Phylogenetic Analysis of Spliceosome SF3a2 in Different Plant Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
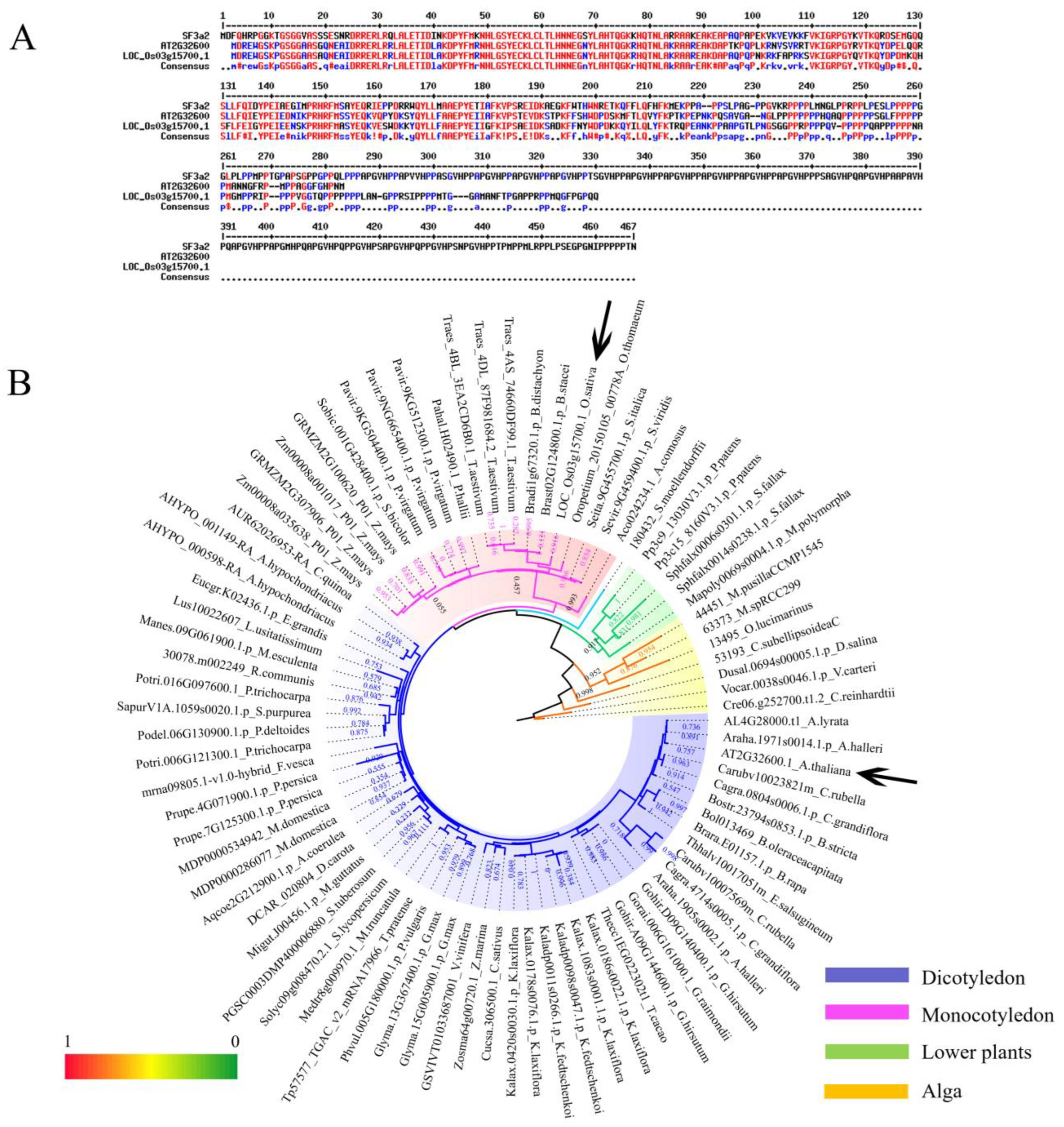
2.1. Identification and Phylogenetic Analysis of SF3a2 Genes in Planta
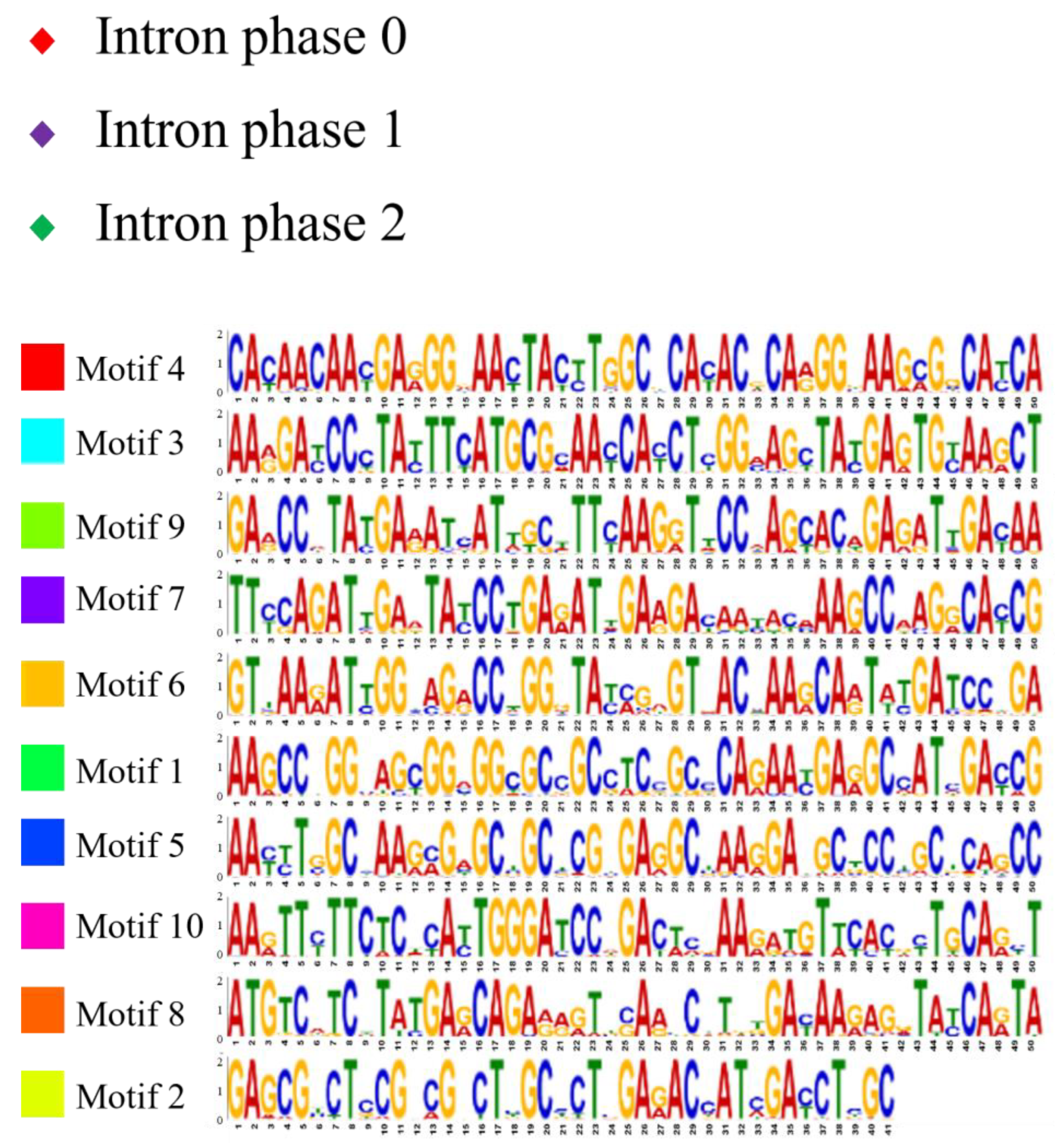
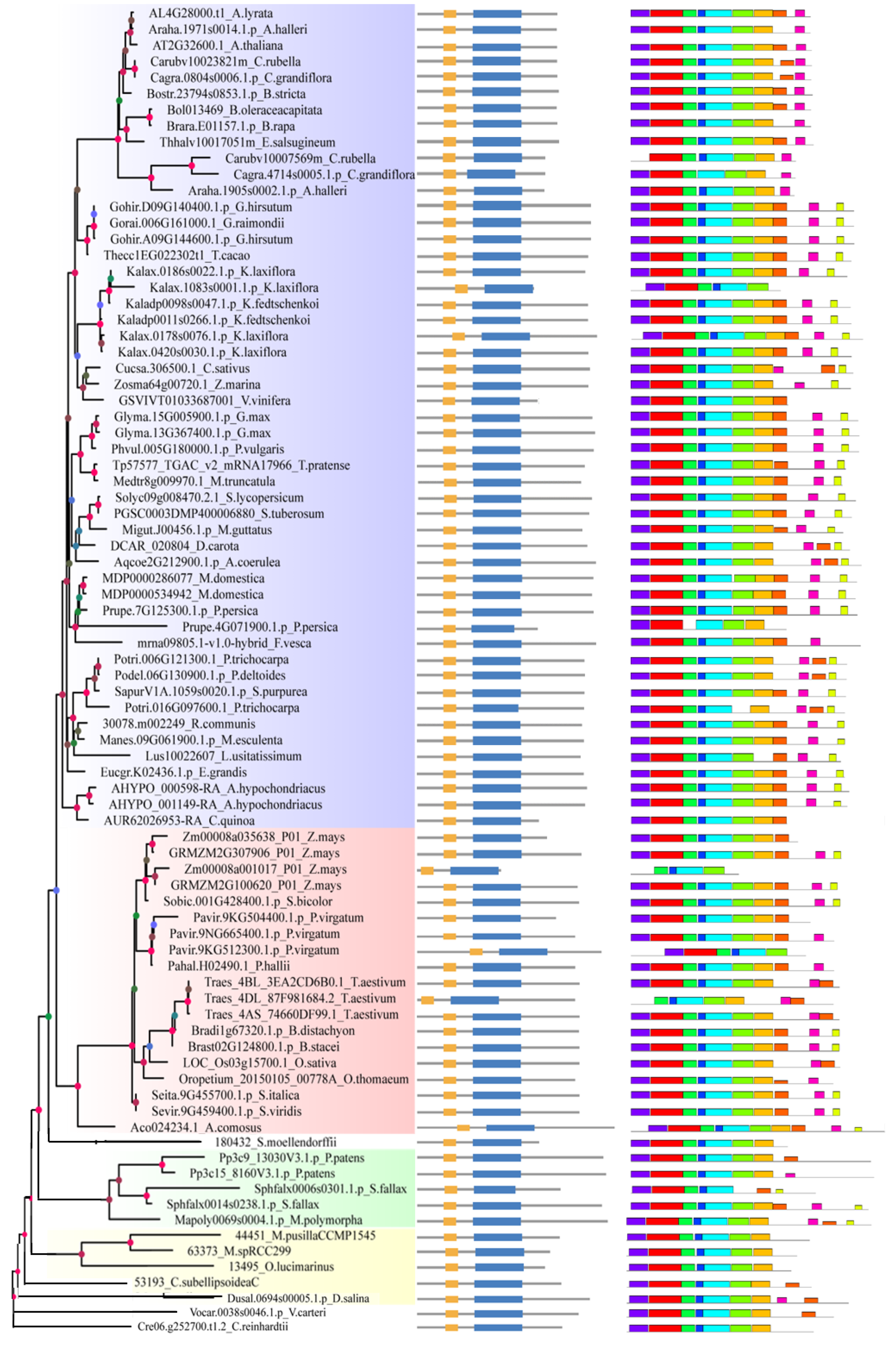
2.2. Analysis of Gene Structure and Peptide Domain
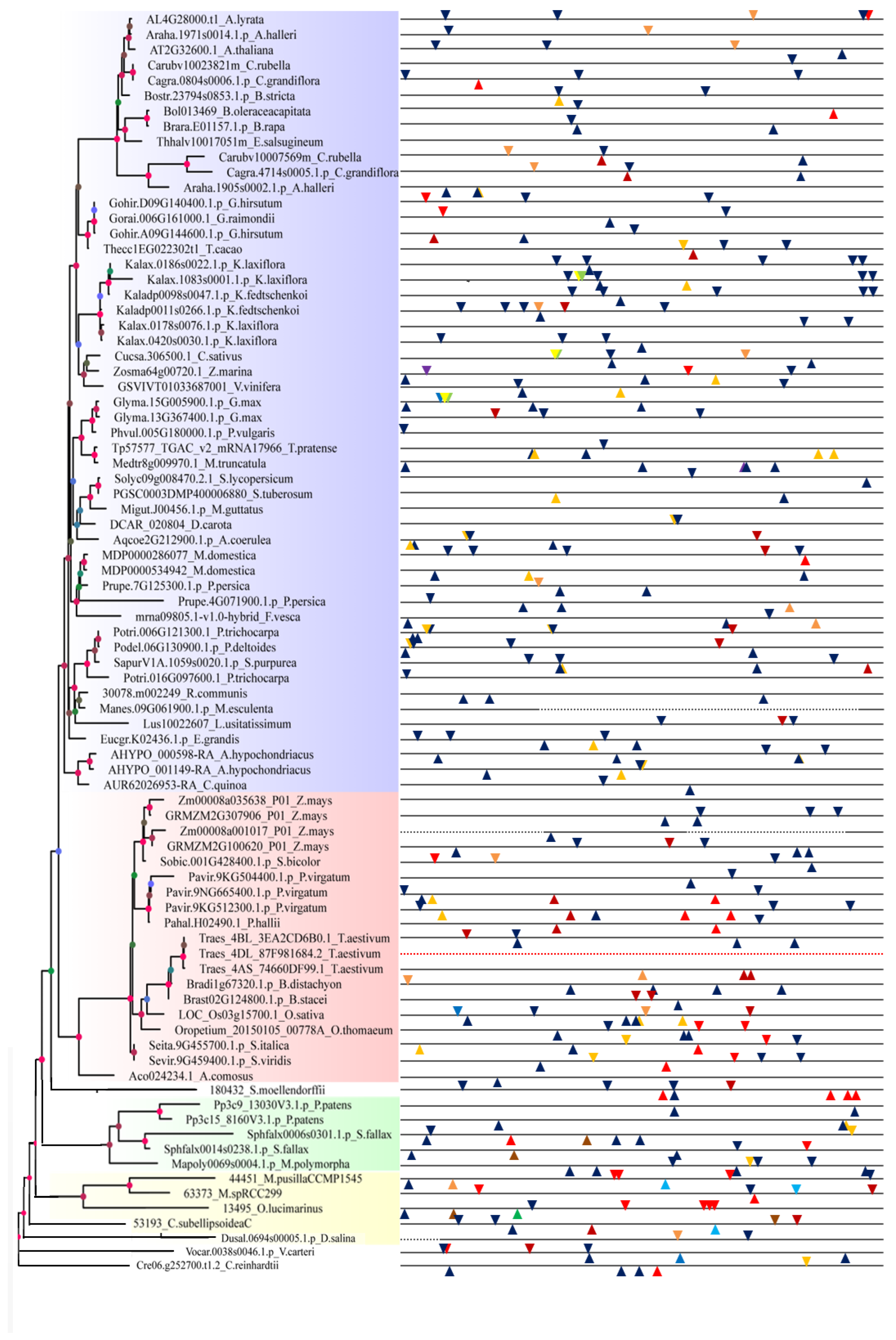
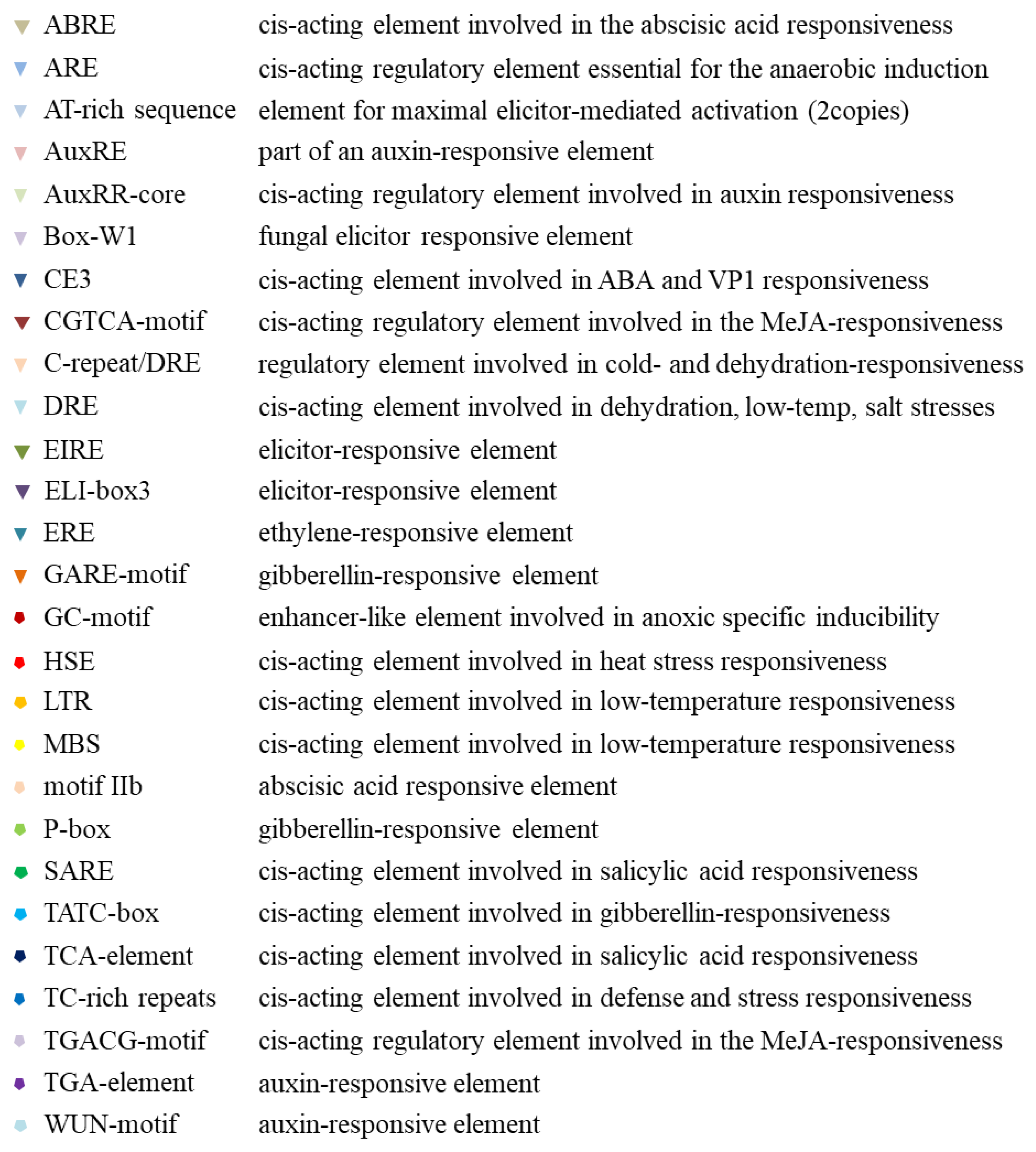
2.3. Promoter Analysis of Tissue, Hormone, and Stress Levels
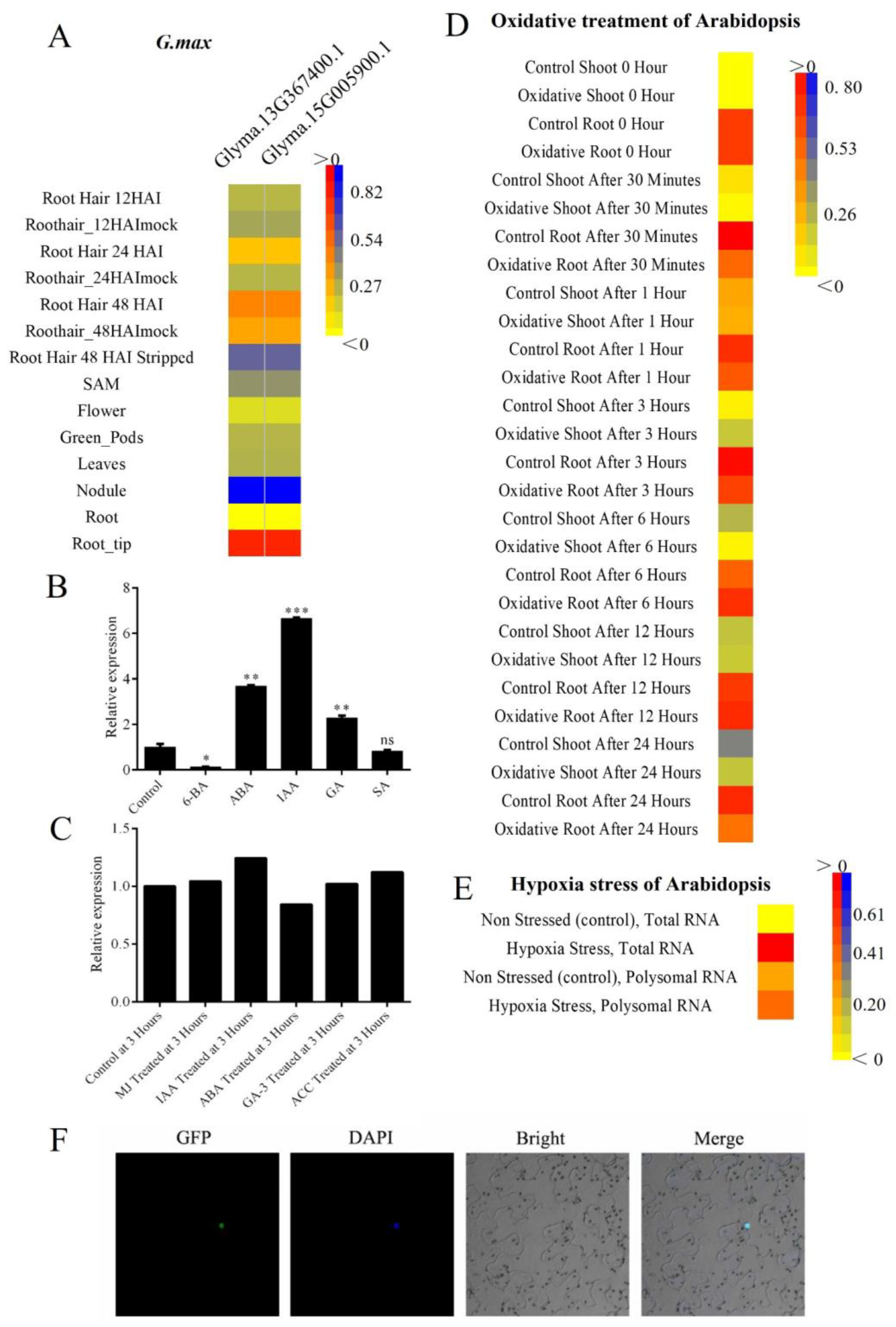
2.4. Expression Pattern for Tissue and Hormone
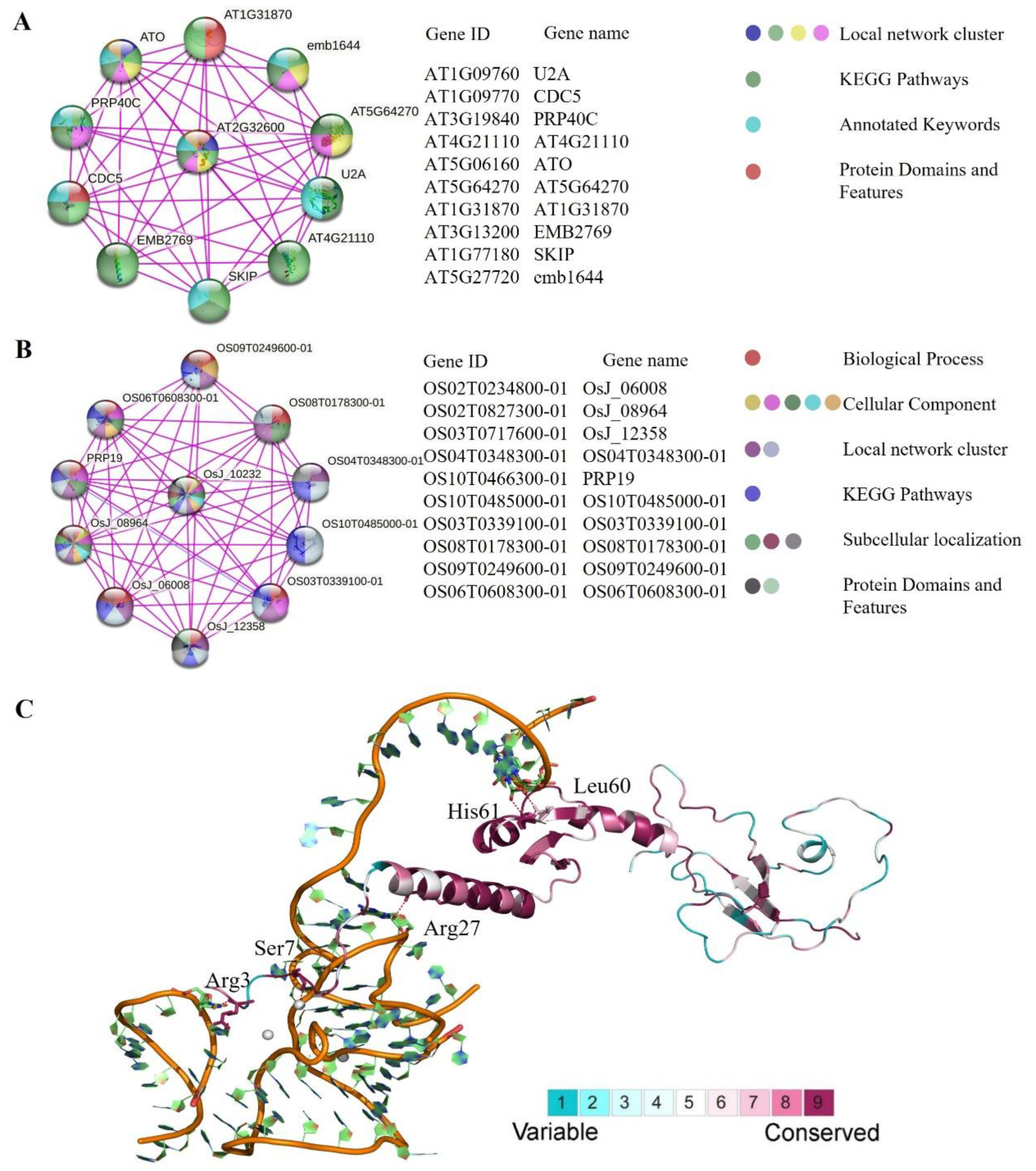
2.5. Analysis of Protein–Protein Interaction and 3D Structure Conservation Analysis
2.6. Genomic Synteny Relationship of SF3a2 Genes
3. Discussion
3.1. The Evolutionary Correlation of SF3a2s Suggests That They May Have Similarities in Function
3.2. Differences between Species Indicate the Functional Differentiation of SF3a2s in Plants
4. Materials and Methods
4.1. Identification and Phylogenetic Analysis of SF3a2 Genes in Planta
4.2. Phylogenetic Analysis of SF3a2 Gene Family in Planta
4.3. Analysis of Gene Structure and Peptide Domain
4.4. Promoter Analysis of Tissue, Hormone, and Stress Levels
4.5. Microarray-Based Expression Analysis
4.6. Plant Materials and Growth Conditions
4.7. Analysis of Protein–Protein Interaction
4.8. Three-Dimensional Protein Analysis
4.9. Vector Construction and Subcellular Localization
4.10. Analysis of Genomic Synteny Relationship
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.; Wan, R.; Wang, L.; Xu, K.; Zhang, Q.; Lei, J.; Shi, Y. Structure of the activated human minor spliceosome. Science 2021, 371, eabg0879. [Google Scholar] [CrossRef]
- Verma, B.; Akinyi, M.V.; Norppa, A.J.; Frilander, M.J. Minor spliceosome and disease. Semin. Cell Dev. Biol. 2018, 79, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Fast, N.M. Intron splicing: U12 spliceosomal introns not so ‘minor’ after all. Curr. Biol. 2021, 31, R912–R914. [Google Scholar] [CrossRef]
- Norppa, A.J.; Frilander, M.J. The integrity of the U12 snRNA 3′ stem-loop is necessary for its overall stability. Nucleic Acids Res. 2021, 49, 2835–2847. [Google Scholar] [CrossRef]
- Tarn, W.Y.; Steitz, J.A. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 1996, 273, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.; Lynch, K.W. Alternative splicing and cancer: Insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020, 34, 1005–1016. [Google Scholar] [CrossRef]
- Jabre, I.; Reddy, A.S.N.; Kalyna, M.; Chaudhary, S.; Khokhar, W.; Byrne, L.J.; Wilson, C.M.; Syed, N.H. Does co-transcriptional regulation of alternative splicing mediate plant stress responses? Nucleic Acids Res. 2019, 47, 2716–2726. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Wu, C. Alternative Splicing and Isoforms: From Mechanisms to Diseases. Genes 2022, 13, 401. [Google Scholar] [CrossRef]
- Xu, S.-J.; Lombroso, S.I.; Fischer, D.K.; Carpenter, M.D.; Marchione, D.M.; Hamilton, P.J.; Lim, C.J.; Neve, R.L.; Garcia, B.A.; Wimmer, M.E.; et al. Chromatin-mediated alternative splicing regulates cocaine-reward behavior. Neuron 2021, 109, 2943–2966.e8. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Ahn, J.H. Regulation of flowering time by ambient temperature: Repressing the repressors and activating the activators. New Phytol. 2021, 230, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Laloum, T.; Martín, G.; Duque, P. Alternative Splicing Control of Abiotic Stress Responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Roychoudhury, A. Gene regulation at transcriptional and post-transcriptional levels to combat salt stress in plants. Physiol. Plant. 2021, 173, 1556–1572. [Google Scholar] [CrossRef]
- Wang, H.L.; Zhang, Y.; Wang, T.; Yang, Q.; Yang, Y.; Li, Z.; Li, B.; Wen, X.; Li, W.; Yin, W.; et al. An alternative splicing variant of PtRD26 delays leaf senescence by regulating multiple NAC transcription factors in Populus. Plant Cell 2021, 33, 1594–1614. [Google Scholar] [CrossRef]
- Xue, X.; Jiao, F.; Xu, H.; Jiao, Q.; Zhang, X.; Zhang, Y.; Du, S.; Xi, M.; Wang, A.; Chen, J.; et al. The role of RNA-binding protein, microRNA and alternative splicing in seed germination: A field need to be discovered. BMC Plant Biol. 2021, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Galej, W.P.; Fica, S.M.; Lin, P.C.; Newman, A.J.; Nagai, K. CryoEM structures of two spliceosomal complexes: Starter and dessert at the spliceosome feast. Curr. Opin. Struct. Biol. 2016, 36, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.E.; Fica, S.M.; Galej, W.P.; Nagai, K. Structural basis for conformational equilibrium of the catalytic spliceosome. Mol. Cell 2021, 81, 1439–1452.e9. [Google Scholar] [CrossRef]
- Wysoczanski, P.; Becker, S.; Zweckstetter, M. Structures of intermediates during RES complex assembly. Sci. Rep. 2015, 5, 12545. [Google Scholar] [CrossRef] [Green Version]
- Nyambega, B.; Helbig, C.; Masiga, D.K.; Clayton, C.; Levin, M.J. Proteins associated with SF3a60 in T. brucei. PLoS ONE 2014, 9, e91956. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Agafonov, D.E.; Schmitzová, J.; Hartmuth, K.; Fabrizio, P.; Lührmann, R. Dynamic Contacts of U2, RES, Cwc25, Prp8 and Prp45 Proteins with the Pre-mRNA Branch-Site and 3′ Splice Site during Catalytic Activation and Step 1 Catalysis in Yeast Spliceosomes. PLoS Genet. 2015, 11, e1005539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Ferfoglia, F.; Raleff, F.; Krämer, A. Interaction domains and nuclear targeting signals in subunits of the U2 small nuclear ribonucleoprotein particle-associated splicing factor SF3a. J. Biol. Chem. 2011, 286, 13106–13114. [Google Scholar] [CrossRef] [Green Version]
- Lorkovic, Z.J.; Lehner, R.; Forstner, C.; Barta, A. Evolutionary conservation of minor U12-type spliceosome between plants and humans. RNA 2005, 11, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gringel, S.; van Bergeijk, J.; Haastert, K.; Grothe, C.; Claus, P. Nuclear fibroblast growth factor-2 interacts specifically with splicing factor SF3a66. Biol. Chem. 2004, 385, 1203–1208. [Google Scholar] [CrossRef]
- Bennett, M.; Reed, R. Correspondence between a mammalian spliceosome component and an essential yeast splicing factor. Science 1993, 262, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Dresser, D.W.; Jamin, S.P.; Atkins, C.J.; Guerrier, D. An expressed GNRP-like gene shares a bi-directional promoter with SF3A2 (SAP62) immediately upstream of AMH. Gene 2001, 277, 163–173. [Google Scholar] [CrossRef]
- Rigo, N.; Sun, C.; Fabrizio, P.; Kastner, B.; Lührmann, R. Protein localisation by electron microscopy reveals the architecture of the yeast spliceosomal B complex. Embo J. 2015, 34, 3059–3073. [Google Scholar] [CrossRef] [Green Version]
- Nesic, D.; Krämer, A. Domains in human splicing factors SF3a60 and SF3a66 required for binding to SF3a120, assembly of the 17S U2 snRNP, and prespliceosome formation. Mol. Cell Biol. 2001, 21, 6406–6417. [Google Scholar] [CrossRef] [Green Version]
- Conant, G.C.; Birchler, J.A.; Pires, J.C. Dosage, duplication, and diploidization: Clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 2014, 19, 91–98. [Google Scholar] [CrossRef]
- Vasco, A.; Moran, R.C.; Ambrose, B.A. The evolution, morphology, and development of fern leaves. Front. Plant Sci. 2013, 4, 345. [Google Scholar] [CrossRef] [Green Version]
- Moyroud, E.; Glover, B.J. The Evolution of Diverse Floral Morphologies. Curr. Biol. 2017, 27, R941–R951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coudert, Y.; Bell, N.E.; Edelin, C.; Harrison, C.J. Multiple innovations underpinned branching form diversification in mosses. New Phytol. 2017, 215, 840–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlad, I.M.; Balaji, V.S.; Vikas, C.R.; Ramani, D.; Larry, S.D. Automatic online tuning for fast Gaussian summation. Adv. Neural Inf. Process. Syst. 2008, 21. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. -Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Das, D.; Li, M.; Song, T.; Yang, J.; Liu, Y. Phylogenetic Analysis of Spliceosome SF3a2 in Different Plant Species. Int. J. Mol. Sci. 2023, 24, 5232. https://doi.org/10.3390/ijms24065232
Tian Y, Das D, Li M, Song T, Yang J, Liu Y. Phylogenetic Analysis of Spliceosome SF3a2 in Different Plant Species. International Journal of Molecular Sciences. 2023; 24(6):5232. https://doi.org/10.3390/ijms24065232
Chicago/Turabian StyleTian, Yuan, Debatosh Das, Min Li, Tao Song, Jingfang Yang, and Yinggao Liu. 2023. "Phylogenetic Analysis of Spliceosome SF3a2 in Different Plant Species" International Journal of Molecular Sciences 24, no. 6: 5232. https://doi.org/10.3390/ijms24065232
APA StyleTian, Y., Das, D., Li, M., Song, T., Yang, J., & Liu, Y. (2023). Phylogenetic Analysis of Spliceosome SF3a2 in Different Plant Species. International Journal of Molecular Sciences, 24(6), 5232. https://doi.org/10.3390/ijms24065232