
Combined Metabolome and Transcriptome Analysis Elucidates Sugar Accumulation in Wucai (Brassica campestris L.)

 ,
,
Abstract
:1. Introduction
2. Results
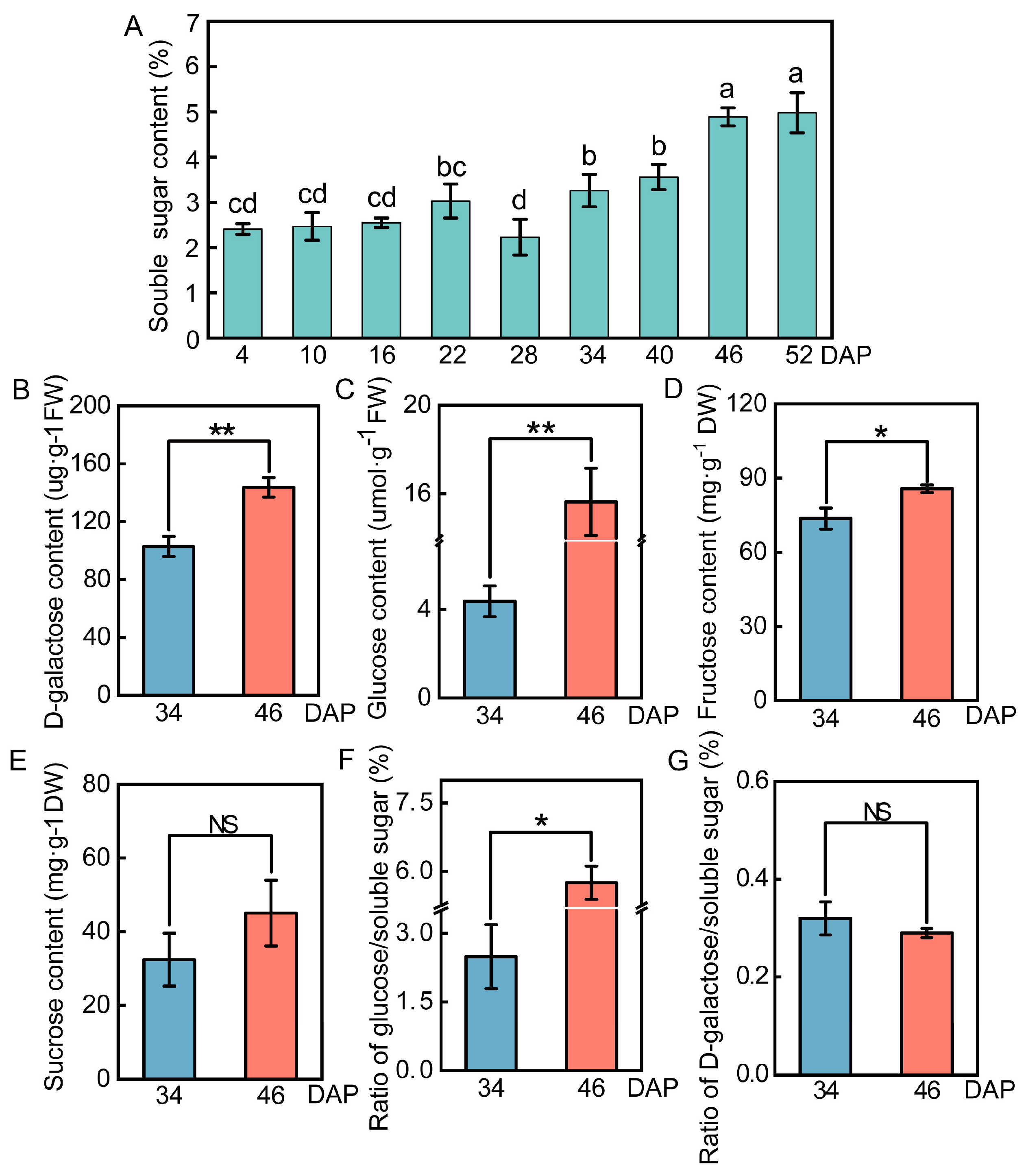
2.1. Changes in Sugar Content in the Wucai Leaves
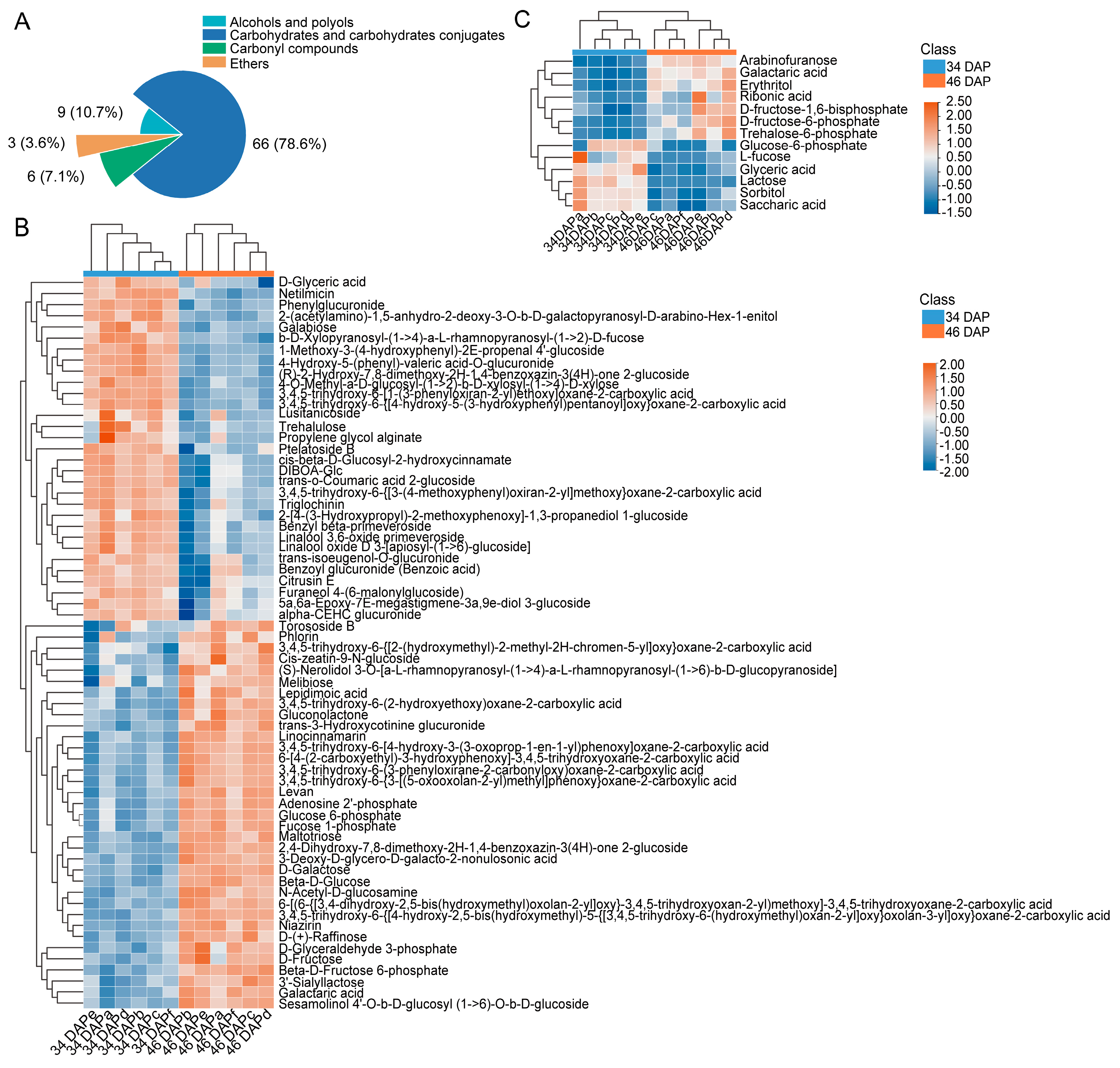
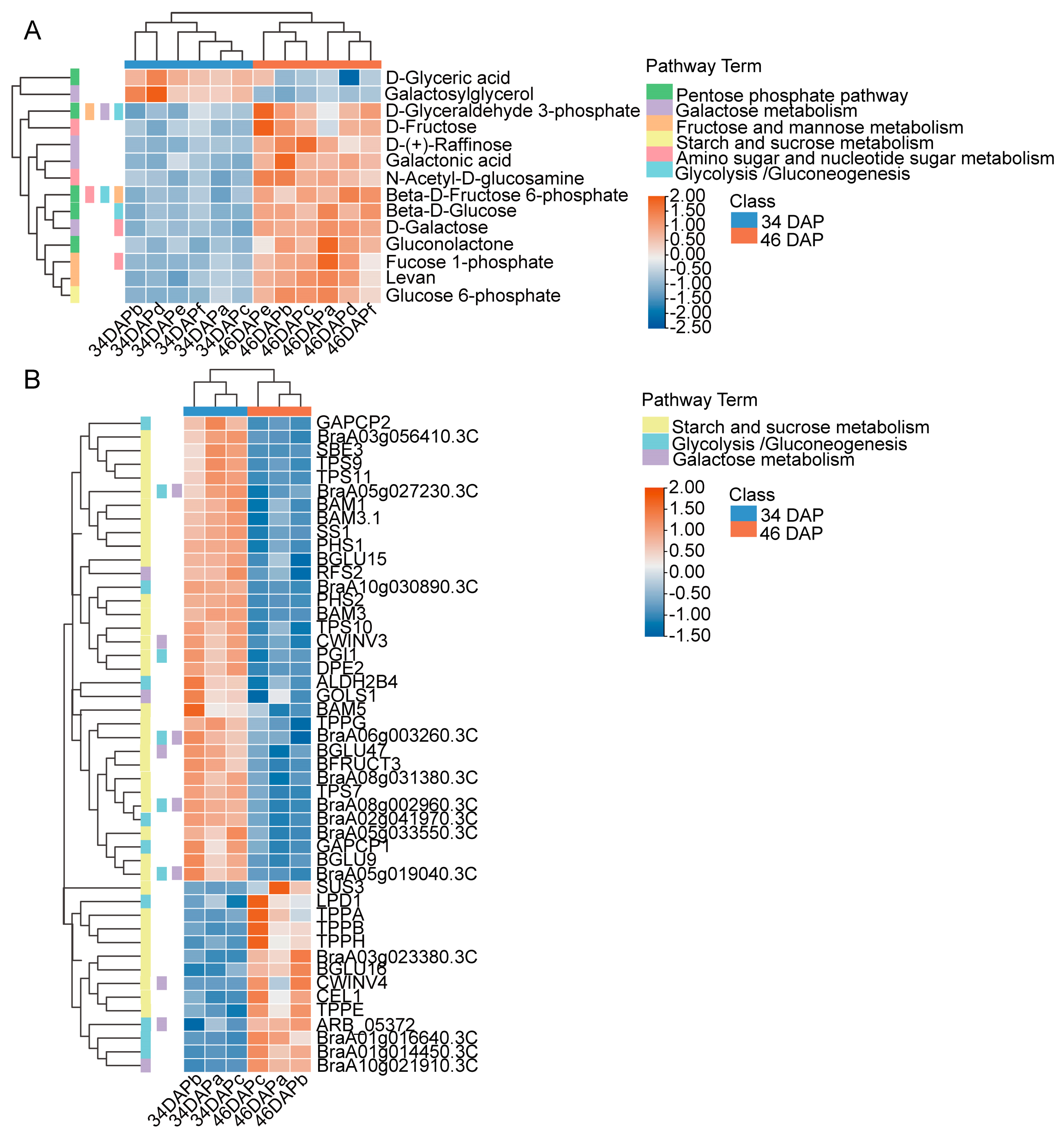
2.2. DAM Analysis in Wucai Leaves
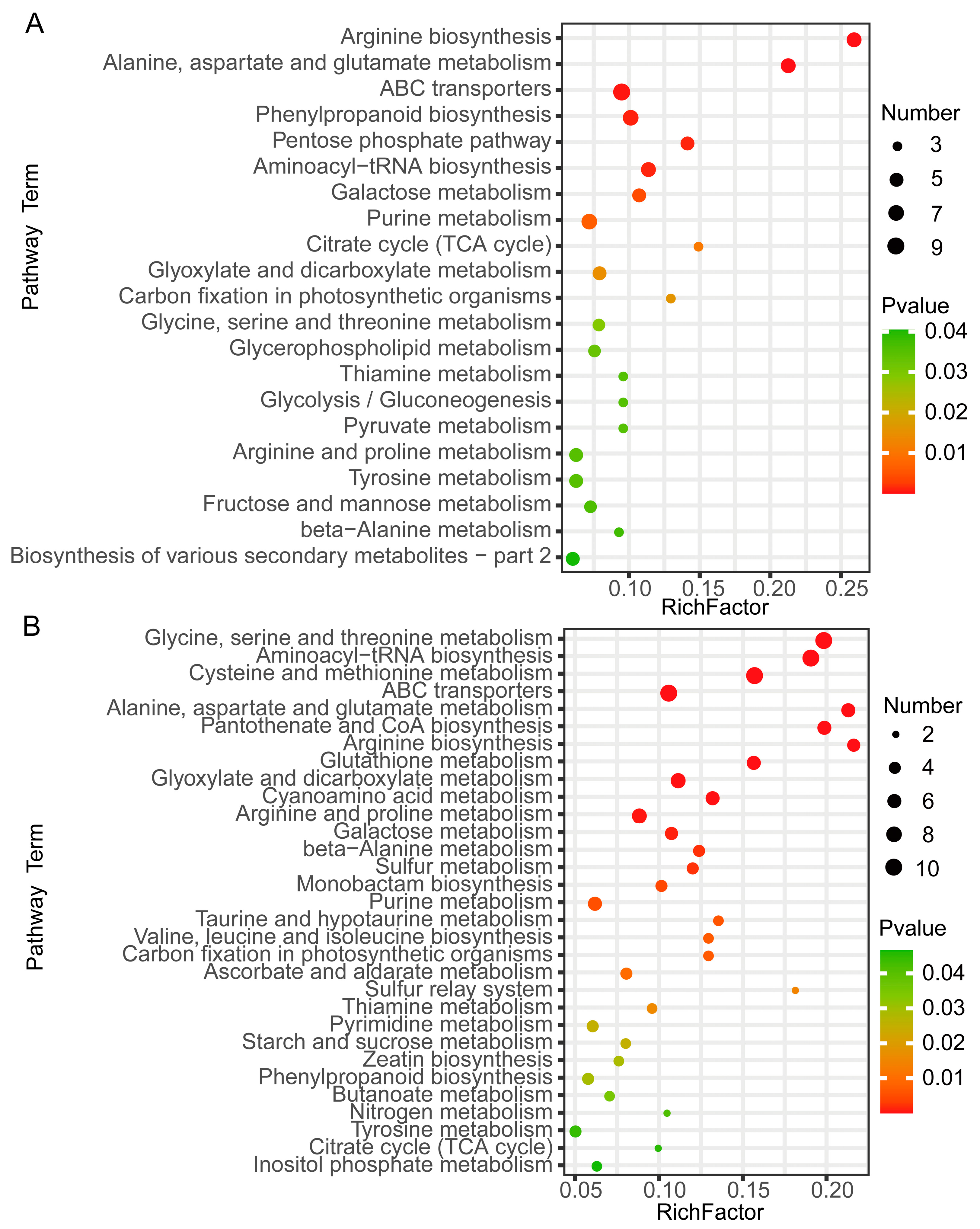
2.3. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DAMs Related to Sugar Accumulation in Wucai Leaves
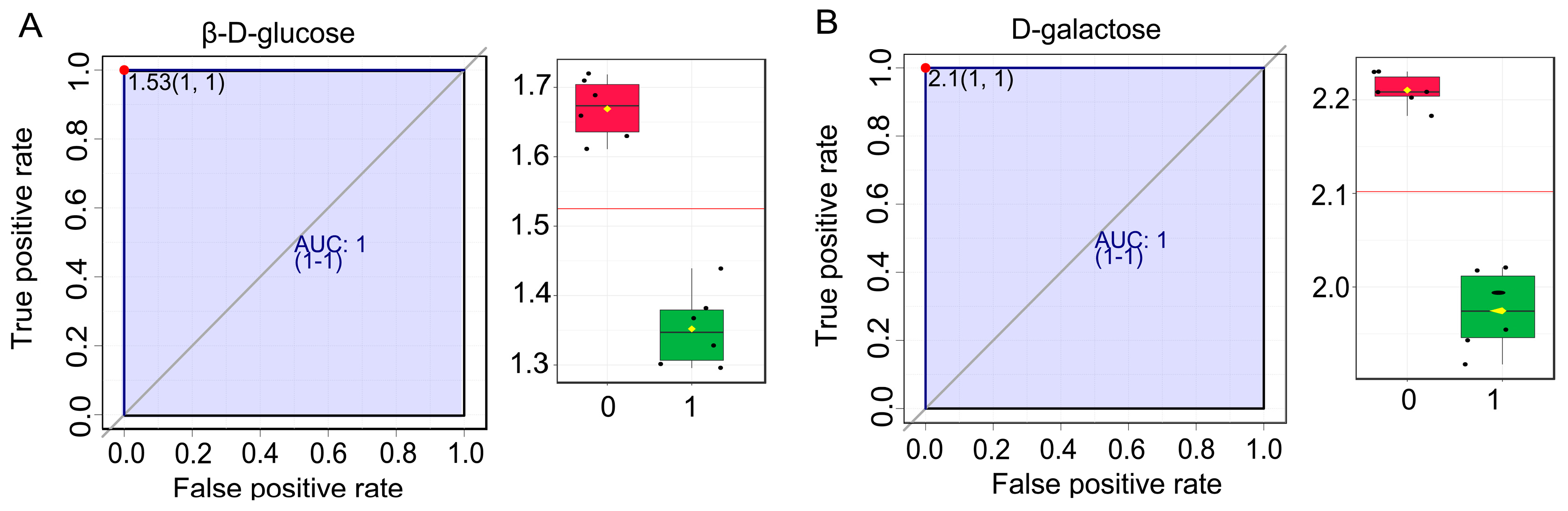
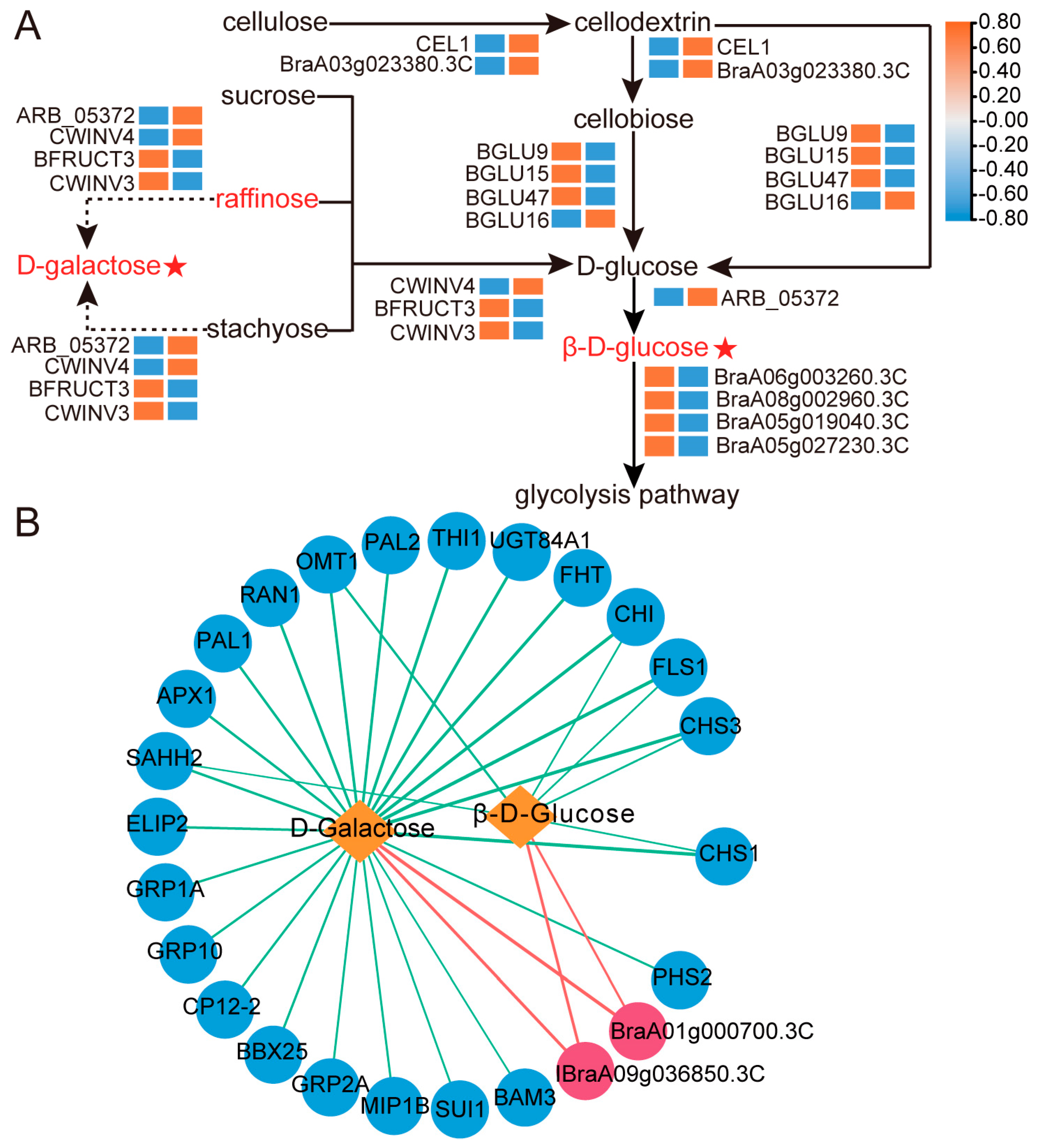
2.4. Biomarkers Analysis Related to Sugar Accumulation
2.5. Transcriptome Analysis
2.6. Coexpression Analysis of Genes Related to D-Galactose and β-D-Glucose Accumulation
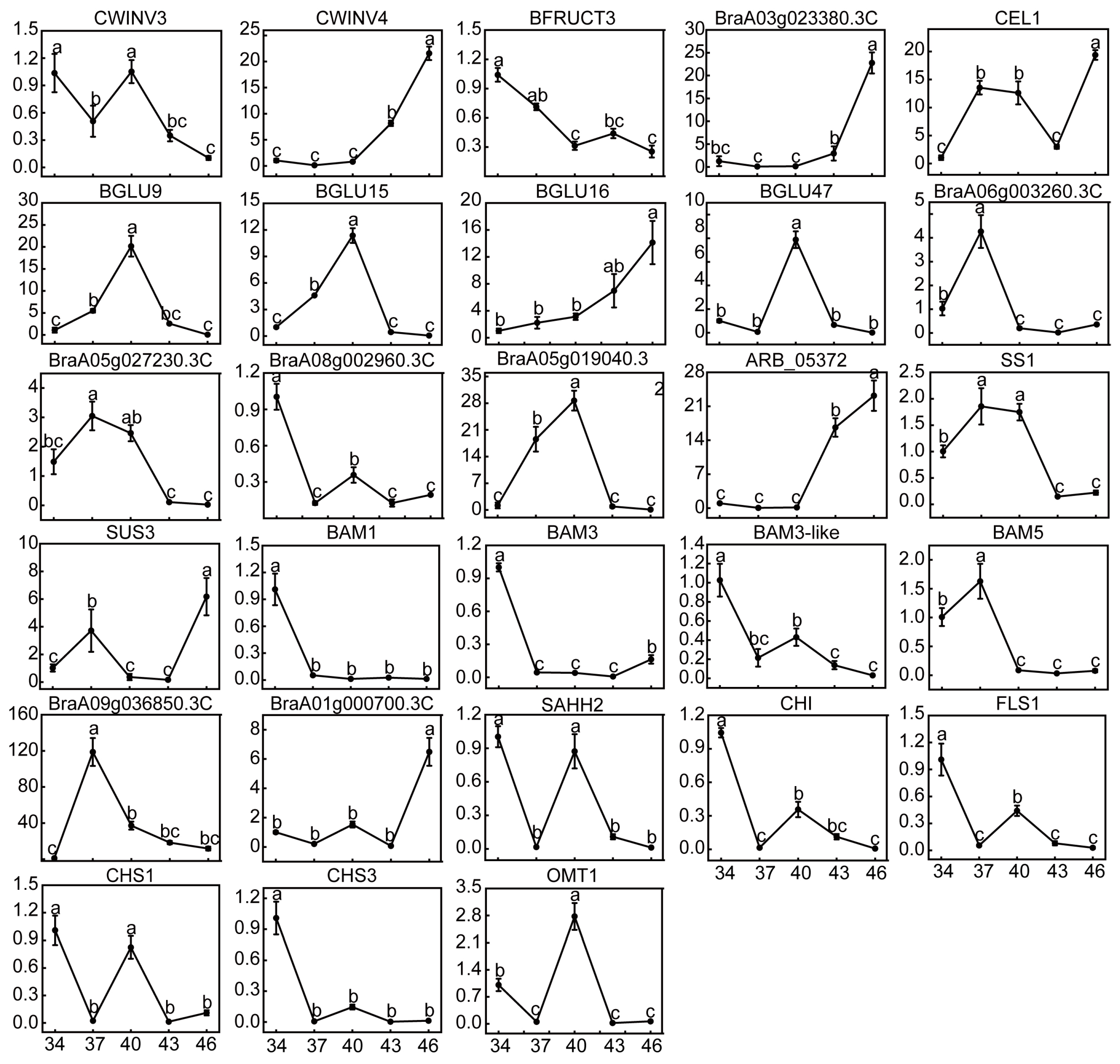
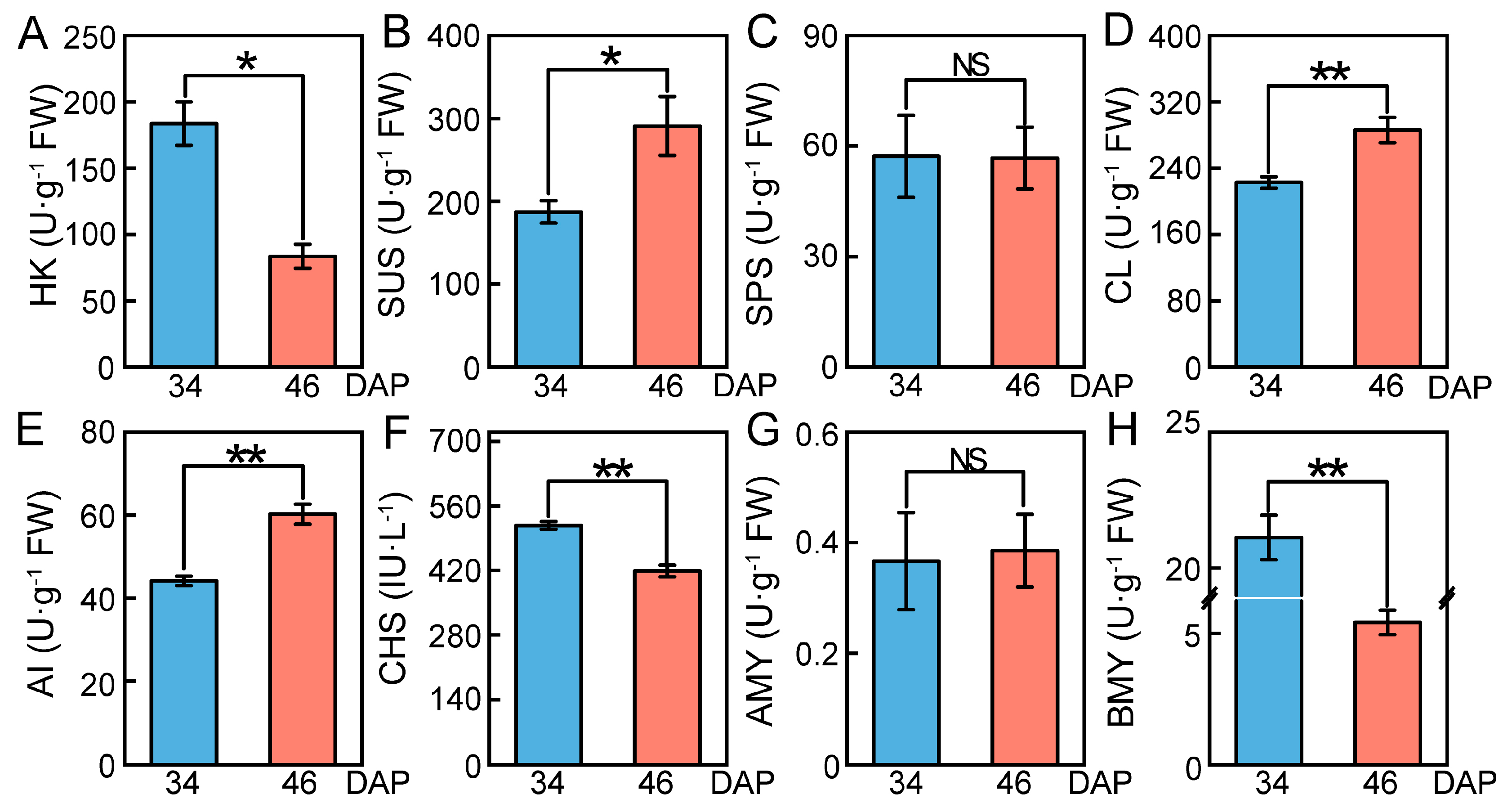
2.7. Changes in Relative Expression Levels of DEGs and Enzyme Activities
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Measurement of Sugar Content and Enzyme Activity
4.3. Metabolomic Analysis
4.4. RNA-Seq Analysis
4.5. qRT-PCR Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, L.; Wang, J.; Xie, S.; Zhao, M.; Nie, L.; Zheng, Y.; Zhu, S.; Hou, J.; Chen, G.; Wang, C. Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype. Int. J. Mol. Sci. 2019, 20, 3760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Liu, S.; Zhu, S.; Chen, G.; Liu, F.; Zou, M.; Wang, C. Comparative response of two wucai (Brassica campestris L.) genotypes to heat stress on antioxidative system and cell ultrastructure in root. Acta Physiol. Plant. 2016, 38, 223. [Google Scholar] [CrossRef]
- Yuan, L.; Nie, L.; Ji, Q.; Zheng, Y.; Zhang, L.; Zhu, S.; Hou, J.; Chen, G.; Wang, C. The effect of exogenous 24-epibrassinolide pretreatment on the quality, antioxidant capacity, and postharvest life of wucai (Brassica campestris L.). Food Sci. Nutr. 2021, 9, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y.; Wang, M.; Han, H.; Luo, Y.; Ding, W.; Xu, W.; Zhong, Y.; Huang, H.; Qu, S. Soluble sugars accumulation and related gene expression during fruit development in Cucurbita maxima Duchesne. Sci. Hortic. 2020, 272, 109520. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Liu, W. Sugar accumulation and characterization of metabolizing enzyme genes in leafy head of Chinese cabbage (Brassica campestris L. ssp. pekinensis). Hortic. Environ. Biotechnol. 2020, 62, 17–29. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Yi, H.; Zhai, W.; Wang, G.; Fu, Q. Transcriptome profiling of Cucumis melo fruit development and ripening. Hortic. Res. 2016, 3, 16014. [Google Scholar] [CrossRef] [Green Version]
- Shammai, A.; Petreikov, M.; Yeselson, Y.; Faigenboim, A.; Moy-Komemi, M.; Cohen, S.; Cohen, D.; Besaulov, E.; Efrati, A.; Houminer, N.; et al. Natural genetic variation for expression of a SWEET transporter among wild species of Solanum lycopersicum (tomato) determines the hexose composition of ripening tomato fruit. Plant J. Cell Mol. Biol. 2018, 96, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.Y.; Ho, L.H.; Neuhaus, H.E.; Guo, W.J. Transporter SlSWEET15 unloads sucrose from phloem and seed coat for fruit and seed development in tomato. Plant Physiol. 2021, 187, 2230–2245. [Google Scholar] [CrossRef]
- Xi, W.; Zheng, H.; Zhang, Q.; Li, W. Profiling Taste and Aroma Compound Metabolism during Apricot Fruit Development and Ripening. Int. J. Mol. Sci. 2016, 17, 998. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xing, Y.; Ramakrishnan, M.; Chen, C.; Xie, F.; Hua, Q.; Chen, J.; Zhang, R.; Zhao, J.; Hu, G.; et al. Transcriptomics-based Identification and Characterization of Genes Related to Sugar Metabolism in ‘Hongshuijing’ Pitaya. Hortic. Plant J. 2022, 8, 450–460. [Google Scholar] [CrossRef]
- Iqbal, S.; Ni, X.; Bilal, M.S.; Shi, T.; Khalil-Ur-Rehman, M.; Zhenpeng, P.; Jie, G.; Usman, M.; Gao, Z. Identification and expression profiling of sugar transporter genes during sugar accumulation at different stages of fruit development in apricot. Gene 2020, 742, 144584. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhao, S.; Lu, X.; He, N.; Zhu, H.; Dou, J.; Liu, W. Comparative transcriptome analysis reveals key genes potentially related to soluble sugar and organic acid accumulation in watermelon. PLoS ONE 2018, 13, e0190096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wu, H.X.; Ma, X.W.; Xu, W.T.; Liang, Q.Z.; Zhan, R.L.; Wang, S.B. Transcriptional mechanism of differential sugar accumulation in pulp of two contrasting mango (Mangifera indica L.) cultivars. Genomics 2020, 112, 4505–4515. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wen, S.; Xiao, S.; Lu, B.; Ma, M.; Bie, Z. Overexpression of the tonoplast sugar transporter CmTST2 in melon fruit increases sugar accumulation. J. Exp. Bot. 2018, 69, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Li, A.-M.; Liao, F.; Qin, C.-X.; Chen, Z.-L.; Zhou, L.; Li, Y.-R.; Li, X.-F.; Lakshmanan, P.; Huang, D.-L. Control of sucrose accumulation in sugarcane (Saccharum spp. hybrids) involves miRNA-mediated regulation of genes and transcription factors associated with sugar metabolism. GCB Bioenergy 2021, 14, 173–191. [Google Scholar] [CrossRef]
- Gerhardt, R.; Stitt, M.; Heldt, H.W. Subcellular Metabolite Levels in Spinach Leaves: Regulation of Sucrose Synthesis during Diurnal Alterations in Photosynthetic Partitioning. Plant Physiol. 1987, 83, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, M.; Cao, Y.; Cheng, X.; Meng, D.; Chen, Y.; Shakoor, A.; Gao, J.; Cai, Y. The Sucrose Synthase Gene Family in Chinese Pear (Pyrus bretschneideri Rehd.): Structure, Expression, and Evolution. Molecules 2018, 23, 1144. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.C. Role of Sucrose-Phosphate Synthase in Partitioning of Carbon in Leaves. Plant Physiol. 1983, 71, 818–821. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.R.; Roy, S.; Sengupta, D.N. A comparative study of cultivar differences in sucrose phosphate synthase gene expression and sucrose formation during banana fruit ripening. Postharvest Biol. Technol. 2009, 54, 15–24. [Google Scholar] [CrossRef]
- Fung, R.W.M.; Langenkämper, G.; Gardner, R.C.; MacRae, E. Differential expression within an SPS gene family. Plant Sci. 2003, 164, 459–470. [Google Scholar] [CrossRef]
- Geigenberger, P.; Reimholz, R.; Deiting, U.; Sonnewald, U.; Stitt, M. Decreased expression of sucrose phosphate synthase strongly inhibits the water stress-induced synthesis of sucrose in growing potato tubers. Plant J. Cell Mol. Biol. 1999, 19, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, W.; Du, L.; Xie, J.; Yao, Y.; Sun, G. Expression Patterns, Activities and Carbohydrate-Metabolizing Regulation of Sucrose Phosphate Synthase, Sucrose Synthase and Neutral Invertase in Pineapple Fruit during Development and Ripening. Int. J. Mol. Sci. 2012, 13, 9460–9477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wongmetha, O.; Ke, L.-S.; Liang, Y.-S. The changes in physical, bio-chemical, physiological characteristics and enzyme activities of mango cv. Jinhwang during fruit growth and development. Chem. Chem. 2015, 72–73, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Feng, F.; Cheng, L. Expression Patterns of Genes Involved in Sugar Metabolism and Accumulation during Apple Fruit Development. PLoS ONE 2012, 7, e33055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.Z.; Hu, X.-M.; Jin, L.-F.; Liu, Y.-Z.; Peng, S.-A. Genome-Wide Identification and Expression Profile Analysis of Citrus Sucrose Synthase Genes: Investigation of Possible Roles in the Regulation of Sugar Accumulation. PLoS ONE 2014, 9, e113623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iraqi, D.; Tremblay, F.M. Analysis of carbohydrate metabolism enzymes and cellular contents of sugars and proteins during spruce somatic embryogenesis suggests a regulatory role of exogenous sucrose in embryo development. J. Exp. Bot. 2001, 52, 2301–2311. [Google Scholar] [CrossRef] [Green Version]
- Burger, Y.; Schaffer, A.A. The contribution of sucrose metabolism enzymes to sucrose accumulation in Cucumis melo. J. Am. Soc. Hortic. Sci. 2007, 132, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.S.; Feng, C.Y.; Wang, M.N.; Li, T.L.; Liu, X.; Jiang, J. Plasma membrane-localized SlSWEET7a and SlSWEET14 regulate sugar transport and storage in tomato fruits. Hortic. Res. 2021, 8, 186. [Google Scholar] [CrossRef]
- De Coninck, B.; Le Roy, K.; Francis, I.; Clerens, S.; Vergauwen, R.; Halliday, A.M.; Smith, S.M.; Van Laere, A.; Van den Ende, W. Arabidopsis AtcwINV3 and 6 are not invertases but are fructan exohydrolases (FEHs) with different substrate specificities. Plant Cell Environ. 2005, 28, 432–443. [Google Scholar] [CrossRef]
- Xu, D.-P.; Sung, S.-J.S.; Black, C.C. Sucrose Metabolism in Lima Bean Seeds. Plant Physiol. 1989, 89, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Dai, N.; Schaffer, A.; Petreikov, M.; Shahak, Y.; Giller, Y.; Ratner, K.; Levine, A.; Granot, D. Overexpression of Arabidopsis Hexokinase in Tomato Plants Inhibits Growth, Reduces Photosynthesis, and Induces Rapid Senescence. Plant Cell 1999, 11, 1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saminathan, T.; García, M.; Ghimire, B.; Lopez, C.; Bodunrin, A.; Nimmakayala, P.; Abburi, V.L.; Levi, A.; Balagurusamy, N.; Reddy, U.K. Metagenomic and Metatranscriptomic Analyses of Diverse Watermelon Cultivars Reveal the Role of Fruit Associated Microbiome in Carbohydrate Metabolism and Ripening of Mature Fruits. Front. Plant Sci. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umer, M.J.; Bin Safdar, L.; Gebremeskel, H.; Zhao, S.J.; Yuan, P.L.; Zhu, H.J.; Kaseb, M.O.; Anees, M.; Lu, X.Q.; He, N.; et al. Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Hortic. Res. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.S.; Zhu, H.J.; Lu, X.Q.; Yang, D.D.; Zhao, S.J.; Umer, M.J.; He, N.; Yuan, P.L.; Anees, M.; Diao, W.N.; et al. An integrated transcriptome and metabolome approach reveals the accumulation of taste-related metabolites and gene regulatory networks during watermelon fruit development. Planta 2021, 254, 35. [Google Scholar] [CrossRef]
- Wang, X.; Rong, L.; Wang, M.; Pan, Y.; Zhao, Y.; Tao, F. Improving the activity of endoglucanase I (EGI) from Saccharomyces cerevisiae by DNA shuffling. RSC Adv. 2017, 7, 46246–46256. [Google Scholar] [CrossRef] [Green Version]
- Sutarlie, L.; Yang, K.L. Hybrid cellulase aggregate with a silica core for hydrolysis of cellulose and biomass. J. Colloid Interface Sci. 2013, 411, 76–81. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Zhou, Y. Transglycosylation, a new role for multifunctional cellulase in overcoming product inhibition during the cellulose hydrolysis. Bioengineered 2017, 8, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Cass, L.G.; Kirven, K.A.; Christoffersen, R.E. Isolation and characterization of a cellulase gene family member expressed during avocado fruit ripening. Mol. Gen. Genet. 1990, 223, 76–86. [Google Scholar] [CrossRef]
- Owino, W.O.; Nakano, R.; Kubo, Y.; Inaba, A. Coordinated expression patterns of genes encoding cell wall modifying enzymes during ripening in distinct anatomical tissue regions of the fig (Ficus carica L.) fruit. Postharvest Biol. Technol. 2004, 32, 253–261. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, G.; Ahmad, T.; Mansoor, S.; Kaur, B. Enzyme Engineering: Current Trends and Future Perspectives. Food Rev. Int. 2021, 37, 121–154. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Li, J.; Zhang, Y.; Zhou, D.; Li, C.; He, L.; Li, H.; Wang, F.; Gao, J. Identification of key genes controlling soluble sugar and glucosinolate biosynthesis in Chinese cabbage by integrating metabolome and genome-wide transcriptome analysis. Front. Plant Sci. 2022, 13, 1043489. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, Y.; Jiao, Y.; Wu, J.; Zhang, Z.; Yu, X.; Ma, Y. Transcriptome profiling of yellow leafy head development during the heading stage in Chinese cabbage (Brassica rapa subsp. pekinensis). Physiol. Plant. 2019, 165, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Wang, C.; Dong, W.; Jiang, Q.; Wang, D.; Li, S.; Chen, M.; Liu, C.; Sun, C.; Chen, K. Transcriptome and metabolome analyses of sugar and organic acid metabolism in Ponkan (Citrus reticulata) fruit during fruit maturation. Gene 2015, 554, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shu, P.; Zhang, C.; Zhang, J.; Chen, Y.; Zhang, Y.; Du, K.; Xie, Y.; Li, M.; Ma, T.; et al. Integrative analyses of metabolome and genome-wide transcriptome reveal the regulatory network governing flavor formation in kiwifruit (Actinidia chinensis). New Phytol. 2022, 233, 373–389. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Kristal, B.S.; Weinshilboum, R.M. Metabolomics: A Global Biochemical Approach to Drug Response and Disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. [Google Scholar] [CrossRef] [Green Version]
- Haenisch, F.; Cooper, J.D.; Reif, A.; Kittel-Schneider, S.; Steiner, J.; Leweke, F.M.; Rothermundt, M.; van Beveren, N.J.M.; Crespo-Facorro, B.; Niebuhr, D.W.; et al. Towards a blood-based diagnostic panel for bipolar disorder. Brain Behav. Immun. 2016, 52, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Kim, J.; Hwang, J.; Song, J.; Kim, K.; Cha, H.-S. Urinary Metabolomic Profiling to Identify Potential Biomarkers for the Diagnosis of Behcet’s Disease by Gas Chromatography/Time-of-Flight-Mass Spectrometry. Int. J. Mol. Sci. 2017, 18, 2309. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.M.; Deng, L.; Wang, X.; Wang, Y.; Pan, L.; Liu, H.; Niu, L.; Lu, Z.; Cui, G.; Zeng, W.; et al. Expression patterns of genes involved in sugar metabolism and accumulation during peach fruit development and ripening. Sci. Hortic. 2019, 257. [Google Scholar] [CrossRef]
- Abbas, H.M.K.; Huang, H.X.; Wang, A.J.; Wu, T.Q.; Xue, S.D.; Ahmad, A.; Xie, D.S.; Li, J.X.; Zhong, Y.J. Metabolic and transcriptomic analysis of two Cucurbita moschata germplasms throughout fruit development. BMC Genom. 2020, 21, 365. [Google Scholar] [CrossRef]
- Dai, N.; Cohen, S.; Portnoy, V.; Tzuri, G.; Harel-Beja, R.; Pompan-Lotan, M.; Carmi, N.; Zhang, G.; Diber, A.; Pollock, S.; et al. Metabolism of soluble sugars in developing melon fruit: A global transcriptional view of the metabolic transition to sucrose accumulation. Plant Mol. Biol. 2011, 76, 1–18. [Google Scholar] [CrossRef]
- Cheng, H.; Kong, W.P.; Tang, T.X.; Ren, K.L.; Zhang, K.L.; Wei, H.X.; Lin, T. Identification of Key Gene Networks Controlling Soluble Sugar and Organic Acid Metabolism During Oriental Melon Fruit Development by Integrated Analysis of Metabolic and Transcriptomic Analyses. Front. Plant Sci. 2022, 13, 830517. [Google Scholar] [CrossRef] [PubMed]
- Darko, E.; Vegh, B.; Khalil, R.; Marcek, T.; Szalai, G.; Pal, M.; Janda, T. Metabolic responses of wheat seedlings to osmotic stress induced by various osmolytes under iso-osmotic conditions. PLoS ONE 2019, 14, e0226151. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.L.; Liu, Q.; Xu, X.B.; He, Y.Q.; Li, Y.Q.; Wang, H.L.; Ye, B.; Huang, C.H.; Zhong, M.; Jia, D.F. Metabolome and Transcriptome Reveal Novel Formation Mechanism of Early Mature Trait in Kiwifruit (Actinidia eriantha). Front. Plant Sci. 2021, 12, 2558. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.S.; Diao, W.N.; Zhu, H.J.; Umer, M.J.; Zhao, S.J.; He, N.; Lu, X.Q.; Yuan, P.L.; Anees, M.; Yang, D.D.; et al. Metabolome and Transcriptome Integration Reveals Insights Into Flavor Formation of “Crimson” Watermelon Flesh During Fruit Development. Front. Plant Sci. 2021, 12, 629361. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Feng, Y.; Tu, M.; Wittich, P.E.; Bate, N.J.; Messing, J. Transcriptome and metabolome reveal distinct carbon allocation patterns during internode sugar accumulation in different sorghum genotypes. Plant Biotechnol. J. 2019, 17, 472–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.W.; Leon, C.; Feil, R.; Lunn, J.E.; Delrot, S.; Gomes, E. Metabolic profiling reveals coordinated switches in primary carbohydrate metabolism in grape berry (Vitis vinifera L.), a non-climacteric fleshy fruit. J. Exp. Bot. 2013, 64, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Bagri, J.; Yadav, A.; Anwar, K.; Dkhar, J.; Singla-Pareek, S.L.; Pareek, A. Metabolic shift in sugars and amino acids regulates sprouting in Saffron corm. Sci. Rep. 2017, 7, 11904. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.X.; Li, Q.T.; Liu, Y.F.; Zhang, F.X.; Ma, B.; Zhang, W.K.; Man, W.Q.; Du, W.G.; Wang, G.D.; Chen, S.Y.; et al. Soybean GmbZIP123 gene enhances lipid content in the seeds of transgenic Arabidopsis plants. J. Exp. Bot. 2013, 64, 4329–4341. [Google Scholar] [CrossRef]
- Wei, W.; Cheng, M.N.; Ba, L.J.; Zeng, R.X.; Luo, D.L.; Qin, Y.H.; Liu, Z.L.; Kuang, J.F.; Lu, W.J.; Chen, J.Y.; et al. Pitaya HpWRKY3 Is Associated with Fruit Sugar Accumulation by Transcriptionally Modulating Sucrose Metabolic Genes HpINV2 and HpSuSy1. Int. J. Mol. Sci. 2019, 20, 1890. [Google Scholar] [CrossRef] [Green Version]
- Peroni, F.H.G.; Koike, C.; Louro, R.P.; Purgatto, E.; do Nascimento, J.R.O.; Lajolo, F.M.; Cordenunsi, B.R. Mango starch degradation. II. The binding of α-amylase and β-amylase to the starch granule. J. Agric. Food Chem. 2008, 56, 7416–7421. [Google Scholar] [CrossRef]
- Chou, H.L.; Dai, Z.Y.; Hsieh, C.W.; Ku, M.S.B. High level expression of Acidothermus cellulolyticus β-1, 4-endoglucanase in transgenic rice enhances the hydrolysis of its straw by cultured cow gastric fluid. Biotechnol. Biofuels Bioprod. 2011, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.L.; Chang, C.K.; Teng, K.H.; Liang, P.H. Construction and characterization of different fusion proteins between cellulases and β-glucosidase to improve glucose production and thermostability. Bioresour. Technol. 2011, 102, 3973–3976. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Jia, Y.; Duan, Y.; Chen, H.; Wang, X.; Zheng, H.; Liu, H.; Wang, J.; Zou, D.; Zhao, H. Integrated Isoform Sequencing and Dynamic Transcriptome Analysis Reveals Diverse Transcripts Responsible for Low Temperature Stress at Anther Meiosis Stage in Rice. Front. Plant Sci. 2021, 12, 795834. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, B.; Gao, S.; Zhang, F.; Terzaghi, W.; Dai, M. Genome-wide association study dissects the genetic bases of salt tolerance in maize seedlings. J. Integr. Plant Biol. 2019, 61, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Park, M.-Y.; Chung, M.-S.; Koh, H.-S.; Lee, D.J.; Ahn, S.-J.; Kim, C.S. Isolation and functional characterization of the Arabidopsis salt-tolerance 32 (AtSAT32) gene associated with salt tolerance and ABA signaling. Physiol. Plant. 2009, 135, 426–435. [Google Scholar] [CrossRef]
- Bevilacqua, A.; Bizzarri, M. Inositols in Insulin Signaling and Glucose Metabolism. Int. J. Endocrinol. 2018, 2018, 1968450. [Google Scholar] [CrossRef] [Green Version]
- Hegeman, C.E.; Good, L.L.; Grabau, E.A. Expression of d-myo-Inositol-3-Phosphate Synthase in Soybean. Implications for Phytic Acid Biosynthesis. Plant Physiol. 2001, 125, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Hu, G.; Li, N.; Habib, S.; Huang, W.; Li, Z. Functional Characterization of SlSAHH2 in Tomato Fruit Ripening. Front. Plant Science. 2017, 8, 1312. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.; Bermejo, A.; Navarro, P.; Forner-Giner, M.Á.; Salvador, A. Rootstock effect on fruit quality, anthocyanins, sugars, hydroxycinnamic acids and flavanones content during the harvest of blood oranges ‘Moro’ and ‘Tarocco Rosso’ grown in Spain. Food Chemistry. 2021, 342, 128305. [Google Scholar] [CrossRef]
- Yang, B.; He, S.; Liu, Y.; Liu, B.; Ju, Y.; Kang, D.; Sun, X.; Fang, Y. Transcriptomics integrated with metabolomics reveals the effect of regulated deficit irrigation on anthocyanin biosynthesis in Cabernet Sauvignon grape berries. Food Chem. 2020, 314, 126170. [Google Scholar] [CrossRef]
- Tsukaya, H.; Ohshima, T.; Naito, S.; Chino, M.; Komeda, Y. Sugar-Dependent Expression of the CHS-A Gene for Chalcone Synthase from Petunia in Transgenic Arabidopsis. Plant Physiol. 1991, 97, 1414–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, S.; Keurentjes, J.; Bentsink, L.; Koornneef, M.; Smeekens, S. Sucrose-Specific Induction of Anthocyanin Biosynthesis in Arabidopsis Requires the MYB75/PAP1 Gene. Plant Physiol. 2005, 139, 1840–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurentin, A.; Edwards, C.A. A microtiter modification of the anthrone-sulfuric acid colorimetric assay for glucose-based carbohydrates. Anal. Biochem. 2003, 315, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chan, C.K.K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. In Plant Bioinformatics: Methods in Molecular Biology; Springer: Cham, Switzerland, 2016; Volume 1374, pp. 339–361. ISBN 978-1-4939-3167-5. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | AUC | T-Test | Log2(FC) |
|---|---|---|---|
| 3,4,5-trihydroxy-6-[4-hydroxy-3-(3-oxoprop-1-en-1-yl) phenoxy] oxane-2-carboxylic acid | 1.0 | 3.9459 × 10−6 | 0.98885 |
| D-Galactose | 1.0 | 1.9657 × 10−7 | 0.94706 |
| 6-[4-(2-carboxyethyl)-3-hydroxyphenoxy]-3,4,5-trihydroxyoxane-2-carboxylic acid | 1.0 | 3.0412 × 10−5 | 0.62342 |
| Pheophorbide a | 1.0 | 3.2816 × 10−7 | 3.142 |
| Quinoline-3-carboxamides | 1.0 | 6.0721 × 10−7 | −2.6307 |
| ibandronate | 1.0 | 3.3144 × 10−9 | 1.3004 |
| 5′-Butyrylphosphouridine | 1.0 | 2.1576 × 10−6 | −1.0662 |
| Phosphatidyl glycerol | 1.0 | 2.0603 × 10−9 | 2.8388 |
| Beta-D-Glucose | 1.0 | 6.6146 × 10−7 | 1.2187 |
| (s)C(S)S-S-Methylcysteine sulfoxide | 1.0 | 2.6679 × 10−7 | 2.2726 |
| 3,4,5-trihydroxy-6-(3-phenyloxirane-2-carbonyloxy) oxane-2-carboxylic acid | 1.0 | 5.1913 × 10−6 | 0.98688 |
| Trehalulose | 1.0 | 9.5148 × 10−4 | −0.77498 |
| Levan | 1.0 | 3.2252 × 10−5 | 0.9449 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zhou, J.; Zhang, S.; Gao, X.; Yang, Y.; Hou, J.; Chen, G.; Tang, X.; Wu, J.; Yuan, L. Combined Metabolome and Transcriptome Analysis Elucidates Sugar Accumulation in Wucai (Brassica campestris L.). Int. J. Mol. Sci. 2023, 24, 4816. https://doi.org/10.3390/ijms24054816
Wang C, Zhou J, Zhang S, Gao X, Yang Y, Hou J, Chen G, Tang X, Wu J, Yuan L. Combined Metabolome and Transcriptome Analysis Elucidates Sugar Accumulation in Wucai (Brassica campestris L.). International Journal of Molecular Sciences. 2023; 24(5):4816. https://doi.org/10.3390/ijms24054816
Chicago/Turabian StyleWang, Chenggang, Jiajie Zhou, Shengnan Zhang, Xun Gao, Yitao Yang, Jinfeng Hou, Guohu Chen, Xiaoyan Tang, Jianqiang Wu, and Lingyun Yuan. 2023. "Combined Metabolome and Transcriptome Analysis Elucidates Sugar Accumulation in Wucai (Brassica campestris L.)" International Journal of Molecular Sciences 24, no. 5: 4816. https://doi.org/10.3390/ijms24054816
APA StyleWang, C., Zhou, J., Zhang, S., Gao, X., Yang, Y., Hou, J., Chen, G., Tang, X., Wu, J., & Yuan, L. (2023). Combined Metabolome and Transcriptome Analysis Elucidates Sugar Accumulation in Wucai (Brassica campestris L.). International Journal of Molecular Sciences, 24(5), 4816. https://doi.org/10.3390/ijms24054816