
Identification of Three Human POLH Germline Variants Defective in Complementing the UV- and Cisplatin-Sensitivity of POLH-Deficient Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
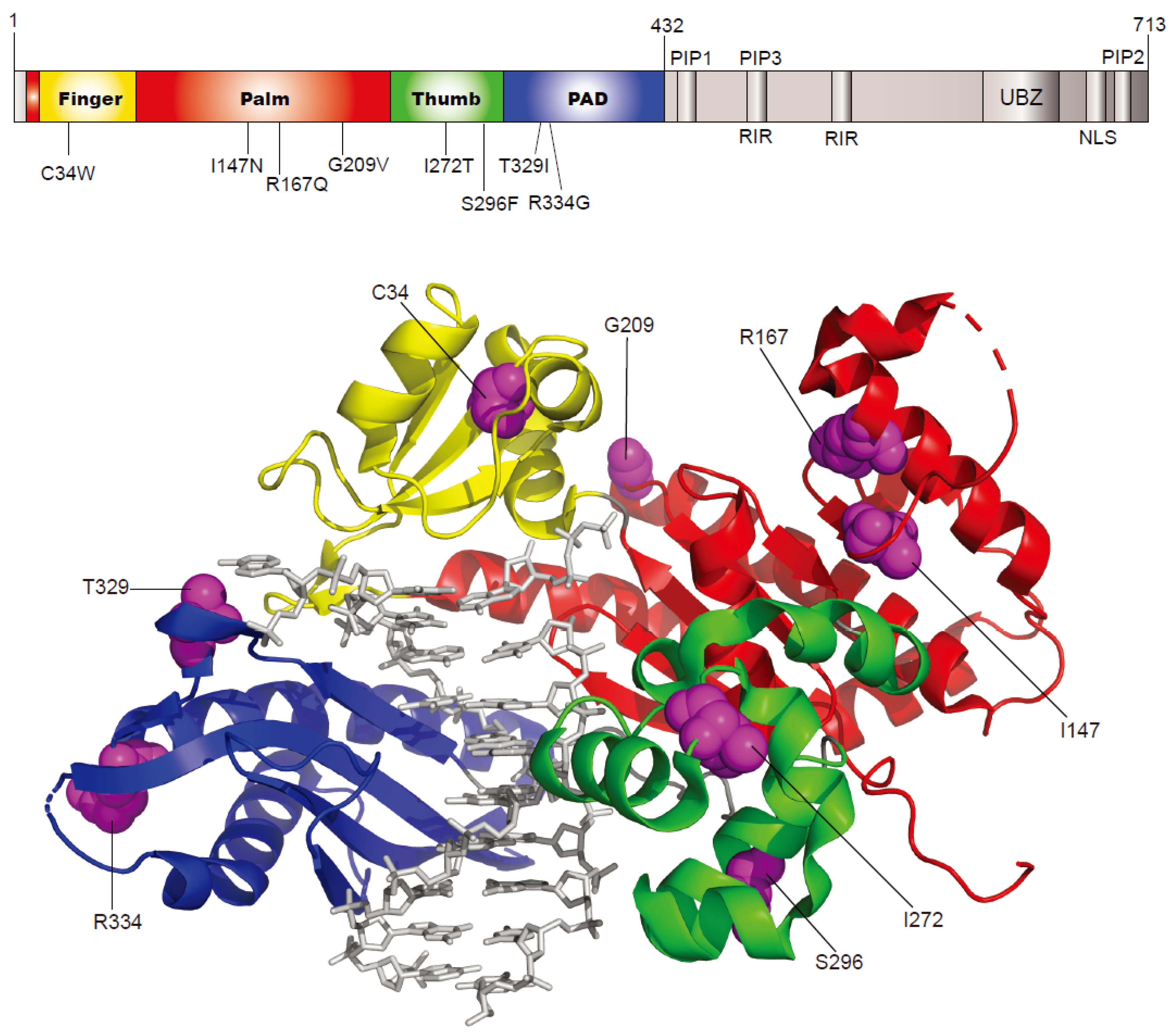
2.1. Selection of Human Germline POLH Gene Variants to Study
2.2. Effects of Eight POLH Variants on Catalytic Activity of Pol η
2.3. Effects of Eight POLH Variants on DNA Substrate Binding of Pol η
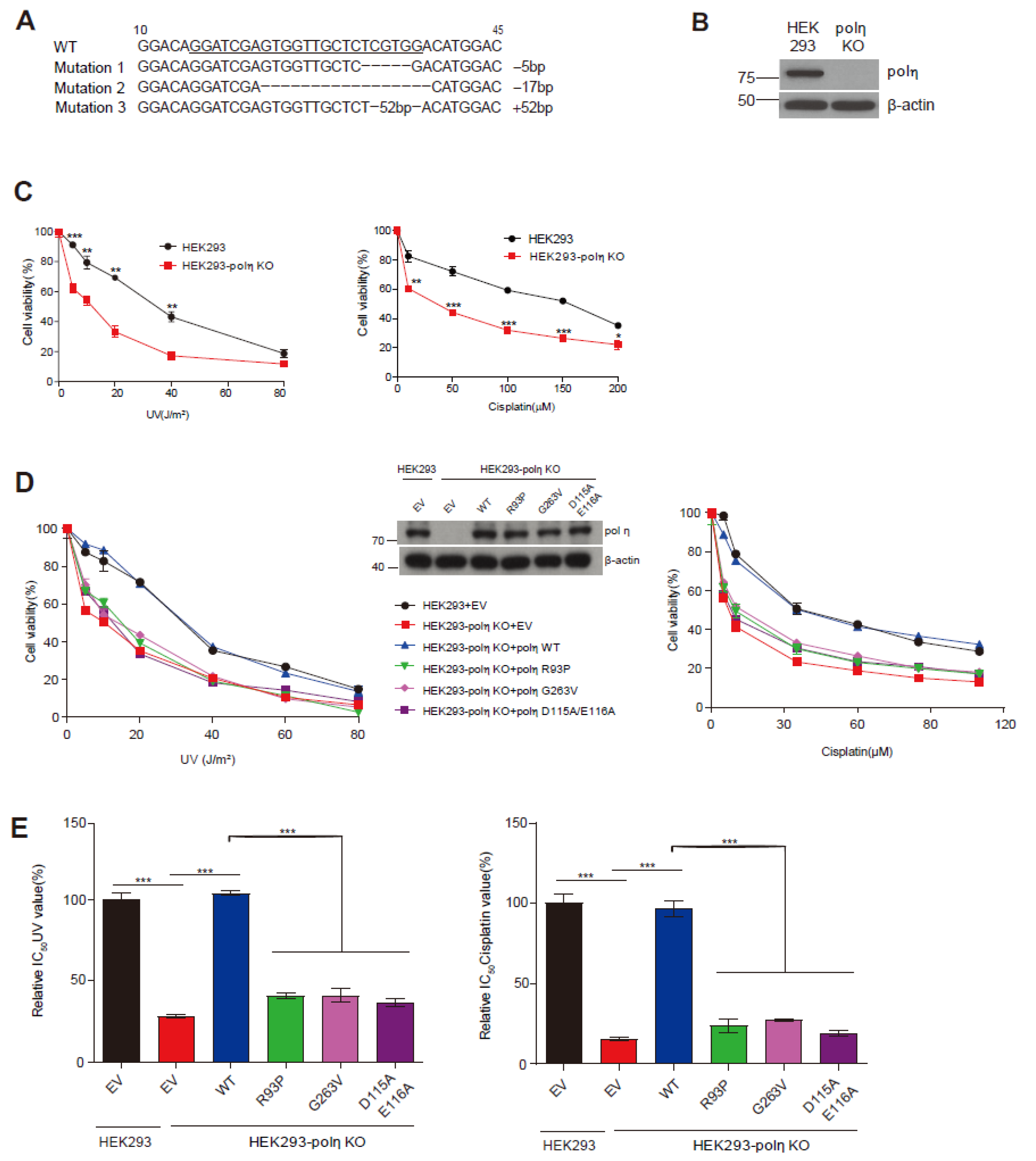
2.4. Complementation of UV and Cisplatin Sensitivity of POLH-KO Cells by Wild-Type Pol η and Mutants D115A/E116A, R93P, and G263V
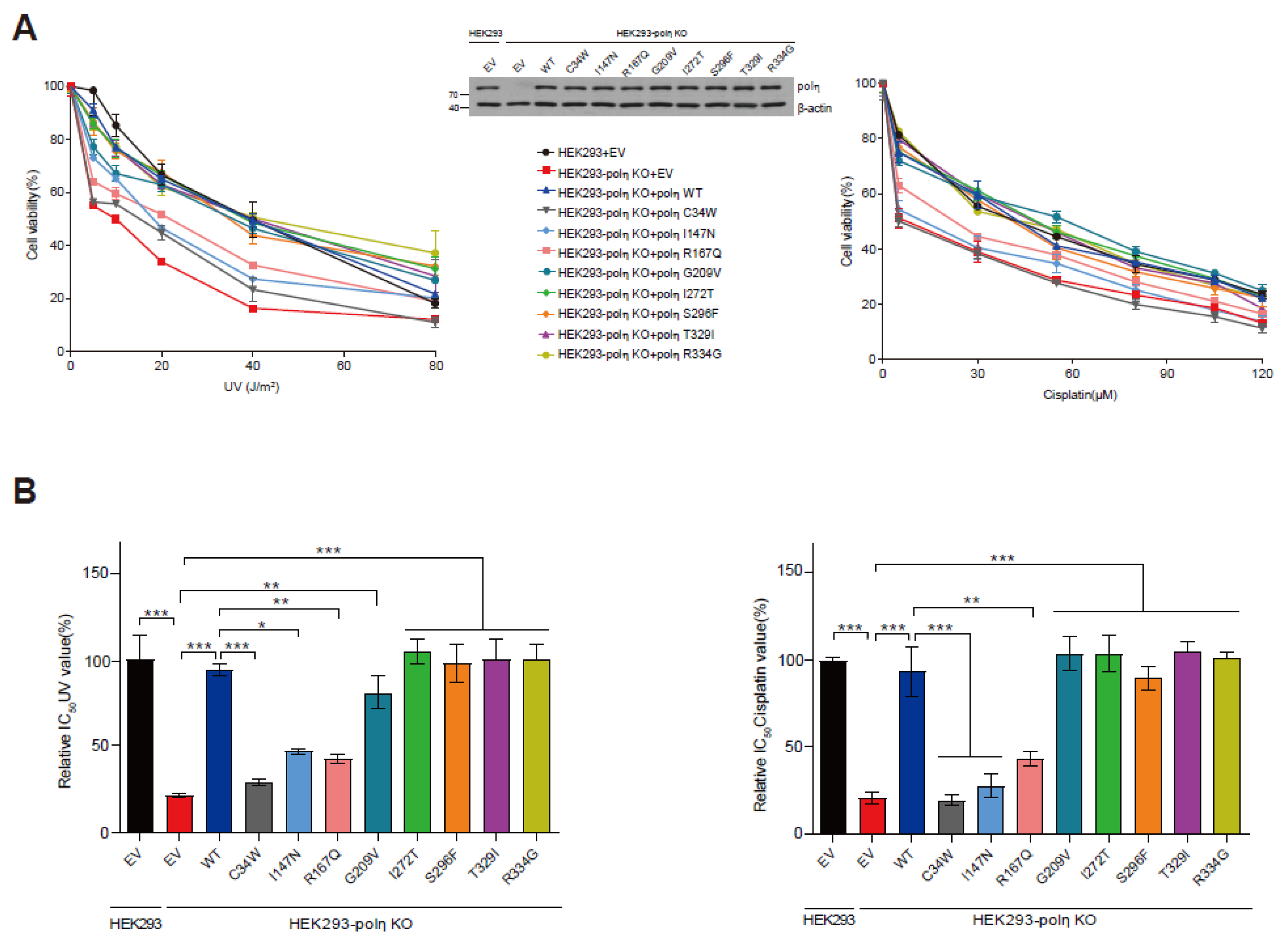
2.5. Capabilities of Eight POLH Variants to Rescue the UV- and Cisplatin-Sensitivity of POLH-KO Cells
3. Discussion
4. Materials and Methods
4.1. DNA Substrates
4.2. Expression Vector Construction for Pol η (1—432) Variants and Protein Purification
4.3. Enzyme Assays and Steady-State Kinetic Analysis
4.4. Fluorescence Polarization
4.5. Mammalian Expression Vector Construction, Cell Culture, and Transfection
4.6. POLH-KO Cell Line Generation and Immunoblotting
4.7. Cell Viability Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CTD | cis-syn cyclobutane thymine dimer |
| EV | empty vector |
| IC50 | concentration that induces 50% inhibition of cell viability |
| KO | knockout |
| TLS | translesion DNA synthesis |
| UV | ultraviolet light |
| WT | wild-type |
References
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase η. Biochemistry 2000, 39, 4575–4580. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Stover, J.S.; Angel, K.C.; Chowdhury, G.; Rizzo, C.J.; Guengerich, F.P. Biochemical basis of genotoxicity of heterocyclic arylamine food mutagens: Human DNA polymerase η selectively produces a two-base deletion in copying the N2-guanyl adduct of 2-amino-3-methylimidazo[4,5-f]quinoline but not the C8 adduct at the NarI G3 site. J. Biol. Chem. 2006, 281, 25297–25306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-Y.; Guengerich, F.P. Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase η. J. Mol. Biol. 2005, 352, 72–90. [Google Scholar] [CrossRef]
- Choi, J.-Y.; Lim, S.; Kim, E.J.; Jo, A.; Guengerich, F.P. Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases α, δ, η, ι, κ, and REV1. J. Mol. Biol. 2010, 404, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Biertumpfel, C.; Zhao, Y.; Kondo, Y.; Ramon-Maiques, S.; Gregory, M.; Lee, J.Y.; Masutani, C.; Lehmann, A.R.; Hanaoka, F.; Yang, W. Structure and mechanism of human DNA polymerase η. Nature 2010, 465, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Biertumpfel, C.; Gregory, M.T.; Hua, Y.J.; Hanaoka, F.; Yang, W. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. USA 2012, 109, 7269–7274. [Google Scholar] [CrossRef] [Green Version]
- Broughton, B.C.; Cordonnier, A.; Kleijer, W.J.; Jaspers, N.G.; Fawcett, H.; Raams, A.; Garritsen, V.H.; Stary, A.; Avril, M.F.; Boudsocq, F.; et al. Molecular analysis of mutations in DNA polymerase η in xeroderma pigmentosum-variant patients. Proc. Natl. Acad. Sci. USA 2002, 99, 815–820. [Google Scholar] [CrossRef] [Green Version]
- Opletalova, K.; Bourillon, A.; Yang, W.; Pouvelle, C.; Armier, J.; Despras, E.; Ludovic, M.; Mateus, C.; Robert, C.; Kannouche, P.; et al. Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations. Hum. Mutat. 2014, 35, 117–128. [Google Scholar] [CrossRef]
- Feltes, B.C.; Menck, C.F.M. Current state of knowledge of human DNA polymerase eta protein structure and disease-causing mutations. Mutat. Res. Rev. Mutat. Res. 2022, 790, 108436. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, M.; Soda, H.; Sadanaga, N.; Taniguchi, H.; Ikeda, T.; Maruta, H.; Dotsu, Y.; Ogawara, D.; Fukuda, Y.; Mukae, H. Alert Regarding Cisplatin-induced Severe Adverse Events in Cancer Patients with Xeroderma Pigmentosum. Intern. Med. 2017, 56, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, A.; Masutani, C.; Iwai, S.; Hanaoka, F. Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase η. Nucleic Acids Res. 2000, 28, 2473–2480. [Google Scholar] [CrossRef] [Green Version]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Song, I.; Jo, A.; Shin, J.-H.; Cho, H.; Eoff, R.L.; Guengerich, F.P.; Choi, J.-Y. Biochemical analysis of six genetic variants of error-prone human DNA polymerase ι involved in translesion DNA synthesis. Chem. Res. Toxicol. 2014, 27, 1837–1852. [Google Scholar] [CrossRef]
- Yeom, M.; Hong, J.K.; Kim, J.K.; Guengerich, F.P.; Choi, J.-Y. Three human pol ι variants with impaired polymerase activity fail to rescue H2O2 sensitivity in POLI-deficient cells. Chem. Res. Toxicol. 2020, 33, 2120–2129. [Google Scholar] [CrossRef]
- Kim, J.-K.; Yeom, M.; Hong, J.-K.; Song, I.; Lee, Y.-S.; Guengerich, F.P.; Choi, J.-Y. Six germline genetic variations impair the translesion synthesis activity of human DNA polymerase κ. Chem. Res. Toxicol. 2016, 29, 1741–1754. [Google Scholar] [CrossRef] [Green Version]
- Yeom, M.; Kim, I.-H.; Kim, J.-K.; Kang, K.; Eoff, R.L.; Guengerich, F.P.; Choi, J.-Y. Effects of twelve germline missense variations on DNA lesion and G-quadruplex bypass activities of human DNA polymerase REV1. Chem. Res. Toxicol. 2016, 29, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Bassett, E.; King, N.M.; Bryant, M.F.; Hector, S.; Pendyala, L.; Chaney, S.G.; Cordeiro-Stone, M. The role of DNA polymerase η in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res. 2004, 64, 6469–6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J., Jr.; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase η is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 2009, 29, 3344–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, C.E.; Kohlstedt, D.; Meisel, C.; Keller, K.; Becker, K.; Mackenroth, L.; Rump, A.; Schrock, E.; Wimberger, P.; Kast, K. BRCA1/2 missense mutations and the value of in-silico analyses. Eur. J. Med. Genet. 2017, 60, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Di Lucca, J.; Guedj, M.; Lacapere, J.J.; Fargnoli, M.C.; Bourillon, A.; Dieude, P.; Dupin, N.; Wolkenstein, P.; Aegerter, P.; Saiag, P.; et al. Variants of the xeroderma pigmentosum variant gene (POLH) are associated with melanoma risk. Eur. J. Cancer 2009, 45, 3228–3236. [Google Scholar] [CrossRef]
- King, N.M.; Nikolaishvili-Feinberg, N.; Bryant, M.F.; Luche, D.D.; Heffernan, T.P.; Simpson, D.A.; Hanaoka, F.; Kaufmann, W.K.; Cordeiro-Stone, M. Overproduction of DNA polymerase η does not raise the spontaneous mutation rate in diploid human fibroblasts. DNA Repair 2005, 4, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Cleaver, J.E.; Hatahet, Z.; Honkanen, R.E.; Chang, J.Y.; Yen, Y.; Chou, K.M. Human DNA polymerase η activity and translocation is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 16578–16583. [Google Scholar] [CrossRef] [Green Version]
- Bienko, M.; Green, C.M.; Sabbioneda, S.; Crosetto, N.; Matic, I.; Hibbert, R.G.; Begovic, T.; Niimi, A.; Mann, M.; Lehmann, A.R.; et al. Regulation of translesion synthesis DNA polymerase η by monoubiquitination. Mol. Cell 2010, 37, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase η with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Lin, Q.; Clark, A.B.; McCulloch, S.D.; Yuan, T.; Bronson, R.T.; Kunkel, T.A.; Kucherlapati, R. Increased susceptibility to UV-induced skin carcinogenesis in polymerase η-deficient mice. Cancer Res. 2006, 66, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rs ID a | Nucleotide Change | Amino Acid Change | Protein Domain | Minor Allele Frequency b | In Silico Prediction | |||
|---|---|---|---|---|---|---|---|---|
| 1000 Genomes | ESP | gnomAD | SIFT | PolyPhen-2 | ||||
| rs371810027 | c.102T > G | C34W | finger | - | 0.00008 | - | deleterious | probably damaging |
| rs200366966 | c.440T > A | I147N | palm | 0.0002 | - | 0.00000398 | deleterious | probably damaging |
| rs201365711 | c.500G > A | R167Q | palm | 0.0002 | - | 0.0000247 | deleterious | possibly damaging |
| rs2307456 | c.626G > T | G209V | palm | 0.0042 | 0.00223 | 0.00364 | deleterious | probably damaging |
| rs147712217 | c.815T > C | I272T | thumb | 0.0008 | 0.00054 | 0.00106 | deleterious | benign |
| rs200149644 | c.887C > T | S296F | thumb | - | - | 0.000163 | deleterious | probably damaging |
| rs35675573 | c.986C > T | T329I | PAD | 0.0046 | 0.00546 | 0.00174 | deleterious | benign |
| rs9333548 | c.1000C > G | R334G | PAD | 0.0014 | 0.00177 | 0.000605 | deleterious | benign |
| DNA Template | Template Base | Pol η (1—432) | Km (μM) | kcat (s−1) | kcat/Km (s−1 μM−1) | Relative Efficiency a | fmis b |
|---|---|---|---|---|---|---|---|
| TT | 3′-T | wild-type | 0.38 ± 0.04 | 0.076 ± 0.002 | 0.20 ± 0.02 | 1 | 0.17 |
| C34W | 1.3 ± 0.2 | 0.032 ± 0.004 | 0.025 ± 0.003 | 0.13 | 0.056 | ||
| I147N | 0.80 ± 0.06 | 0.0096 ± 0.0002 | 0.012 ± 0.001 | 0.06 | 0.36 | ||
| R167Q | 0.26 ± 0.04 | 0.018 ± 0.001 | 0.069 ± 0.010 | 0.35 | 0.14 | ||
| G209V | 0.20 ± 0.05 | 0.023 ± 0.002 | 0.12 ± 0.01 | 0.60 | 0.27 | ||
| I272T | 0.081 ± 0.019 | 0.041 ± 0.002 | 0.51 ± 0.10 | 2.6 | 0.084 | ||
| S296F | 0.12 ± 0.02 | 0.052 ± 0.002 | 0.43 ± 0.06 | 2.2 | 0.10 | ||
| T329I | 0.17 ± 0.03 | 0.051 ± 0.003 | 0.30 ± 0.05 | 1.5 | 0.16 | ||
| R334G | 0.15 ± 0.04 | 0.073 ± 0.007 | 0.49 ± 0.14 | 2.5 | 0.12 | ||
| 5′-T | wild-type | 0.087 ± 0.017 | 0.096 ± 0.005 | 1.1 ± 0.2 | 1 | 0.17 | |
| C34W | 0.11 ± 0.01 | 0.030 ± 0.001 | 0.27 ± 0.02 | 0.25 | 0.056 | ||
| I147N | 0.084 ± 0.008 | 0.021 ± 0.001 | 0.25 ± 0.02 | 0.23 | 0.11 | ||
| R167Q | 0.074 ± 0.010 | 0.032 ± 0.001 | 0.43 ± 0.05 | 0.39 | 0.095 | ||
| G209V | 0.048 ± 0.010 | 0.053 ± 0.002 | 1.1 ± 0.2 | 1.0 | 0.21 | ||
| I272T | 0.011 ± 0.001 | 0.029 ± 0.001 | 2.6 ± 0.2 | 2.4 | 0.24 | ||
| S296F | 0.04 ± 0.01 | 0.056 ± 0.002 | 1.4 ± 0.2 | 1.3 | 0.41 | ||
| T329I | 0.011 ± 0.001 | 0.032 ± 0.001 | 2.9 ± 0.3 | 2.6 | 0.13 | ||
| R334G | 0.016 ± 0.002 | 0.038 ± 0.002 | 2.4 ± 0.3 | 2.2 | 0.20 | ||
| CTD | 3′-T | wild-type | 0.39 ± 0.05 | 0.066 ± 0.003 | 0.17 ± 0.02 | 1 | 0.14 |
| C34W | 0.72 ± 0.07 | 0.0083 ± 0.0002 | 0.012 ± 0.001 | 0.07 | 0.066 | ||
| I147N | 0.42 ± 0.07 | 0.0075 ± 0.0003 | 0.018 ± 0.002 | 0.11 | 0.24 | ||
| R167Q | 0.48 ± 0.05 | 0.023 ± 0.001 | 0.048 ± 0.005 | 0.28 | 0.085 | ||
| G209V | 0.23 ± 0.05 | 0.061 ± 0.005 | 0.27 ± 0.06 | 1.6 | 0.063 | ||
| I272T | 0.025 ± 0.007 | 0.017 ± 0.001 | 0.68 ± 0.16 | 4.0 | 0.050 | ||
| S296F | 0.11 ± 0.01 | 0.034 ± 0.001 | 0.31 ± 0.02 | 1.8 | 0.11 | ||
| T329I | 0.054 ± 0.009 | 0.038 ± 0.002 | 0.70 ± 0.11 | 4.1 | 0.031 | ||
| R334G | 0.13 ± 0.02 | 0.077 ± 0.006 | 0.59 ± 0.09 | 3.5 | 0.049 | ||
| 5′-T | wild-type | 0.065 ± 0.005 | 0.044 ± 0.005 | 0.68 ± 0.04 | 1 | 0.053 | |
| C34W | 0.10 ± 0.02 | 0.013 ±0.001 | 0.13 ± 0.02 | 0.19 | 0.016 | ||
| I147N | 0.060 ± 0.006 | 0.011 ± 0.0003 | 0.18 ± 0.01 | 0.26 | 0.018 | ||
| R167Q | 0.11 ± 0.01 | 0.028 ± 0.001 | 0.25 ± 0.02 | 0.37 | 0.052 | ||
| G209V | 0.030 ± 0.003 | 0.032 ± 0.001 | 1.1 ± 0.1 | 1.6 | 0.032 | ||
| I272T | 0.0081 ± 0.0008 | 0.015 ± 0.0004 | 1.9 ± 0.2 | 2.8 | 0.022 | ||
| S296F | 0.017 ± 0.003 | 0.030 ± 0.001 | 1.8 ± 0.3 | 2.6 | 0.037 | ||
| T329I | 0.013 ± 0.001 | 0.020 ± 0.001 | 1.5 ± 0.1 | 2.2 | 0.080 | ||
| R334G | 0.020 ± 0.002 | 0.023 ± 0.001 | 1.2 ± 0.1 | 1.8 | 0.067 |
| pol η (1—432) | Kd (nM) | |
|---|---|---|
| 13-FAM-mer/25-TT-mer | 13-FAM-mer/25-CTD-mer | |
| wild-type | 10 ± 1 | 11 ± 2 |
| C34W | 6.1 ± 0.6 | 5.9 ± 0.8 |
| I147N | 6.7 ± 1.2 | 7.0 ± 1.5 |
| R167Q | 9.5 ± 1.2 | 18 ± 3 |
| G209V | 8.4 ± 1.0 | 11 ± 2 |
| I272T | 8.5 ± 1.9 | 11 ± 2 |
| S296F | 10 ± 2 | 14 ± 2 |
| T329I | 5.0 ± 0.9 | 5.9 ± 0.7 |
| R334G | 11 ± 2 | 12 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeom, M.; Hong, J.-K.; Shin, J.-H.; Lee, Y.; Guengerich, F.P.; Choi, J.-Y. Identification of Three Human POLH Germline Variants Defective in Complementing the UV- and Cisplatin-Sensitivity of POLH-Deficient Cells. Int. J. Mol. Sci. 2023, 24, 5198. https://doi.org/10.3390/ijms24065198
Yeom M, Hong J-K, Shin J-H, Lee Y, Guengerich FP, Choi J-Y. Identification of Three Human POLH Germline Variants Defective in Complementing the UV- and Cisplatin-Sensitivity of POLH-Deficient Cells. International Journal of Molecular Sciences. 2023; 24(6):5198. https://doi.org/10.3390/ijms24065198
Chicago/Turabian StyleYeom, Mina, Jin-Kyung Hong, Joo-Ho Shin, Yunjong Lee, Frederick Peter Guengerich, and Jeong-Yun Choi. 2023. "Identification of Three Human POLH Germline Variants Defective in Complementing the UV- and Cisplatin-Sensitivity of POLH-Deficient Cells" International Journal of Molecular Sciences 24, no. 6: 5198. https://doi.org/10.3390/ijms24065198
APA StyleYeom, M., Hong, J. -K., Shin, J. -H., Lee, Y., Guengerich, F. P., & Choi, J. -Y. (2023). Identification of Three Human POLH Germline Variants Defective in Complementing the UV- and Cisplatin-Sensitivity of POLH-Deficient Cells. International Journal of Molecular Sciences, 24(6), 5198. https://doi.org/10.3390/ijms24065198