, I.; Yang, H.; Ma, F.; Montalban-Bravo, G.; Thongon, N.; Marchica, V.; Richard-Carpentier, G.; Chien, K.; Manyam, G.; Wang, F.; et al. Stem Cell Architecture Drives Myelodysplastic Syndrome Progression and Predicts Response to Venetoclax-Based Therapy. Nat. Med. 2022, 28, 557–567. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Ball, B.J.; Famulare, C.A.; Stein, E.M.; Tallman, M.S.; Derkach, A.; Roshal, M.; Gill, S.I.; Manning, B.M.; Koprivnikar, J.; Mccloskey, J.; et al. Venetoclax and Hypomethylating Agents (HMAs) Induce High Response Rates in MDS, Including Patients after HMA Therapy Failure. Blood Adv. 2020, 4, 2866–2870. [Google Scholar] [CrossRef]
- Jelinek, T.; Mihalyova, J.; Kascak, M.; Duras, J.; Hajek, R. PD-1/PD-L1 Inhibitors in Haematological Malignancies: Update 2017. Immunology 2017, 152, 357–371. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, S.; Rowley, J.D. Chromosome 14 Translocations in Non-burkitt Lymphomas. Int. J. Cancer 1978, 22, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Arulananda, S.; Lee, E.F.; Fairlie, W.D.; John, T. The Role of BCL-2 Family Proteins and Therapeutic Potential of BH3-Mimetics in Malignant Pleural Mesothelioma. Expert Rev. Anticancer Ther. 2021, 21, 413–424. [Google Scholar] [CrossRef]
- Kiliszek, P.; Juszczyński, P. Deregulacja Rodziny Białek BCL2 w Chłoniakach B-Komórkowych—Implikacje Molekularne, Patogenetyczne, Kliniczne i Terapeutyczne Deregulation of BCL2 Family Proteins in B-Cell Lymphomas—Molecular, Pathogenetic, Clinical and Therapeutic Implications. Hematol. Clin. Pract. 2012, 3, 288–301. [Google Scholar]
- Bednarek, J.; Wesierska-Gadek, J.; Kiliańska, Z.M. New Face of Antiapoptotic Proteins. I. Protein Mcl-Postepy Biochem. 2007, 53, 228–238. [Google Scholar]
- McBride, A.; Houtmann, S.; Wilde, L.; Vigil, C.; Eischen, C.M.; Kasner, M.; Palmisiano, N. The Role of Inhibition of Apoptosis in Acute Leukemias and Myelodysplastic Syndrome. Front. Oncol. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty Years of BCL-2: Translating Cell Death Discoveries into Novel Cancer Therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Choudhury, S. A Comparative Analysis of BCL-2 Family. Bioinformation 2019, 15, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Shi, J.; Sun, W.; Ruan, X.; Guo, Y.; Zhao, L.; Wang, J.; Li, B. Genetic Polymorphisms of Bcl-2 Promoter in Cancer Susceptibility and Prognosis: A Meta-Analysis. Oncotarget 2018, 9, 12351–12364. [Google Scholar] [CrossRef] [Green Version]
- Gasperin-s, H.; Ben, C.G.; Zamorano-carrillo, A. Exploring the Conformational Space of Bcl-2 Protein Variants: Dynamic Contributions of the Flexible Loop Domain and Transmembrane Region. Molecules 2019, 24, 3896. [Google Scholar]
- Pan, W.; Yang, J.; Wei, J.; Chen, H.; Ge, Y.; Zhang, J.; Wang, Z.; Zhou, C.; Yuan, Q.; Zhou, L.; et al. Functional BCL-2 Regulatory Genetic Variants Contribute to Susceptibility of Esophageal Squamous Cell Carcinoma. Sci. Rep. 2015, 5, 11833. [Google Scholar] [CrossRef]
- Zhang, X.; Weng, W.; Xu, W.; Wang, Y.; Yu, W.; Tang, X.; Ma, L.; Pan, Q.; Wang, J.; Sun, F. Role of Bcl-2-938 C>A Polymorphism in Susceptibility and Prognosis of Cancer: A Meta-Analysis. Sci. Rep. 2014, 4, 7241. [Google Scholar] [CrossRef] [Green Version]
- Hasle, H.; Niemeyer, C.M.; Chessells, J.M.; Baumann, I.; Bennett, J.M.; Kerndrup, G.; Head, D.R. A Pediatric Approach to the WHO Classification of Myelodysplastic and Myeloproliferative Diseases. Leukemia 2003, 17, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Kurotaki, H.; Tsushima, Y.; Nagai, K.; Yagihashi, S. Apoptosis, Bcl-2 Expression and P53 Accumulation in Myelodysplastic Syndrome, Myelodysplastic-Syndrome-Derived Acute Myelogenous Leukemia and de Novo Acute Myelogenous Leukemia. Acta Haematol. 1999, 102, 115–123. [Google Scholar] [CrossRef]
- Boudard, D.; Vasselon, C.; Berthéas, M.F.; Jaubert, J.; Mounier, C.; Reynaud, J.; Viallet, A.; Chautard, S.; Guyotat, D.; Campos, L. Expression and Prognostic Significance of Bcl-2 Family Proteins in Myelodysplastic Syndromes. Am. J. Hematol. 2002, 70, 115–125. [Google Scholar] [CrossRef]
- Mittelman, M.; Oster, H.S.; Hoffman, M.; Neumann, D. The Lower Risk MDS Patient at Risk of Rapid Progression. Leuk. Res. 2010, 34, 1551–1555. [Google Scholar] [CrossRef]
- Davis, R.E.; Greenberg, P.L. Bcl-2 Expression by Myeloid Precursors in Myelodysplastic Syndromes: Relation to Disease Progression. Leuk. Res. 1998, 22, 767–777. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in Hematologic Malignancies: Rationale and Progress. Blood Rev. 2020, 176, 139–148. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in Cancer: Current Status and Perspectives. J. Hematol. Oncol. 2021, 14, 1–18. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 Is a Mechanism of Intrinsic Resistance to ABT-199 Which Can Be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Llambi, F.; Wang, Y.M.; Victor, B.; Yang, M.; Schneider, D.M.; Gingras, S.; Parsons, M.J.; Zheng, J.H.; Brown, S.A.; Pelletier, S.; et al. BOK Is a Non-Canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell 2016, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Moravcikova, E.; Krepela, E.; Donnenberg, V.S.; Donnenberg, A.D.; Benkova, K.; Rabachini, T.; Fernandez-Marrero, Y.; Bachmann, D.; Kaufmann, T. BOK Displays Cell Death-Independent Tumor Suppressor Activity in Non-Small Cell Lung Carcinoma. Int. J. Cancer. 2017, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.H.; Perales, O.; Michaud, M.; Katz, S.G. BOK Promotes Erythropoiesis in a Mouse Model of Myelodysplastic Syndrome. Ann. Hematol. 2019 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Hamada, Y.; Tamura, H.; Ishibashi, M.; Okuyama, N.; Kondo, A.; Moriya, K.; Ogata, K.; Kakumoto, K.; Miyazaki, Y.; Matsuda, A.; et al. CD7 Expression On Blasts Of Myelodysplastic Syndromes Is Associated With Apoptosis Resistance With Decreased Expression Of The Proapoptotic Protein Bad and An Independent Unfavorable Prognostic Factor Together With The Revised IPSS Score In Patients. Blood 2013, 122, 2799. [Google Scholar] [CrossRef]
- Tyczewska, A.; Twardowski, T. Choroby Cywilizacyjne—Terapeutyczne Zastosowania Strategii Antysensu. Nauka 2008, 1, 45–62. [Google Scholar]
- Walker, A.R.; Marcucci, G.; Yin, J.; Blum, W.; Stock, W.; Kohlschmidt, J.; Mrózek, K.; Carroll, A.J.; Eisfeld, A.K.; Wang, E.S.; et al. Phase 3 Randomized Trial of Chemotherapy with or without Oblimersen in Older AML Patients: CALGB 10201 (Alliance). Blood Adv. 2021, 5, 2775–2787. [Google Scholar] [CrossRef]
- Wu, X.; Hu, Z.; Nizzero, S.; Zhang, G.; Ramirez, M.R.; Shi, C.; Zhou, J.; Ferrari, M.; Shen, H. Bone-Targeting Nanoparticle to Co-Deliver Decitabine and Arsenic Trioxide for Effective Therapy of Myelodysplastic Syndrome with Low Systemic Toxicity. J. Control. Release 2017, 268, 92–101. [Google Scholar] [CrossRef]
- Wu, P.; Liu, L.; Weng, J.; Geng, S.; Deng, C.; Lu, Z.; Luo, C.; Du, X. The Synergistic Effects of Decitabine Combined with Arsenic Trioxide (ATO) in the Human Myelodysplastic Syndrome Cell Line SKM-1. Indian J. Hematol. Blood Transfus. 2016, 32, 412–417. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wang, Y.; Tong, H.; Qian, W.; Jin, J. Downregulation of HTERT: An Important As2O3 Induced Mechanism of Apoptosis in Myelodysplastic Syndrome. PLoS ONE 2014, 9, e113199. [Google Scholar] [CrossRef] [Green Version]
- Galimberti, S.; Guerrini, F.; Salvi, F.; Petrini, I.; Gioia, D.; Messa, E.; Palumbo, G.A.; Cilloni, D.; Petrini, M.; Levis, A. Arsenic Trioxide and Ascorbic Acid Interfere with the BCL2 Family Genes in Patients with Myelodysplastic Syndromes: An Ex-Vivo Study. J. Hematol. Oncol. 2012, 5, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Liu, Z.; Jiang, H.; Li, L.; Fu, R. Decitabine Shows Synergistic Effects with Arsenic Trioxide against Myelodysplastic Syndrome Cells via Endoplasmic Reticulum Stress-Related Apoptosis. J. Investig. Med. 2019, 67, 1067–1075. [Google Scholar] [CrossRef]
- Anether, G.; Tinhofer, I.; Senfter, M.; Greil, R. Tetrocarcin-A-Induced ER Stress Mediates Apoptosis in B-CLL Cells via a Bcl-2-Independent Pathway. Blood 2003, 101, 4561–4568. [Google Scholar] [CrossRef]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Bélec, L.; Billot, X.; et al. Small Molecule Obatoclax (GX15-070) Antagonizes MCL-1 and Overcomes MCL-1-Mediated Resistance to Apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [Green Version]
- Ades, L.; Boehrer, S.; Braun, T.; Grosjean, J.; Fabre, C.; Fenaux, P.; Kroemer, G. Disruption of Bcl-2 Heterodimerization by the BH3 Mimetic ABT-737 Restores Spontaneous Apoptosis and Induces Differentiation in High Risk MDS and AML. Blood 2007, 110, 398. [Google Scholar] [CrossRef]
- Gorombei, P.; Guidez, F.; Ganesan, S.; Chiquet, M.; Pellagatti, A.; Goursaud, L.; Tekin, N.; Beurlet, S.; Patel, S.; Guerenne, L.; et al. Bcl-2 Inhibitor Abt-737 Effectively Targets Leukemia-Initiating Cells with Differential Regulation of Relevant Genes Leading to Extended Survival in a Nras/Bcl-2 Mouse Model of High Risk-Myelodysplastic Syndrome. Int. J. Mol. Sci. 2021, 22, 10658. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a Targeted High-Affinity Inhibitor of BCL-2, in Lymphoid Malignancies: A Phase 1 Dose-Escalation Study of Safety, Pharmacokinetics, Pharmacodynamics, and Antitumour Activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro De Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients with Relapsed Small Cell Lung Cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [Green Version]
- Jilg, S.; Kauschinger, J.; Reidel, V.; Müller-Thomas, C.; Hauch, R.; Schauwecker, J.; Schmidt, B.; Höckendorf, U.; Peschel, C.; Götze, K.; et al. Combination of 5-Azacytidine and ABT-199 Has a Synergistic Apoptotic Effect in High-Risk MDS/SAML after HMA Failure. Blood 2016, 128, 4297. [Google Scholar] [CrossRef]
- Jilg, S.; Reidel, V.; Müller-Thomas, C.; König, J.; Schauwecker, J.; Höckendorf, U.; Huberle, C.; Gorka, O.; Schmidt, B.; Burgkart, R.; et al. Blockade of BCL-2 Proteins Efficiently Induces Apoptosis in Progenitor Cells of High-Risk Myelodysplastic Syndromes Patients. Leukemia 2016, 30, 112–123. [Google Scholar] [CrossRef]
- Jilg, S.; Hauch, R.T.; Kauschinger, J.; Buschhorn, L.; Odinius, T.O.; Dill, V.; Thomas, C.M.; Herold, T.; Prodinger, P.M.; Schmidt, B.; et al. Venetoclax with Azacitidine Targets Refractory MDS but Spares Healthy Hematopoiesis at Tailored Dose. Exp. Hematol. Oncol. 2019, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Garcia, J.S.; Borate, U.; Fong, C.Y.; Baer, M.R.; Nolte, F.; Peterlin, P.; Jurcic, J.G.; Garcia-Manero, G.; Hong, W.-J.; et al. A Phase 1b Study Evaluating the Safety and Efficacy of Venetoclax in Combination with Azacitidine in Treatment-Naïve Patients with Higher-Risk Myelodysplastic Syndrome. Blood 2019, 134, 568. [Google Scholar] [CrossRef]
- Hecker, J.S.; Pachzelt, L.; Götze, K.S. Are Myelodysplastic Syndromes Ready for Venetoclax? Exploring Future Potential and Considerations. Expert Rev. Hematol. 2021, 14, 789–793. [Google Scholar] [CrossRef]
- Garcia, J.S.; Kim, H.T.; Murdock, H.M.; Cutler, C.S.; Brock, J.; Gooptu, M.; Ho, V.T.; Koreth, J.; Nikiforow, S.; Romee, R.; et al. Adding Venetoclax to Fludarabine/Busulfan RIC Transplant for High-Risk MDS and AML Is Feasible, Safe, and Active. Blood Adv. 2021, 5, 5536–5545. [Google Scholar] [CrossRef]
- Reidel, V.; Kauschinger, J.; Hauch, R.T.; Müller-Thomas, C.; Nadarajah, N.; Burgkart, R.; Schmidt, B.; Hempel, D.; Jacob, A.; Slotta-Huspenina, J.; et al. Selective Inhibition of BCL-2 Is a Promising Target in Patients with High-Risk Myelodysplastic Syndromes and Adverse Mutational Profile. Oncotarget 2018, 9, 17270–17281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.V.; Chhetri, R.; Dholakia, R.; Kok, C.H.; Gangat, N.; Alkhateeb, H.B.; Al-Kali, A.; Patnaik, M.M.; Baranwal, A.; Greipp, P.T.; et al. Outcomes Following Venetoclax-Based Treatment in Therapy-Related Myeloid Neoplasms. Am. J. Hematol. 2022, 97, 1013–1022. [Google Scholar] [CrossRef]
- Chandhok, N.S.; Boddu, P.C.; Gore, S.D.; Prebet, T. What Are the Most Promising New Agents in Myelodysplastic Syndromes? Curr. Opin. Hematol. 2019, 26, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.W.; Wang, H.Q.; Ban, W.W.; Chang, Z.; Chen, H.Z.; Jia, L.; Liu, F.T. Novel HDAC Inhibitor Chidamide Synergizes with Rituximab to Inhibit Diffuse Large B-Cell Lymphoma Tumour Growth by Upregulating CD20. Cell Death Dis. 2020, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Zhao, S.; Guo, J.; Zhao, Y.; Fei, C.; Gu, S.; Li, X. Chidamide, a Novel Histone Deacetylase Inhibitor, Displays Potent Antitumor Activity Against MDS Cells Mainly through JAK2/STAT3 Signaling Inhibition. Blood 2015, 126, 5233. [Google Scholar] [CrossRef]
- Liu, Z.; ding, K.; Li, L.; Liu, H.; Wang, Y.; Liu, C.; Fu, R. A Novel Histone Deacetylase Inhibitor Chidamide Induces G0/G1 Arrest and Apoptosis in Myelodysplastic Syndromes. Biomed. Pharmacother. 2016, 83, 1032–1037. [Google Scholar] [CrossRef]
- Mao, J.; Li, S.; Zhao, H.; Zhu, Y.; Hong, M.; Zhu, H.; Qian, S.X.; Li, J.Y. Effects of Chidamide and Its Combination with Decitabine on Proliferation and Apoptosis of Leukemia Cell Lines. Am. J. Transl. Res. 2018, 10, 2567–2578. [Google Scholar]
- Li, G.; Li, D.; Yuan, F.; Cheng, C.; Chen, L.; Wei, X. Synergistic Effect of Chidamide and Venetoclax on Apoptosis in Acute Myeloid Leukemia Cells and Its Mechanism. Ann. Transl. Med. 2021, 9, 1575. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 Immunoreceptor Inhibits B Cell Receptor-Mediated Signaling by Recruiting Src Homology 2-Domain-Containing Tyrosine Phosphatase 2 to Phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Ma, L.; Zhang, X.; Huang, L.; Wei, J. Targeting PD-1/PD-L1 Pathway in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Exp. Hematol. Oncol. 2022, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3ζ Signalosome and Downstream Signaling to PKCθ. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkazanc, D.; Yoyen-Ermis, D.; Tavukcuoglu, E.; Buyukasik, Y.; Esendagli, G. Functional Exhaustion of CD4+ T Cells Induced by Co-Stimulatory Signals from Myeloid Leukaemia Cells. Immunology 2016, 149, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-Cell Exhaustion: Characteristics, Causes and Conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Freeman, G.J.; Wherry, E.J.; Ahmed, R.; Sharpe, A.H. Reinvigorating Exhausted HIV-Specific T Cells via PD-1-PD-1 Ligand Blockade. J. Exp. Med. 2006, 203, 2223–2227. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune Dilated Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Grzywnowicz, M.; Giannopoulos, K. The Role of Receptor Programmed Death-1 and Its Ligands in Immune System and Tumors. Acta Haematol. Pol. 2012, 43, 132–145. [Google Scholar] [CrossRef]
- Wang, J.; Yoshida, T.; Nakaki, F.; Hiai, H.; Okazaki, T.; Honjo, T. Establishment of NOD-Pdcd1−/− Mice as an Efficient Animal Model of Type I Diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 11823–11828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quatrini, L.; Vacca, P.; Tumino, N.; Besi, F.; Di Pace, A.L.; Scordamaglia, F.; Martini, S.; Munari, E.; Mingari, M.C.; Ugolini, S.; et al. Glucocorticoids and the Cytokines IL-12, IL-15, and IL-18 Present in the Tumor Microenvironment Induce PD-1 Expression on Human Natural Killer Cells. J. Allergy Clin. Immunol. 2021, 147, 349–360. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 Regulates PD-1 Expression upon T Cell Activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef] [Green Version]
- Yoyen-Ermis, D.; Tunali, G.; Tavukcuoglu, E.; Horzum, U.; Ozkazanc, D.; Sutlu, T.; Buyukasik, Y.; Esendagli, G. Myeloid Maturation Potentiates STAT3-Mediated Atypical IFN-γ Signaling and Upregulation of PD-1 Ligands in AML and MDS. Sci. Rep. 2019, 9, 11697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmaninejad, A.; Khoramshahi, V.; Azani, A.; Soltaninejad, E.; Aslani, S.; Zamani, M.R.; Zal, M.; Nesaei, A.; Hosseini, S.M. PD-1 and Cancer: Molecular Mechanisms and Polymorphisms. Immunogenetics 2018, 70, 73–86. [Google Scholar] [CrossRef]
- Studies, C. Association between PD-1 and PD-L1 Polymorphisms. Cancers 2019, 11, 1150. [Google Scholar]
- Zak, K.M.; Grudnik, P.; Magiera, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef]
- Philips, E.A.; Garcia-España, A.; Tocheva, A.S.; Ahearn, I.M.; Adam, K.R.; Pan, R.; Mor, A.; Kong, X.P. The Structural Features That Distinguish PD-L2 from PD-L1 Emerged in Placental Mammals. J. Biol. Chem. 2020, 295, 4372–4380. [Google Scholar] [CrossRef] [Green Version]
- Flies, D.B.; Chen, L. The New B7s: Playing a Pivotal Role in Tumor Immunity. J. Immunother. 2007, 30, 251–260. [Google Scholar] [CrossRef]
- Lázár-Molnár, E.; Yan, Q.; Cao, E.; Ramagopal, U.; Nathenson, S.G.; Almo, S.C. Crystal Structure of the Complex between Programmed Death-1 (PD-1) and Its Ligand PD-L. Proc. Natl. Acad. Sci. USA 2008, 105, 10483–10488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The Bone-Marrow Niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Graf, J.R.; Forster, S.; Bruehl, F.K.; Banz, Y.; Hallal, M.; Brodard, J.; Bacher, V.U.; Allam, R.; Schürch, C.M.; Bonadies, N. Diagnostic and Prognostic Implications of Caspase-1 and Pd-l1 Co-expression Patterns in Myelodysplastic Syndromes. Cancers 2021, 13, 5712. [Google Scholar] [CrossRef]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone Progenitor Dysfunction Induces Myelodysplasia and Secondary Leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Li, A.J.; Calvi, L.M. The Microenvironment in Myelodysplastic Syndromes: Niche-Mediated Disease Initiation and Progression. Exp. Hematol. 2017, 55, 3–18. [Google Scholar] [CrossRef]
- Pronk, E.; Raaijmakers, M.H.G.P. The Mesenchymal Niche in MDS. Blood 2019, 133, 1031–1038. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 Inflammasome Functions as a Driver of the Myelodysplastic Syndrome Phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of Myelodysplasia by Myeloid-Derived Suppressor Cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Eksioglu, E.A.; Chen, X.; Kandell, W.; Le Trinh, T.; Cen, L.; Qi, J.; Sallman, D.A.; Zhang, Y.; Tu, N.; et al. S100A9-Induced Overexpression of PD-1/PD-L1 Contributes to Ineffective Hematopoiesis in Myelodysplastic Syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef]
- Kouroukli, O.; Symeonidis, A.; Foukas, P.; Maragkou, M.K.; Kourea, E.P. Bone Marrow Immune Microenvironment in Myelodysplastic Syndromes. Cancers 2022, 14, 5656. [Google Scholar] [CrossRef]
- Dong, Y.; Han, Y.; Huang, Y.; Jiang, S.; Huang, Z.; Chen, R.; Yu, Z.; Yu, K.; Zhang, S. PD-L1 Is Expressed and Promotes the Expansion of Regulatory T Cells in Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 1710. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 10–12. [Google Scholar] [CrossRef]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-γ and Tumor Necrosis Factor-α Induce an Immunoinhibitory Molecule, B7-H1, via Nuclear Factor-ΚB Activation in Blasts in Myelodysplastic Syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 Mutations in Myelodysplastic Syndromes and Secondary AML Confer an Immunosuppressive Phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Moskorz, W.; Cosmovici, C.; Jäger, P.S.; Cadeddu, R.P.; Timm, J.; Haas, R. Myelodysplastic Syndrome Patients Display Alterations in Their Immune Status Reflected by Increased PD-L1-Expressing Stem Cells and Highly Dynamic Exhausted T-Cell Frequencies. Br. J. Haematol. 2021, 193, 941–945. [Google Scholar] [CrossRef]
- Montes, P.; Bernal, M.; Campo, L.N.; González-Ramírez, A.R.; Jiménez, P.; Garrido, P.; Jurado, M.; Garrido, F.; Ruiz-Cabello, F.; Hernández, F. Tumor Genetic Alterations and Features of the Immune Microenvironment Drive Myelodysplastic Syndrome Escape and Progression. Cancer Immunol. Immunother. 2019, 68, 2015–2027. [Google Scholar] [CrossRef]
- Tcvetkov, N.Y.; Morozova, E.V.; Epifanovskaya, O.S.; Babenko, E.V.; Barabanshikova, M.V.; Lepik, K.V.; Bakin, E.A.; Vlasova, J.J.; Smirnova, A.G.; Zander, A.R.; et al. Profile of Checkpoint Molecules Expression on Bone Marrow Cell Populations in Patients with High-Risk Myelodysplastic Syndrome. Blood 2020, 136, 43–44. [Google Scholar] [CrossRef]
- Coats, T.; Smith, A.e; Mourikis, T.P.; Irish, J.M.; Kordasti, S.; Mufti, G.J. Mass Cytometry Reveals PD1 Upregulation Is an Early Step in MDS Disease Progression. Blood 2016, 128, 4296. [Google Scholar] [CrossRef]
- Meng, F.; Li, L.; Lu, F.; Yue, J.; Liu, Z.; Zhang, W.; Fu, R. Overexpression of TIGIT in NK and T Cells Contributes to Tumor Immune Escape in Myelodysplastic Syndromes. Front. Oncol. 2020, 10, 1595. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in Myelodysplastic Syndromes Is Enhanced by Treatment with Hypomethylating Agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenborn, J.R.; Wilson, C.B. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Van de Loosdrecht, A.A.; Alhan, C.; Ossenkoppele, G.J.; Westers, T.M.; Bontkes, H.J. Role of Immune Responses in the Pathogenesis of Low-Risk MDS and High-Risk MDS: Implications for Immunotherapy. Br. J. Haematol. 2011, 153, 568–581. [Google Scholar] [CrossRef]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and Up-Regulation of PD-1 in T Cells by Azacytidine in MDS/AML Patients: A Rationale for Combined Targeting of PD-1 and DNA Methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Manero, G.; Tallman, M.S.; Martinelli, G.; Ribrag, V.; Yang, H.; Balakumaran, A.; Chlosta, S.; Zhang, Y.; Smith, B.D. Pembrolizumab, a PD-1 Inhibitor, in Patients with Myelodysplastic Syndrome (MDS) after Failure of Hypomethylating Agent Treatment. Blood 2016, 128, 345. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Daver, N.G.; Jabbour, E.J.; Alvarado, Y.; DiNardo, C.D.; Ravandi, F.; Borthakur, G.; Bose, P.; et al. A Phase II Study of Nivolumab or Ipilimumab with or without Azacitidine for Patients with Myelodysplastic Syndrome (MDS). Blood 2018, 132, 465. [Google Scholar] [CrossRef]
- Santini, V. How I Treat MDS after Hypomethylating Agent Failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Chien, K.S.; Kim, K.; Nogueras-Gonzalez, G.M.; Borthakur, G.; Naqvi, K.; Daver, N.G.; Montalban-Bravo, G.; Cortes, J.E.; DiNardo, C.D.; Jabbour, E.; et al. Phase II Study of Azacitidine with Pembrolizumab in Patients with Intermediate-1 or Higher-Risk Myelodysplastic Syndrome. Br. J. Haematol. 2021, 195, 378–387. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Cavenagh, J.; Voso, M.T.; Taussig, D.; Tormo, M.; Boss, I.; Copeland, W.B.; Gray, V.E.; Previtali, A.; O’Connor, T.; et al. Efficacy and Safety of Azacitidine (AZA) in Combination with the Anti-PD-L1 Durvalumab (Durva) for the Front-Line Treatment of Older Patients (Pts) with Acute Myeloid Leukemia (AML) Who Are Unfit for Intensive Chemotherapy (IC) and Pts with Higher-Risk My. Blood 2019, 134, 829. [Google Scholar] [CrossRef]
- Takayama, S.; Sato, T.; Krajewski, S.; Kochel, K.; Irie, S.; Milian, J.A.; Reed, J.C. Cloning and Functional Analysis of BAG-1: A Novel Bcl-2-Binding Protein with Anti-Cell Death Activity. Cell 1995, 80, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Adam, A.; Zhao, C.; Chen, H. Recent Advancements in the Mechanisms Underlying Resistance to Pd-1/Pd-L1 Blockade Immunotherapy. Cancers 2021, 13, 663. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Shen, J.; Lan, Q.; Zhang, K.; Xu, Y.; Duah, M.; Xu, K.; Pan, B. Blockade of PD-1/PD-L1 Increases Effector T Cells and Aggravates Murine Chronic Graft-versus-Host Disease. Int. Immunopharmacol. 2022, 110, 109051. [Google Scholar] [CrossRef] [PubMed]
- Dronca, R.S.; Mansfield, A.S.; Park, S.S.; Dong, H. BCL-2-Interacting Mediator of Cell Death (Bim) Is a Novel Biomarker for Response to Anti-PD-1 Therapy in Patients with Advanced Melanoma. Immunotherapy 2016, 8, 1351–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhapp, F.J.; Haribhai, D.; Mathew, R.; Duggan, R.; Ellis, P.A.; Wang, R.; Lasater, E.A.; Shi, Y.; Dave, N.; Riehm, J.J.; et al. Venetoclax Increases Intratumoral Effector t Cells and Antitumor Efficacy in Combination with Immune Checkpoint Blockade. Cancer Discov. 2021, 11, 68–79. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference/Identifier | Trial Phase | Population Characteristics | Molecular Target | Treatment | Results/Status |
|---|---|---|---|---|---|
| Galimberti et al. [44] | Phase II Single-arm | N = 12 with MDS | BCL2L10, BAD, BAX | ATO + ascorbic acid | An increased expression of pro-apoptotic BAD and BAX and decreased anti-apoptotic BCL2L10. |
| McBride et al. [19] | Phase I Single-arm | N = 14 with MDS | BIM, BAK, BCL-2, MCL-1 | Obatoclax | Three patients achieved improvement of transfusion dependence. |
| McBride et al. [19] | Phase II Single-arm | N = 24 with MDS | BIM, BAK, BCL-2, MCL-1 | Obatoclax | The response rate was 8% with thrombocytopenia, anemia and pneumonia among Grades 3–4. |
| Wei et al. [55] | Phase IB Single-arm | N = 59 with HR-MDS | BCL-2 | Venetoclax + AZA | CR = 18, mCR = 22, DS =11 and progression (PD) =2 |
| Jilg et al. [54] | Phase IB Single-arm | N = 78 with HR-MDS | BCL-2 | Venetoclax + AZA | CR = 42%, mCR = 35% |
| Hecker et al. [56] | Phase III Multi-arms | Ongoing | BCL-2 | Venetoclax/placebo | Ongoing |
| NCT02966782 | Phase IB Multi-arms | Ongoing | BCL-2 | Venetoclax/Venetoclax + AZA | Ongoing |
| NCT03404193 | Phase II Signle-arm | Ongoing | BCL-2 | Venetoclax + Decitabine | Ongoing |
| Reference/Identifier | Trial Phase | Population Characteristics | Molecular Target | Treatment | Results/ Status |
|---|---|---|---|---|---|
| Garcia-Manero et al. [106] | Phase Ib Single-arm | N = 28 with MDS | PD-1 | embrolizumab | ORR = 4%, OS in 24 months= 49% |
| Garcia-Manero et al. [107] | Phase II Multi-arms | N = 35 with MDS, lost response to HMA | PD-1/CTLA-4 | Nivolumab/ipilimumab | Nivolumab: No CR, Ipilimumab CR = 15% |
| Chien et al. [109] | Phase II Single-arm | N = 17 with untreated MDS | PD-1 | Pembrolizumab + AZA | ORR = 76%, CR = 18%. |
| Chien et al. [109] | Phase II Single-arm | N = 20 with MDS, lost response to HMA | PD-1 | Pembrolizumab + AZA | ORR = 25%, CR = 5%. |
| Zeidan et al. [110] | Phase II Multi-arm | N = 84 with MDS | PD-L1 | AZA/AZA + durvalumab | No statistically significant differences in ORR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuszczak, B.; Wróbel, T.; Wicherska-Pawłowska, K.; Rybka, J. The Role of BCL-2 and PD-1/PD-L1 Pathway in Pathogenesis of Myelodysplastic Syndromes. Int. J. Mol. Sci. 2023, 24, 4708. https://doi.org/10.3390/ijms24054708
Kuszczak B, Wróbel T, Wicherska-Pawłowska K, Rybka J. The Role of BCL-2 and PD-1/PD-L1 Pathway in Pathogenesis of Myelodysplastic Syndromes. International Journal of Molecular Sciences. 2023; 24(5):4708. https://doi.org/10.3390/ijms24054708
Chicago/Turabian StyleKuszczak, Bartłomiej, Tomasz Wróbel, Katarzyna Wicherska-Pawłowska, and Justyna Rybka. 2023. "The Role of BCL-2 and PD-1/PD-L1 Pathway in Pathogenesis of Myelodysplastic Syndromes" International Journal of Molecular Sciences 24, no. 5: 4708. https://doi.org/10.3390/ijms24054708
APA StyleKuszczak, B., Wróbel, T., Wicherska-Pawłowska, K., & Rybka, J. (2023). The Role of BCL-2 and PD-1/PD-L1 Pathway in Pathogenesis of Myelodysplastic Syndromes. International Journal of Molecular Sciences, 24(5), 4708. https://doi.org/10.3390/ijms24054708