

Ligand-Induced Activation of GPR110 (ADGRF1) to Improve Visual Function Impaired by Optic Nerve Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
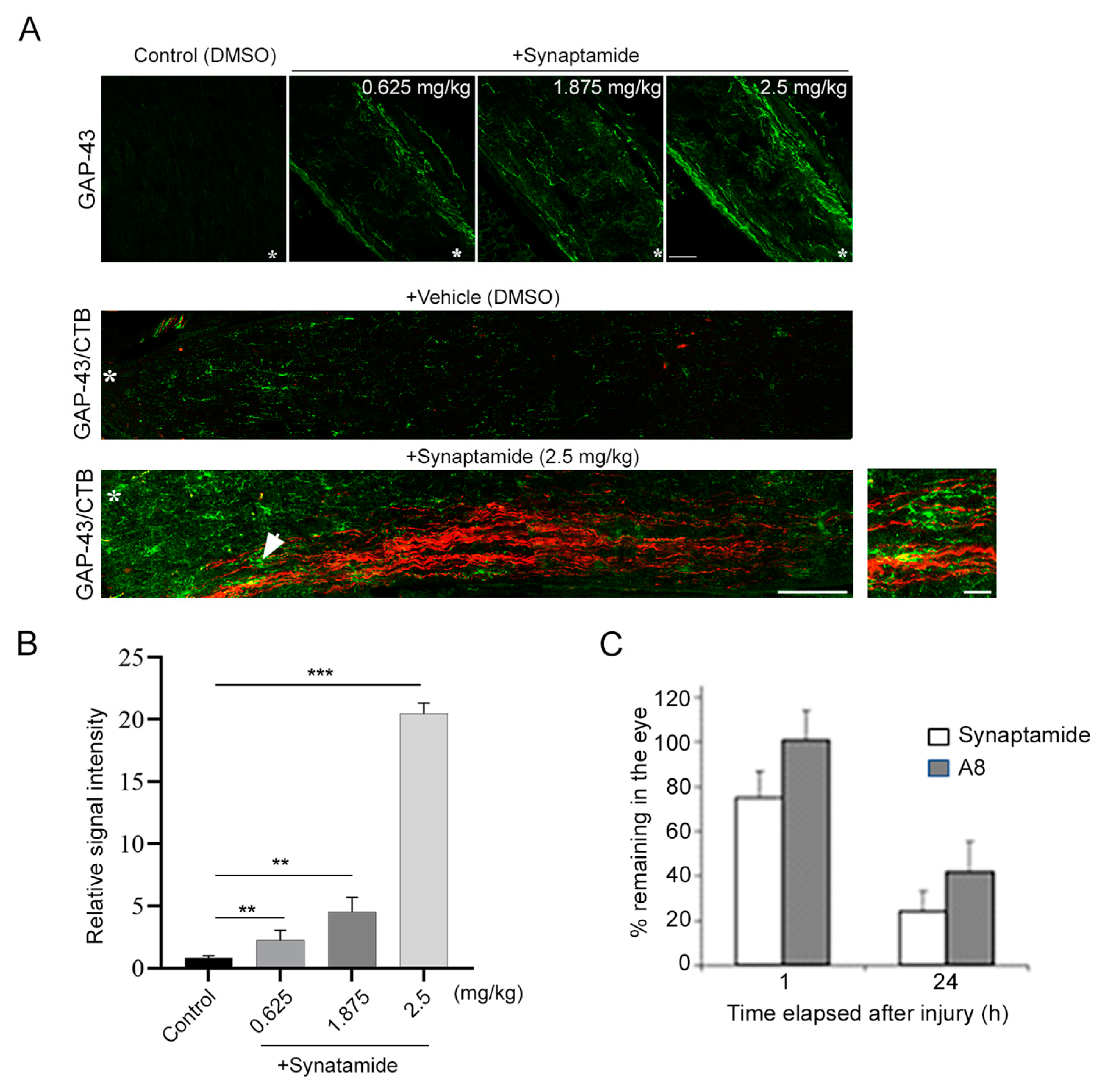
2.1. Synaptamide Stimulates Axon Regeneration after ONC
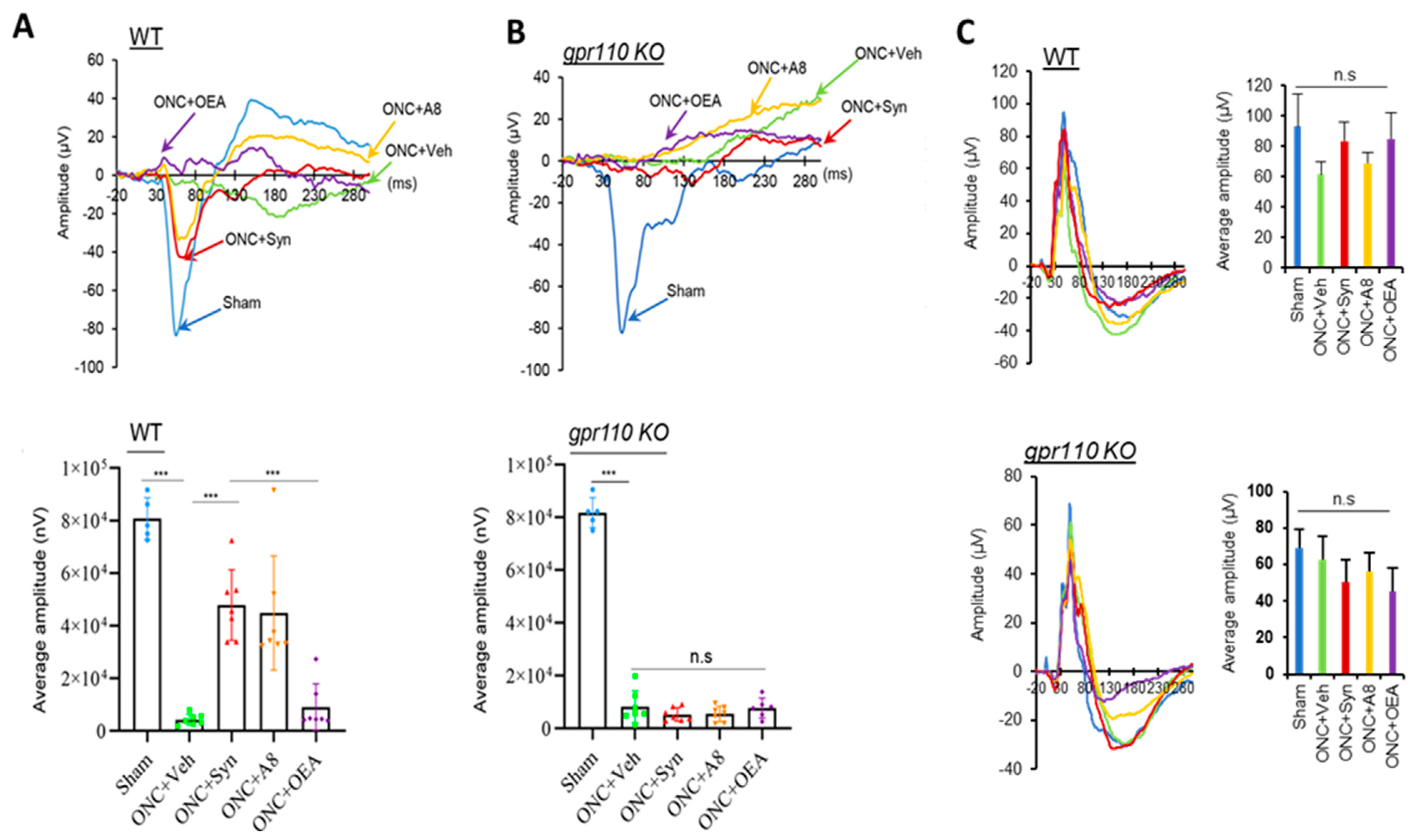
2.2. GPR110 Activation by Synaptamide or A8 Treatment Leads to Partial Restoration of Visual Activity Impaired by ONC
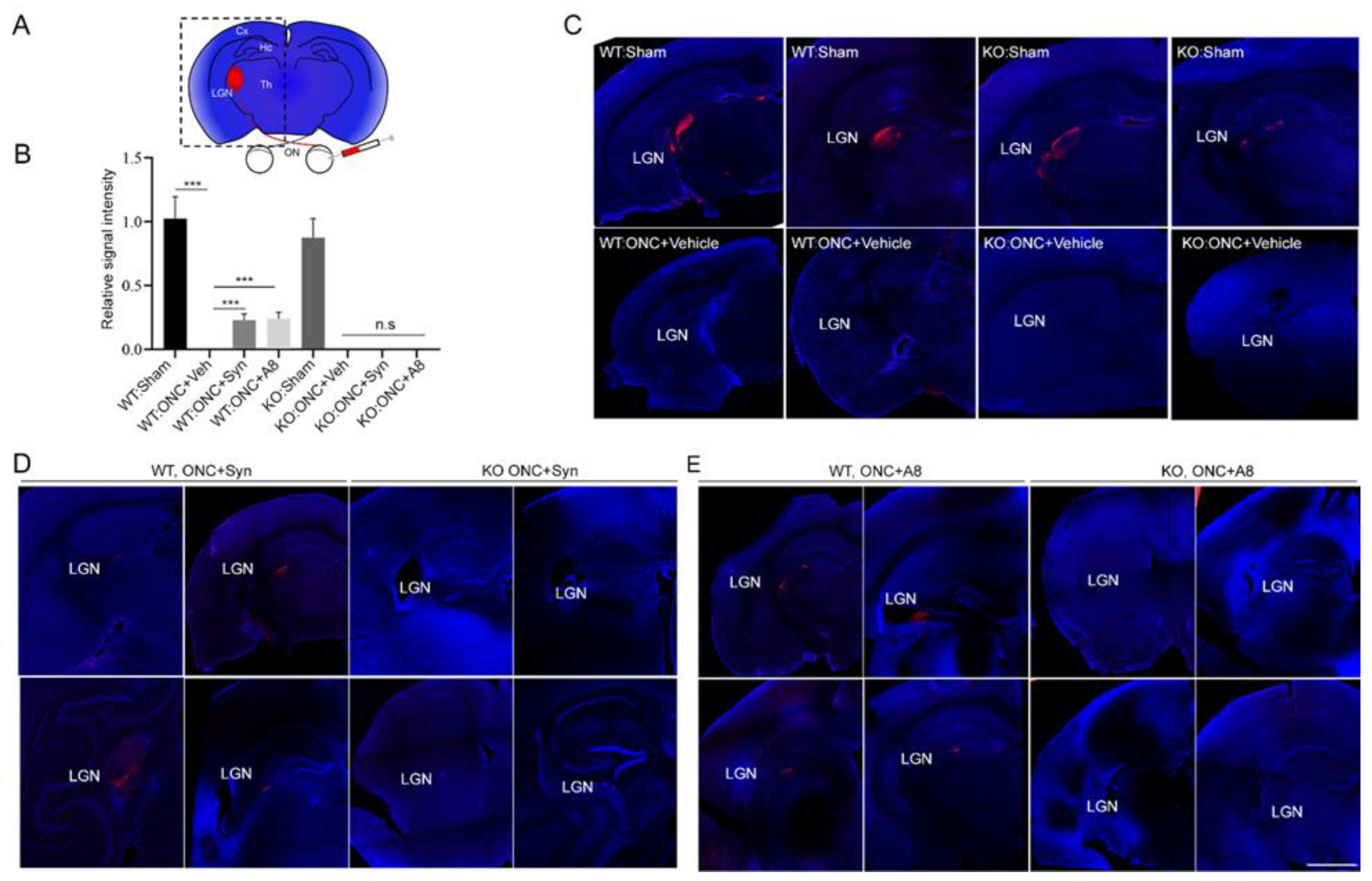
2.3. ONC-Induced Loss of RGC Axons at the Brain Target was Partly Prevented by the Treatment with GPR110 Ligands
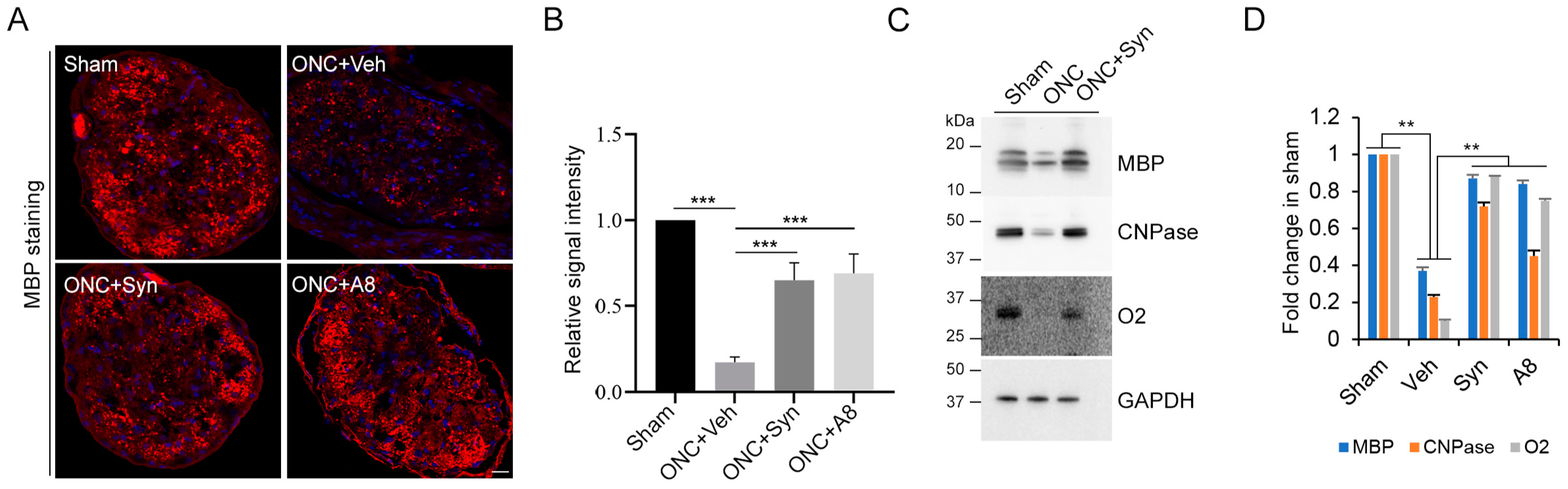
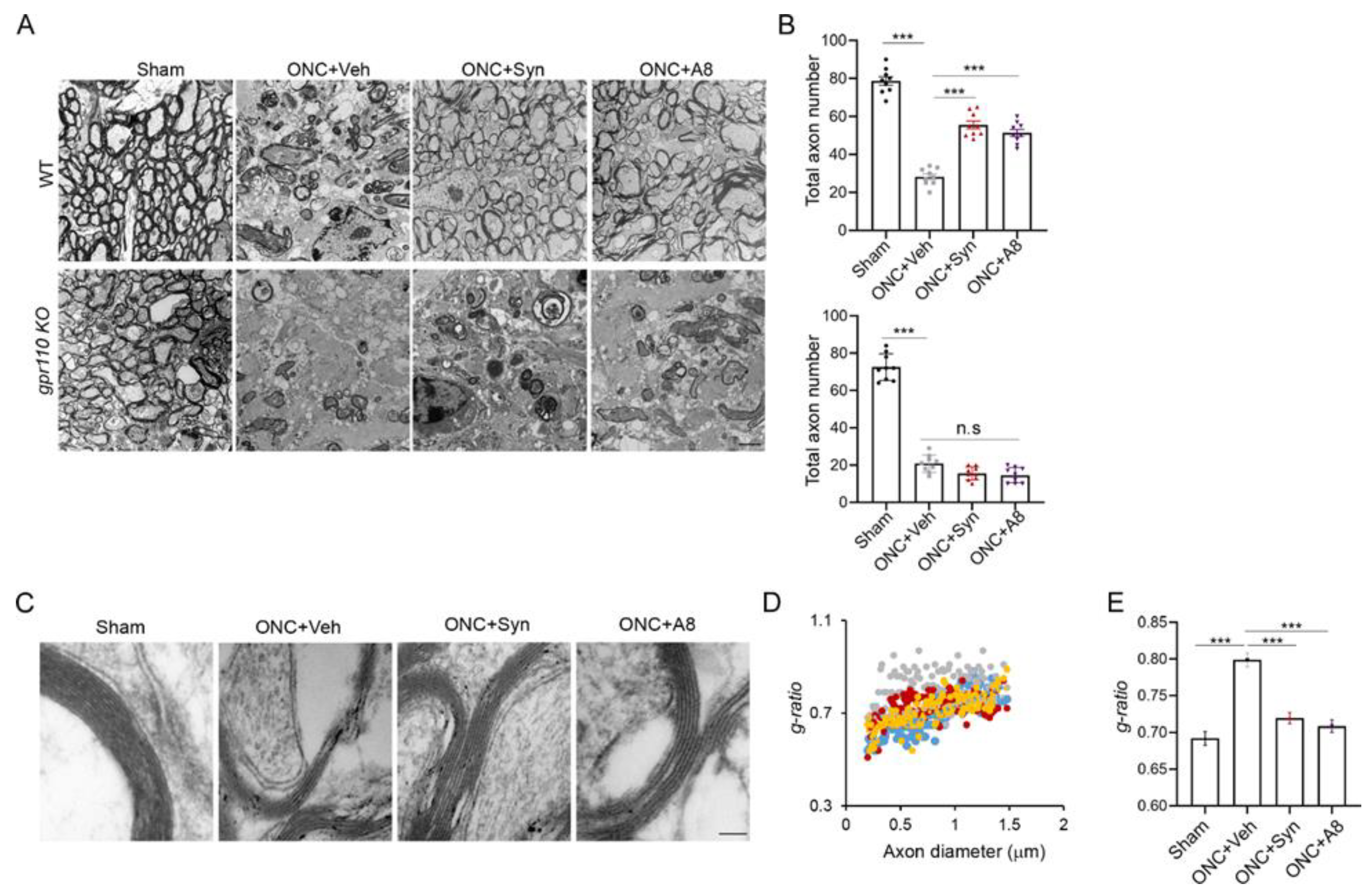
2.4. Optic Nerve Myelination Status Degraded after ONC Was Improved by Synaptamide or A8 Treatment
2.5. GPR110 Ligands Improved Structural Integrity of Axons Deteriorated by ONC
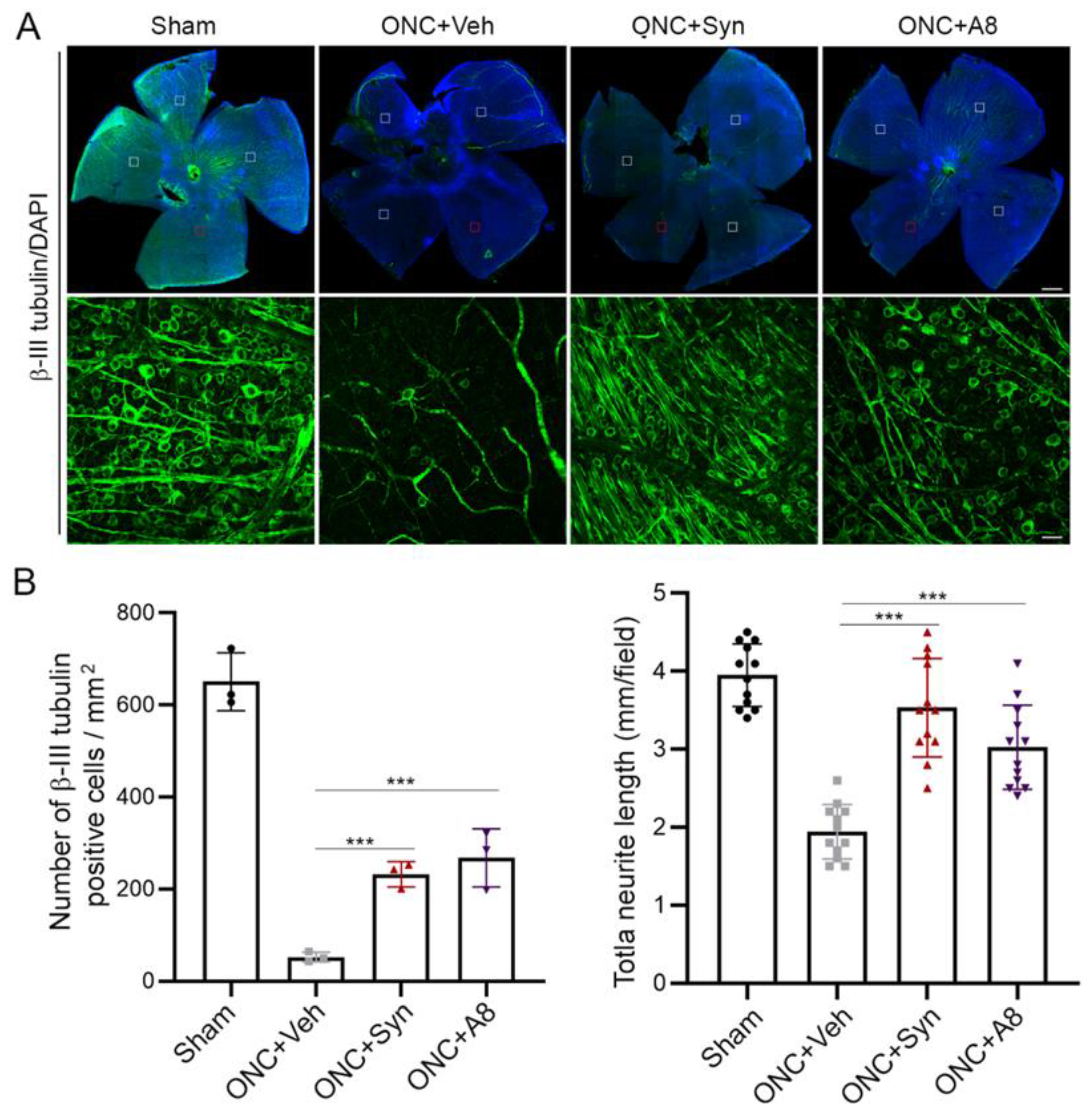
2.6. RGC Loss after ONC Was Alleviated by Synaptamide or A8 Treatment
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Optic Nerve Crush (ONC)
4.3. Intravitreal Administration of Synaptamide or A8
4.4. Anterograde Labeling
4.5. Visual-Evoked Potentials (VEP) and Electroretinogram (ERG)
4.6. Electron Microscopic Analysis
4.7. Whole Mount Retina Staining and RGC Survival Quantification
4.8. Immunohistochemistry
4.9. Western Blot
4.10. Determination of the Synaptamide and A8 Level
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fischer, D.; Leibinger, M. Promoting optic nerve regeneration. Prog. Retin. Eye Res. 2012, 31, 688–701. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Jin, Y. Intrinsic Control of Axon Regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [PubMed] [Green Version]
- Berkelaar, M.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [PubMed] [Green Version]
- Goldberg, J.L.; Barres, B.A. The relationship between neuronal survival and regeneration. Annu. Rev. Neurosci. 2000, 23, 579–612. [Google Scholar] [CrossRef]
- McGee, A.W.; Yang, Y.; Fischer, Q.S.; Daw, N.W.; Strittmatter, S.M. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 2005, 309, 2222–2226. [Google Scholar] [CrossRef] [Green Version]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [Green Version]
- Benowitz, L.I.; He, Z.; Goldberg, J.L. Reaching the brain: Advances in optic nerve regeneration. Exp. Neurol. 2017, 287 Pt 3, 365–373. [Google Scholar] [CrossRef]
- Laha, B.S.; Stafford, B.K.; Huberman, A. Regenerating optic pathways from the eye to the brain. Science 2017, 356, 1031–1034. [Google Scholar] [CrossRef]
- Gao, Y.; Deng, K.W.; Hou, J.W.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, M.; Harvey, A.R. Cyclic AMP and the regeneration of retinal ganglion cell axons. Int. J. Biochem. Cell Biol. 2014, 56, 66–73. [Google Scholar] [CrossRef]
- Batty, N.J.; Fenrich, K.K.; Fouad, K. The role of cAMP and its downstream targets in neurite growth in the adult nervous system. Neurosci. Lett. 2017, 652, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Huang, B.X.; Kwon, H.; Rashid, M.A.; Kharebava, G.; Desai, A.; Patnaik, S.; Marugan, J.; Kim, H.Y. Orphan GPR110 (ADGRF1) targeted by N-docosahexaenoylethanolamine in development of neurons and cognitive function. Nat. Commun. 2016, 7, 13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.X.; Hu, X.; Kwon, H.S.; Fu, C.; Lee, J.W.; Southall, N.; Marugan, J.; Kim, H.Y. Synaptamide activates the adhesion GPCR GPR110 (ADGRF1) through GAIN domain binding. Commun. Biol. 2020, 3, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.K.; Kevala, K.; Hu, X.; Patnaik, S.; Marugan, J.; Kim, H.Y. Ligand-Induced GPR110 Activation Facilitates Axon Growth after Injury. Int. J. Mol. Sci. 2021, 22, 3386. [Google Scholar] [CrossRef]
- Chen, H.K.; Kevala, K.; Aflaki, E.; Marugan, J.; Kim, H.Y. GPR110 ligands reduce chronic optic tract gliosis and visual deficit following repetitive mild traumatic brain injury in mice. J. Neurotrauma 2021, 18, 157. [Google Scholar]
- Dorfman, L.J.; Gaynon, M.; Ceranski, J.; Louis, A.A.; Howard, J.E. Visual electrical evoked potentials: Evaluation of ocular injuries. Neurology 1987, 37, 123–128. [Google Scholar] [CrossRef]
- Couto, L.A.; Narciso, M.S.; Hokoc, J.N.; Martinez, A.M.B. Calpain inhibitor 2 prevents axonal degeneration of opossum optic nerve fibers. J. Neurosci. Res. 2004, 77, 410–419. [Google Scholar] [CrossRef]
- Dutta, D.J.; Woo, D.H.; Lee, P.R.; Pajevic, S.; Bukalo, O.; Huffman, W.C.; Wake, H.; Basser, P.J.; SheikhBahaei, S.; Lazarevic, V.; et al. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 11832–11837. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Migallon, M.C.; Valiente-Soriano, F.J.; Salinas-Navarro, M.; Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Vidal-Sanz, M.; Agudo-Barriuso, M. Nerve fibre layer degeneration and retinal ganglion cell loss long term after optic nerve crush or transection in adult mice. Exp. Eye Res. 2018, 170, 40–50. [Google Scholar] [CrossRef]
- de Lima, S.; Koriyama, Y.; Kurimoto, T.; Oliveira, J.T.; Yin, Y.; Li, Y.; Gilbert, H.Y.; Fagiolini, M.; Martinez, A.M.; Benowitz, L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc. Natl. Acad. Sci. USA 2012, 109, 9149–9154. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Spector, A.A. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol. Asp. Med. 2018, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Shen, Y.; De Bellard, M.; Tang, S.; Filbin, M.T. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron 1999, 22, 89–101. [Google Scholar] [PubMed] [Green Version]
- Cai, D.; Qiu, J.; Cao, Z.; McAtee, M.; Bregman, B.S.; Filbin, M.T. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J. Neurosci. 2001, 21, 4731–4739. [Google Scholar]
- Templeton, J.P.; Geisert, E.E. A practical approach to optic nerve crush in the mouse. Mol. Vis. 2012, 18, 2147–2152. [Google Scholar]
- Rydel, R.E.; Greene, L.A. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc. Natl. Acad. Sci. USA 1988, 85, 1257–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, E.J.; Moon, L.D.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef]
- Ruschel, J.; Hellal, F.; Flynn, K.C.; Dupraz, S.; Elliott, D.A.; Tedeschi, A.; Bates, M.; Sliwinski, C.; Brook, G.; Dobrindt, K.; et al. Axonal regeneration. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science 2015, 348, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.C.H.; Kim, H.Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflammation 2019, 16, 225. [Google Scholar] [CrossRef]
- Desai, A.; Chen, H.; Kim, H. Multiple mild closed head injuries lead to visual dysfunction in a mouse model. J. Neurotrauma 2019, 37, 286–294. [Google Scholar]
- Benchorin, G.; Calton, M.A.; Beaulieu, M.O.; Vollrath, D. Assessment of murine retinal function by electroretinography. Bio Protoc. 2017, 7, e2218. [Google Scholar]
- Abu-Asab, M. AA Concise Practical Manual of Transmission Electron Microscopy: For Biological & Clinical Specimens; Kindle Direct Publishing: Seattle, WA, USA, 2021. [Google Scholar]
- Friede, R.L.; Beuche, W. Combined scatter diagrams of sheath thickness and fibre calibre in human sural nerves: Changes with age and neuropathy. J. Neurol. Neurosurg. Psychiatry 1985, 48, 749–756. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, H.-S.; Kevala, K.; Qian, H.; Abu-Asab, M.; Patnaik, S.; Marugan, J.; Kim, H.-Y. Ligand-Induced Activation of GPR110 (ADGRF1) to Improve Visual Function Impaired by Optic Nerve Injury. Int. J. Mol. Sci. 2023, 24, 5340. https://doi.org/10.3390/ijms24065340
Kwon H-S, Kevala K, Qian H, Abu-Asab M, Patnaik S, Marugan J, Kim H-Y. Ligand-Induced Activation of GPR110 (ADGRF1) to Improve Visual Function Impaired by Optic Nerve Injury. International Journal of Molecular Sciences. 2023; 24(6):5340. https://doi.org/10.3390/ijms24065340
Chicago/Turabian StyleKwon, Heung-Sun, Karl Kevala, Haohua Qian, Mones Abu-Asab, Samarjit Patnaik, Juan Marugan, and Hee-Yong Kim. 2023. "Ligand-Induced Activation of GPR110 (ADGRF1) to Improve Visual Function Impaired by Optic Nerve Injury" International Journal of Molecular Sciences 24, no. 6: 5340. https://doi.org/10.3390/ijms24065340
APA StyleKwon, H. -S., Kevala, K., Qian, H., Abu-Asab, M., Patnaik, S., Marugan, J., & Kim, H. -Y. (2023). Ligand-Induced Activation of GPR110 (ADGRF1) to Improve Visual Function Impaired by Optic Nerve Injury. International Journal of Molecular Sciences, 24(6), 5340. https://doi.org/10.3390/ijms24065340