
COVID-19-Induced Myocarditis: Pathophysiological Roles of ACE2 and Toll-like Receptors

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
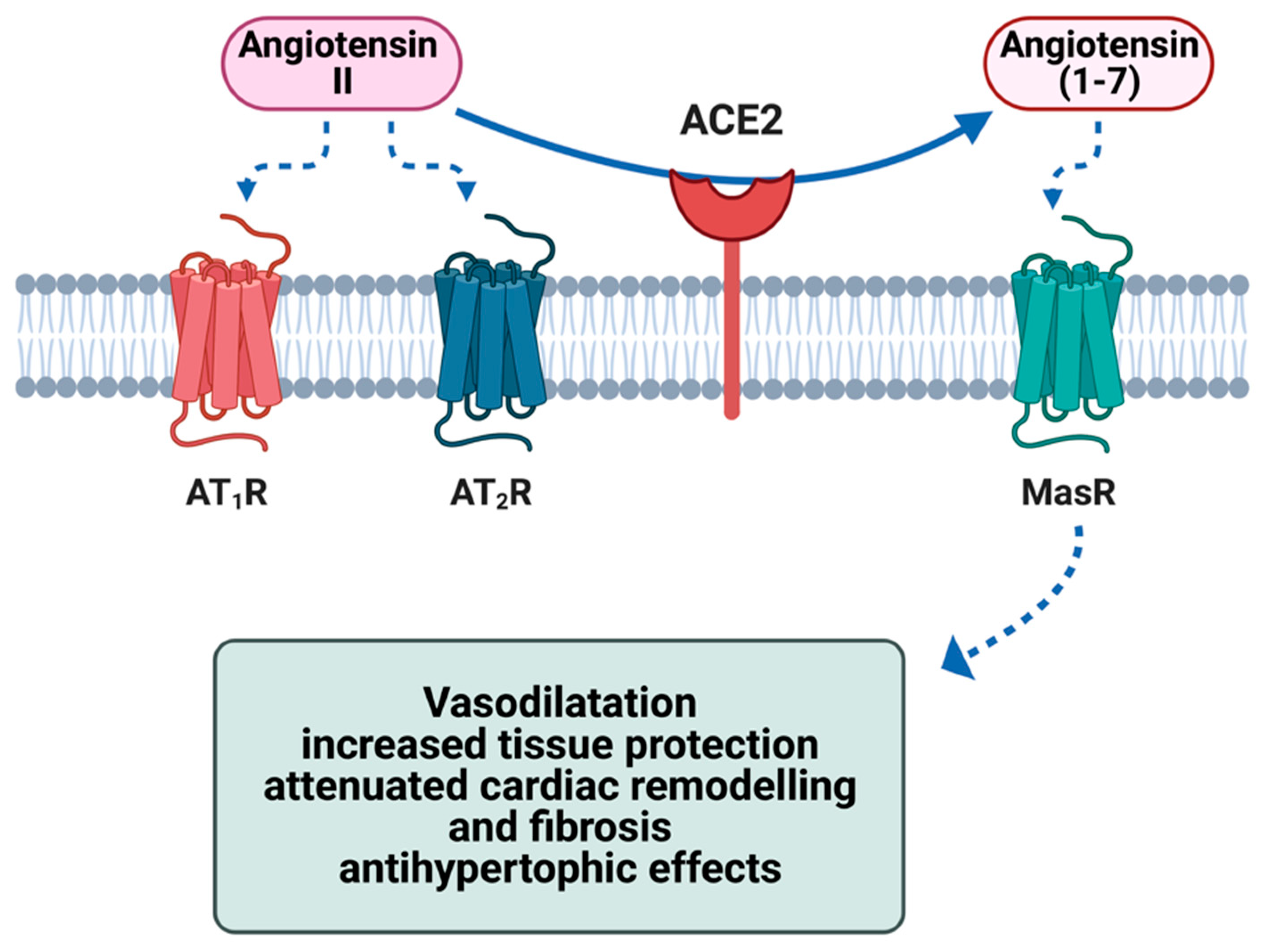
2. ACE2 Biology and Physiological Functions
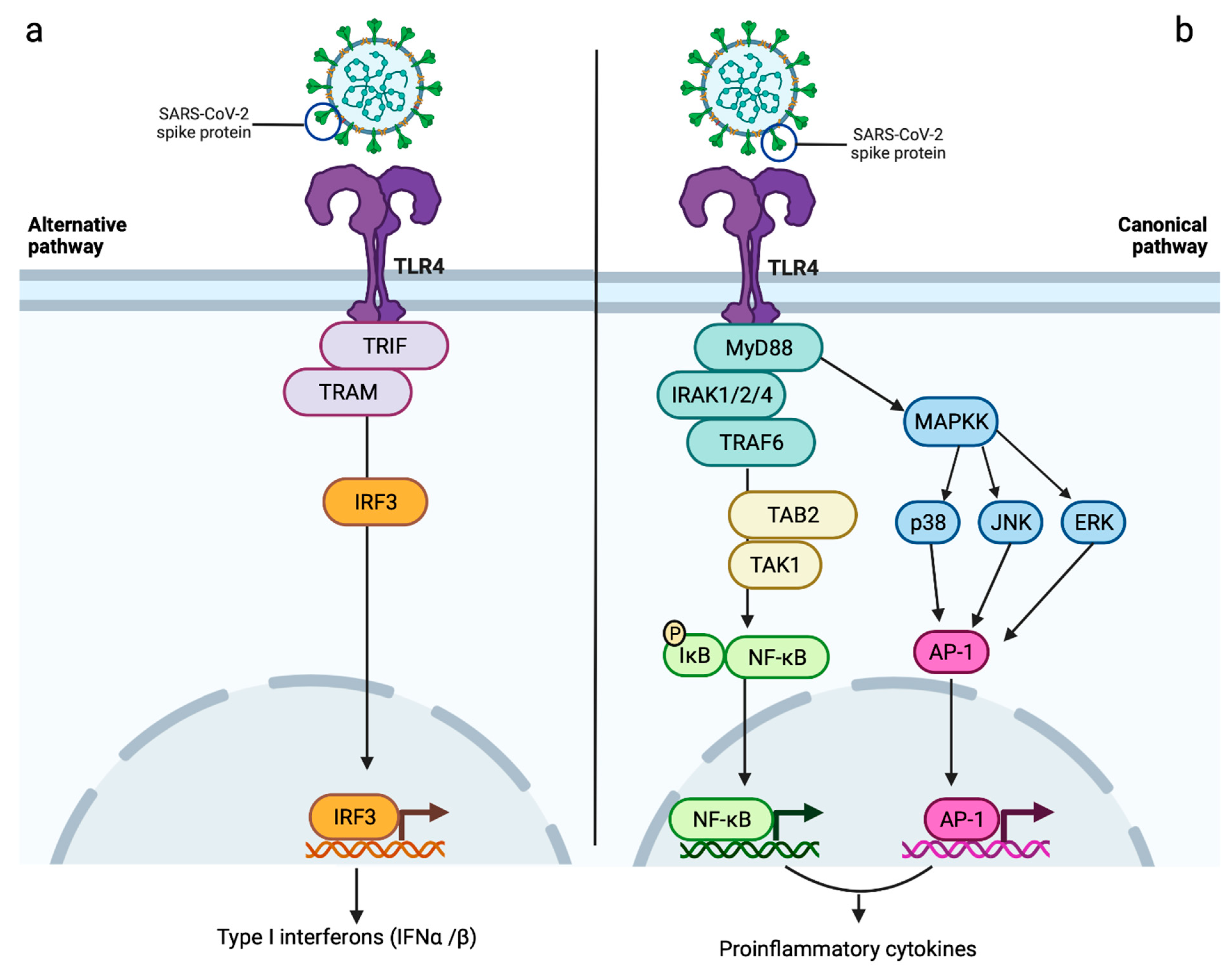
3. Toll-like Receptors: Biology and Physiological Role
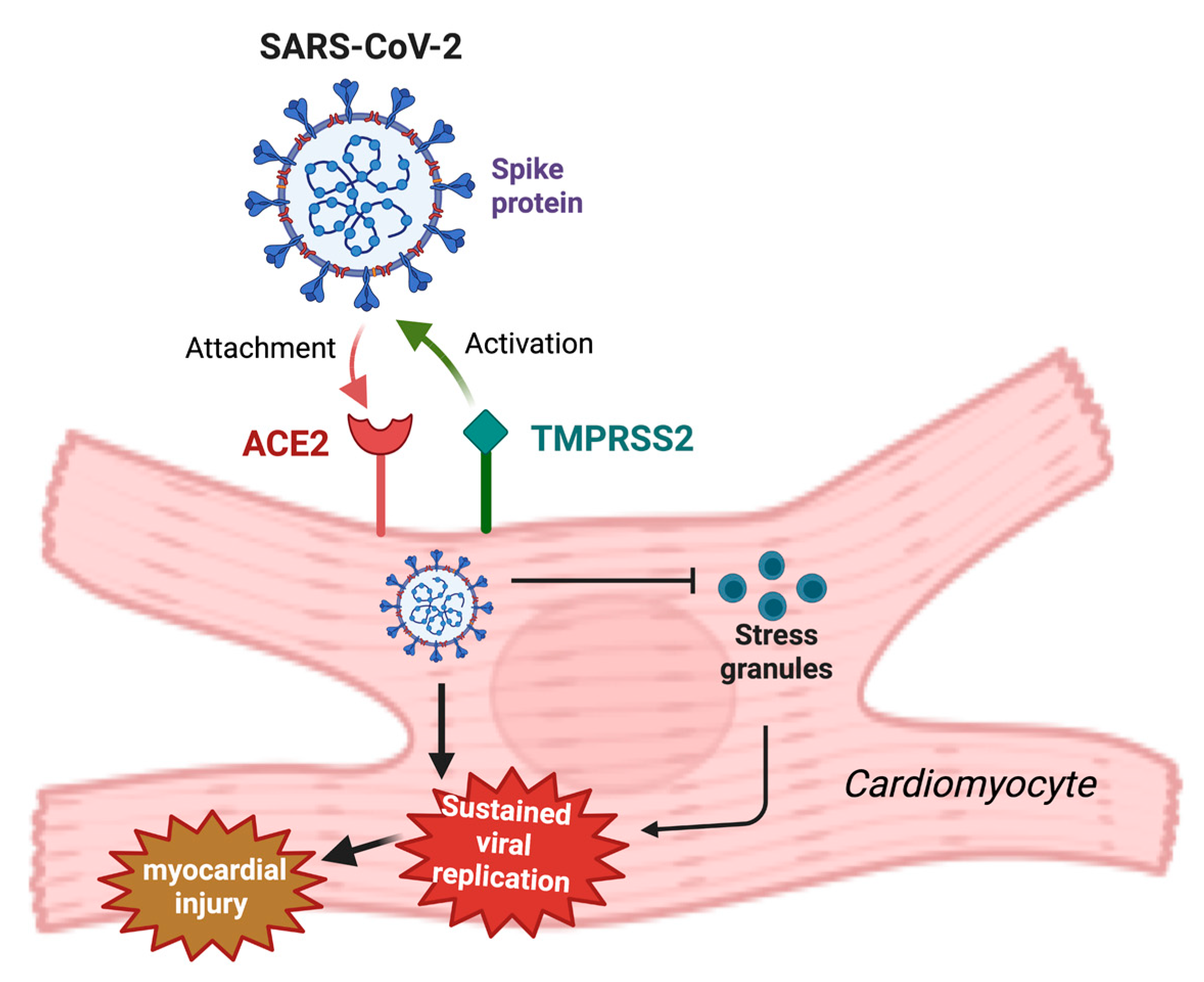
4. Virus-Induced Myocarditis and Its Pathophysiology
5. Role of Cardiovascular Factors (ACE2 and RAAS) in the Pathogenesis of COVID-19-Related Myocarditis
6. Role of Immune System Components (Toll-like Receptors)
7. Myocarditis as a Consequence of mRNA COVID-19 Vaccination
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| ADAM-17 | a disintegrin and metalloprotease 17 |
| Ang (1-7) | angiotensin (1-7) |
| Ang (1-9) | angiotensin (1-9) |
| Ang II | angiotensin II |
| AP-1 | activator protein 1 |
| APCs | antigen-presenting cells |
| cAMP | cyclic adenosine monophosphate |
| CD4+ | helper T cells |
| CD8+ | cytotoxic T cells |
| COVID-19 | coronavirus disease 2019 |
| DAMPs | damage-associated molecular patterns |
| DCM | dilated cardiomyopathy |
| dsRNA | double stranded RNA |
| dUTP | deoxyuridine, triphosphate |
| ERK | extracellular signal-regulated kinase |
| IFNAR1 | interferon α and β receptor subunit 1 |
| IFNAR2 | interferon α and β receptor subunit 2 |
| IFNα | interferon alpha |
| IFNγ | interferon gamma |
| IL-1 | interleukin 1 |
| IL-12 | interleukin 12 |
| IL-18 | interleukin 18 |
| IL-1α | interleukin 1 alpha |
| IL-1β | interleukin 1 beta |
| IL-6 | interleukin 6 |
| iNOS | inducible nitric oxide synthase |
| ISGs | interferon-stimulated genes |
| IκB | inhibitor of nuclear factor kappa B |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LRR | leucine-rich repeat |
| MasR | Mas receptor |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MHC class I | major histocompatibility complex class I |
| mRNA | messenger ribonucleic acid |
| MyD88 | myeloid differentiation factor 88 |
| NF-kB | nuclear factor kappa B |
| NK | natural killer |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NOD | nucleotide-binding and oligomerization domain |
| p38 | p38 mitogen-activated protein kinases |
| PAMPs | pathogen-associated molecular patterns |
| PKA | protein kinase A |
| RAAS | renin-angiotensin-aldosterone system |
| RBD | receptor-binding domain |
| ROS | reactive oxygen species |
| SARS-CoV-1 | severe acute respiratory syndrome coronavirus 1 |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SCD | sudden cardiac death |
| SG | stress granule |
| ssRNA | single stranded RNA |
| STAT | signal transducer and activator of transcription |
| TAB2 | TGF-beta activated kinase 1 binding protein 2 |
| TAK1 | transforming growth factor-β activated kinase 1 |
| TIR | Toll/IL-1 receptor homologous domain |
| TLR | Toll-like receptor |
| TMPRSS2 | transmembrane serine protease 2 |
| TNFα | tumour necrosis factor alpha |
| TNFβ | tumour necrosis factor beta |
| TRAF6 | TNF receptor associated factor 6 |
| TRAM | TRIF-related adaptor molecule |
| TRIF | TIR-domain-containing adapter-inducing interferon β |
| TUNEL | terminal deozynucleotidyl transferase dUTP nick end labelling |
| UTR | untranslated region |
| α-MHC | α-myosin heavy chain |
| β-MHC | β-myosin heavy chain |
References
- Sagar, S.; Liu, P.P.; Cooper, L.T. Myocarditis. Lancet 2012, 379, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [PubMed]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648, 2648a–2648d. [Google Scholar] [CrossRef] [PubMed]
- Dennert, R.; Crijns, H.J.; Heymans, S. Acute viral myocarditis. Eur. Heart J. 2008, 29, 2073–2082. [Google Scholar] [CrossRef]
- Leone, O.; Pieroni, M.; Rapezzi, C.; Olivotto, I. The spectrum of myocarditis: From pathology to the clinics. Virchows Arch. 2019, 475, 279–301. [Google Scholar] [CrossRef]
- Lynge, T.H.; Nielsen, T.S.; Gregers Winkel, B.; Tfelt-Hansen, J.; Banner, J. Sudden cardiac death caused by myocarditis in persons aged 1–49 years: A nationwide study of 14,294 deaths in Denmark. Forensic Sci. Res. 2019, 4, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, M.; Binzenhöfer, L.; Mahrholdt, H.; Johannes Schindler, M.; Esefeld, K.; Tschöpe, C. Myocarditis in athletes: A clinical perspective. Eur. J. Prev. Cardiol. 2020, 28, 1050–1057. [Google Scholar] [CrossRef]
- Olejniczak, M.; Schwartz, M.; Webber, E.; Shaffer, A.; Perry, T.E. Viral Myocarditis-Incidence, Diagnosis and Management. J. Cardiothorac. Vasc. Anesth. 2020, 34, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Luo, H.; Qiu, Y.; Yang, D.; McManus, B. Myocarditis. Circ. Res. 2016, 118, 496–514. [Google Scholar] [CrossRef]
- Basso, C. Myocarditis. New Engl. J. Med. 2022, 387, 1488–1500. [Google Scholar] [CrossRef]
- Cooper, L.T., Jr. Myocarditis. New Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [Green Version]
- Esfandiarei, M.; McManus, B.M. Molecular Biology and Pathogenesis of Viral Myocarditis. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 127–155. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper, L.T., Jr.; Chahal, C.A.A. Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 2020, 17, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Patone, M.; Mei, X.W.; Handunnetthi, L.; Dixon, S.; Zaccardi, F.; Shankar-Hari, M.; Watkinson, P.; Khunti, K.; Harnden, A.; Coupland, C.A.C.; et al. Risks of myocarditis, pericarditis, and cardiac arrhythmias associated with COVID-19 vaccination or SARS-CoV-2 infection. Nat. Med. 2022, 28, 410–422. [Google Scholar] [CrossRef]
- Ali, M.; Shiwani, H.A.; Elfaki, M.Y.; Hamid, M.; Pharithi, R.; Kamgang, R.; Egom, C.B.; Oyono, J.L.E.; Egom, E.E.-A. COVID-19 and myocarditis: A review of literature. Egypt. Heart J. 2022, 74, 23. [Google Scholar] [CrossRef]
- Alhogbani, T. Acute myocarditis associated with novel Middle east respiratory syndrome coronavirus. Ann. Saudi Med. 2016, 36, 78–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, A.F.; Afsahi, A.M.; Gupta, A.; Gholamrezanezhad, A. Severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS), influenza, and COVID-19, beyond the lungs: A review article. Radiol. Med. 2021, 126, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Klingel, K.; Tschöpe, C. SARS-CoV-2-related myocarditis-like syndromes Shakespeare’s question: What’s in a name? Eur. J. Heart Fail. 2020, 22, 922–925. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Babapoor-Farrokhran, S.; Gill, D.; Walker, J.; Rasekhi, R.T.; Bozorgnia, B.; Amanullah, A. Myocardial injury and COVID-19: Possible mechanisms. Life Sci. 2020, 253, 117723. [Google Scholar] [CrossRef]
- Maiese, A.; Frati, P.; Del Duca, F.; Santoro, P.; Manetti, A.C.; La Russa, R.; Di Paolo, M.; Turillazzi, E.; Fineschi, V. Myocardial Pathology in COVID-19-Associated Cardiac Injury: A Systematic Review. Diagnostics 2021, 11, 1647. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Bozkurt, B.; Kamat, I.; Hotez, P.J. Myocarditis With COVID-19 mRNA Vaccines. Circulation 2021, 144, 471–484. [Google Scholar] [CrossRef]
- Witberg, G.; Barda, N.; Hoss, S.; Richter, I.; Wiessman, M.; Aviv, Y.; Grinberg, T.; Auster, O.; Dagan, N.; Balicer, R.D.; et al. Myocarditis after Covid-19 Vaccination in a Large Health Care Organization. New Engl. J. Med. 2021, 385, 2132–2139. [Google Scholar] [CrossRef]
- Abu Mouch, S.; Roguin, A.; Hellou, E.; Ishai, A.; Shoshan, U.; Mahamid, L.; Zoabi, M.; Aisman, M.; Goldschmid, N.; Berar Yanay, N. Myocarditis following COVID-19 mRNA vaccination. Vaccine 2021, 39, 3790–3793. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Li, Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm. Sin. B 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Ertl, G.; Bauersachs, J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat. Clin. Pr. Cardiovasc. Med. 2007, 4, 444–454. [Google Scholar] [CrossRef]
- Vallejo, J.G. Role of toll-like receptors in cardiovascular diseases. Clin. Sci. 2011, 121, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Favere, K.; Bosman, M.; Klingel, K.; Heymans, S.; Van Linthout, S.; Delputte, P.L.; De Sutter, J.; Heidbuchel, H.; Guns, P.J. Toll-Like Receptors: Are They Taking a Toll on the Heart in Viral Myocarditis? Viruses 2021, 13, 1003. [Google Scholar] [CrossRef] [PubMed]
- Becher, P.M.; Hinrichs, S.; Fluschnik, N.; Hennigs, J.K.; Klingel, K.; Blankenberg, S.; Westermann, D.; Lindner, D. Role of Toll-like receptors and interferon regulatory factors in different experimental heart failure models of diverse etiology: IRF7 as novel cardiovascular stress-inducible factor. PLoS ONE 2018, 13, e0193844. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Kishore, R. Cardiovascular Manifestations of COVID-19 Infection. Cells 2020, 9, 2508. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.-h.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-Converting Enzyme 2 Suppresses Pathological Hypertrophy, Myocardial Fibrosis, and Cardiac Dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef]
- Bader, M.; Turner, A.J.; Alenina, N. ACE2, a multifunctional protein—From cardiovascular regulation to COVID-19. Clin. Sci. 2020, 134, 3229–3232. [Google Scholar] [CrossRef] [PubMed]
- Zamorano Cuervo, N.; Grandvaux, N. ACE2: Evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. Elife 2020, 9, e61390. [Google Scholar] [CrossRef]
- Turner, A.J. ACE2 Cell Biology, Regulation, and Physiological Functions. Prot. Arm Renin Angiotensin Syst. (RAS) 2015, 2015, 185–189. [Google Scholar]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-COV-2. Front. Cell. Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Lu, X. Regulation of Angiotensin-Converting Enzyme 2: A Potential Target to Prevent COVID-19? Front. Endocrinol. 2021, 12, 1284. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Cooper, S.L.; Boyle, E.; Jefferson, S.R.; Heslop, C.R.A.; Mohan, P.; Mohanraj, G.G.J.; Sidow, H.A.; Tan, R.C.P.; Hill, S.J.; Woolard, J. Role of the Renin-Angiotensin-Aldosterone and Kinin-Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID. Int. J. Mol. Sci. 2021, 22, 8255. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Guo, L.; Zhang, Y.; Li, H.; Han, S.; Shen, D. The ACE2-Ang (1–7)-Mas receptor axis attenuates cardiac remodeling and fibrosis in post-myocardial infarction. Mol. Med. Rep. 2017, 16, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Rabelo, L.A.; Todiras, M.; Nunes-Souza, V.; Qadri, F.; Szijártó, I.A.; Gollasch, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Genetic Deletion of ACE2 Induces Vascular Dysfunction in C57BL/6 Mice: Role of Nitric Oxide Imbalance and Oxidative Stress. PLoS ONE 2016, 11, e0150255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksova, A.; Gagno, G.; Sinagra, G.; Beltrami, A.P.; Janjusevic, M.; Ippolito, G.; Zumla, A.; Fluca, A.L.; Ferro, F. Effects of SARS-CoV-2 on Cardiovascular System: The Dual Role of Angiotensin-Converting Enzyme 2 (ACE2) as the Virus Receptor and Homeostasis Regulator-Review. Int. J. Mol. Sci. 2021, 22, 4526. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, U.; Ertl, G.; Frantz, S. Toll-like receptors as potential therapeutic targets in cardiac dysfunction. Expert Opin. Ther. Targets 2011, 15, 753–765. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 2009, 420, 1–16. [Google Scholar] [CrossRef] [Green Version]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed. Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Kaisho, T.; Akira, S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006, 117, 979–987. [Google Scholar] [CrossRef]
- Botos, I.; Segal, D.M.; Davies, D.R. The structural biology of Toll-like receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, J.H.; Mathur, S.; Wang, Y.; Bateman, R.M.; Walley, K.R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc. Res. 2006, 72, 384–393. [Google Scholar] [CrossRef]
- Kindermann, I.; Barth, C.; Mahfoud, F.; Ukena, C.; Lenski, M.; Yilmaz, A.; Klingel, K.; Kandolf, R.; Sechtem, U.; Cooper, L.T.; et al. Update on Myocarditis. J. Am. Coll. Cardiol. 2012, 59, 779–792. [Google Scholar] [CrossRef]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis--diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef]
- Liu, P.P.; Mason, J.W. Advances in the Understanding of Myocarditis. Circulation 2001, 104, 1076–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.P.; Opavsky, M.A. Viral Myocarditis. Circ. Res. 2000, 86, 253–254. [Google Scholar] [CrossRef] [Green Version]
- Saraste, A.; Arola, A.; Vuorinen, T.; Kytö, V.; Kallajoki, M.; Pulkki, K.; Voipio-Pulkki, L.M.; Hyypiä, T. Cardiomyocyte apoptosis in experimental coxsackievirus B3 myocarditis. Cardiovasc. Pathol. 2003, 12, 255–262. [Google Scholar] [CrossRef]
- McManus, B.M.; Chow, L.H.; Wilson, J.E.; Anderson, D.R.; Gulizia, J.M.; Gauntt, C.J.; Klingel, K.E.; Beisel, K.W.; Kandolf, R. Direct myocardial injury by enterovirus: A central role in the evolution of murine myocarditis. Clin. Immunol. Immunopathol. 1993, 68, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Badorff, C.; Lee, G.H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral protease 2A cleaves dystrophin: Evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Maisch, B.; Ristić, A.D.; Hufnagel, G.; Pankuweit, S. Pathophysiology of viral myocarditis: The role of humoral immune response. Cardiovasc. Pathol. 2002, 11, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.L.; Colston, J.T.; Zabalgoitia, M.; Chandrasekar, B. Contractile depression and expression of proinflammatory cytokines and iNOS in viral myocarditis. Am. J. Physiol. 1998, 274, H249–H258. [Google Scholar] [CrossRef]
- Matsumori, A.; Yamada, T.; Suzuki, H.; Matoba, Y.; Sasayama, S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br. Heart J. 1994, 72, 561–566. [Google Scholar] [CrossRef]
- Massilamany, C.; Huber, S.A.; Cunningham, M.W.; Reddy, J. Relevance of molecular mimicry in the mediation of infectious myocarditis. J. Cardiovasc. Transl. Res. 2014, 7, 165–171. [Google Scholar] [CrossRef]
- Caforio, A.L.; Mahon, N.J.; Tona, F.; McKenna, W.J. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: Pathogenetic and clinical significance. Eur. J. Heart Fail. 2002, 4, 411–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallukat, G.; Schimke, I. Agonistic autoantibodies directed against G-protein-coupled receptors and their relationship to cardiovascular diseases. Semin. Immunopathol. 2014, 36, 351–363. [Google Scholar] [CrossRef]
- Caforio, A.L.; Grazzini, M.; Mann, J.M.; Keeling, P.J.; Bottazzo, G.F.; McKenna, W.J.; Schiaffino, S. Identification of alpha- and beta-cardiac myosin heavy chain isoforms as major autoantigens in dilated cardiomyopathy. Circulation 1992, 85, 1734–1742. [Google Scholar] [CrossRef] [Green Version]
- Wolff, P.G.; Kühl, U.; Schultheiss, H.-P. Laminin distribution and autoantibodies to laminin in dilated cardiomyopathy and myocarditis. Am. Heart J. 1989, 117, 1303–1309. [Google Scholar] [CrossRef]
- Limas, C.J.; Goldenberg, I.F.; Limas, C. Autoantibodies against beta-adrenoceptors in human idiopathic dilated cardiomyopathy. Circ. Res. 1989, 64, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Limas, C.J.; Goldenberg, I.F.; Limas, C. Influence of anti-beta-receptor antibodies on cardiac adenylate cyclase in patients with idiopathic dilated cardiomyopathy. Am. Heart J. 1990, 119, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Goldman, J.H.; Haven, A.J.; Baig, K.M.; Libera, L.D.; McKenna, W.J. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur. Heart J. 1997, 18, 270–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, Y.; Wang, D.W. SARS-CoV-2: A potential novel etiology of fulminant myocarditis. Herz 2020, 45, 230–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehmer, T.K.; Kompaniyets, L.; Lavery, A.M.; Hsu, J.; Ko, J.Y.; Yusuf, H.; Romano, S.D.; Gundlapalli, A.V.; Oster, M.E.; Harris, A.M. Association Between COVID-19 and Myocarditis Using Hospital-Based Administrative Data—United States, March 2020–January 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Dmytrenko, O.; Lavine, K.J. Cardiovascular Tropism and Sequelae of SARS-CoV-2 Infection. Viruses 2022, 14, 1137. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.-P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Sharma, R.K.; Stevens, B.R.; Obukhov, A.G.; Grant, M.B.; Oudit, G.Y.; Li, Q.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. ACE2 (Angiotensin-Converting Enzyme 2) in Cardiopulmonary Diseases. Hypertension 2020, 76, 651–661. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-f.; Xu, W.; Liu, S.-w. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Örd, M.; Faustova, I.; Loog, M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 16944. [Google Scholar] [CrossRef]
- Zheng, Z.-Q.; Wang, S.-Y.; Xu, Z.-S.; Fu, Y.-Z.; Wang, Y.-Y. SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov. 2021, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, D.; Palmeira, J.d.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin Converting Enzyme 2. Circulation 2020, 142, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Palau, V.; Riera, M.; Soler, M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transpl. 2020, 35, 1071–1072. [Google Scholar] [CrossRef]
- Sharma, R.K.; Li, J.; Krishnan, S.; Richards, E.M.; Raizada, M.K.; Mohandas, R. Angiotensin-converting enzyme 2 and COVID-19 in cardiorenal diseases. Clin. Sci. 2021, 135, 1–17. [Google Scholar] [CrossRef]
- Rice, G.I.; Jones, A.L.; Grant, P.J.; Carter, A.M.; Turner, A.J.; Hooper, N.M. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006, 48, 914–920. [Google Scholar] [CrossRef]
- Almengló, C.; Couselo-Seijas, M.; Agra, R.M.; Varela-Román, A.; García-Acuña, J.M.; González-Peteiro, M.; González-Juanatey, J.R.; Eiras, S.; Álvarez, E. Soluble angiotensin-converting enzyme levels in heart failure or acute coronary syndrome: Revisiting its modulation and prognosis value. J. Mol. Med. 2021, 99, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Lindbäck, J.; Eriksson, N.; Hijazi, Z.; Eikelboom, J.W.; Ezekowitz, M.D.; Granger, C.B.; Lopes, R.D.; Yusuf, S.; Oldgren, J.; et al. Angiotensin-converting enzyme 2 (ACE2) levels in relation to risk factors for COVID-19 in two large cohorts of patients with atrial fibrillation. Eur. Heart J. 2020, 41, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.-P.; Zhou, R.; Chan, K.-H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228. [Google Scholar] [CrossRef]
- Gonzalez, S.M.; Siddik, A.B.; Su, R.-C. Regulated Intramembrane Proteolysis of ACE2: A Potential Mechanism Contributing to COVID-19 Pathogenesis? Front. Immunol. 2021, 12, 612807. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; An, Y. ACE2 Shedding and the Role in COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 789180. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Martens, C.R.; Accornero, F. Viruses in the Heart: Direct and Indirect Routes to Myocarditis and Heart Failure. Viruses 2021, 13, 1924. [Google Scholar] [CrossRef] [PubMed]
- Komarowska, I.; Coe, D.; Wang, G.; Haas, R.; Mauro, C.; Kishore, M.; Cooper, D.; Nadkarni, S.; Fu, H.; Steinbruchel, D.A.; et al. Hepatocyte Growth Factor Receptor c-Met Instructs T Cell Cardiotropism and Promotes T Cell Migration to the Heart via Autocrine Chemokine Release. Immunity 2015, 42, 1087–1099. [Google Scholar] [CrossRef] [Green Version]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Akhmerov, A.; Marbán, E. COVID-19 and the Heart. Circ. Res. 2020, 126, 1443–1455. [Google Scholar] [CrossRef] [Green Version]
- Aboudounya, M.M.; Holt, M.R.; Heads, R.J. SARS-CoV-2 Spike S1 glycoprotein is a TLR4 agonist, upregulates ACE2 expression and induces pro-inflammatory M1 macrophage polarisation. BioRxiv 2021, 1, 455921. [Google Scholar]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Mabrey, F.L.; Morrell, E.D.; Wurfel, M.M. TLRs in COVID-19: How they drive immunopathology and the rationale for modulation. Innate Immun. 2021, 27, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Barhoum, P.; Chambrun, M.P.d.; Dorgham, K.; Kerneis, M.; Burrel, S.; Quentric, P.; Parizot, C.; Chommeloux, J.; Bréchot, N.; Moyon, Q.; et al. Heterogeneity of Fulminant COVID-19--Related Myocarditis in Adults. J. Am. Coll. Cardiol. 2022, 80, 299–312. [Google Scholar] [CrossRef]
- Anjum, F.R.; Anam, S.; Abbas, G.; Mahmood, M.S.; Rahman, S.U.; Goraya, M.U.; Abdullah, R.M.; Luqman, M.; Ali, A.; Akram, M.K.; et al. Type I IFNs: A Blessing in Disguise or Partner in Crime in MERS-CoV-, SARS-CoV-, and SARS-CoV-2-Induced Pathology and Potential Use of Type I IFNs in Synergism with IFN-γ as a Novel Antiviral Approach Against COVID-19. Viral Immunol. 2021, 34, 321–329. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Onabajo, O.O.; Banday, A.R.; Stanifer, M.L.; Yan, W.; Obajemu, A.; Santer, D.M.; Florez-Vargas, O.; Piontkivska, H.; Vargas, J.M.; Ring, T.J.; et al. and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet. 2020, 52, 1283–1293. [Google Scholar] [CrossRef]
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome—With a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.; Rauseo, A.M.; Liu, Z.; Sahu, S.K.; et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. 2016, 7, e2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vukusic, K.; Thorsell, A.; Muslimovic, A.; Jonsson, M.; Dellgren, G.; Lindahl, A.; Sandstedt, J.; Hammarsten, O. Overexpression of the SARS-CoV-2 receptor angiotensin converting enzyme 2 in cardiomyocytes of failing hearts. Sci. Rep. 2022, 12, 965. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Oster, M.E.; Shay, D.K.; Su, J.R.; Gee, J.; Creech, C.B.; Broder, K.R.; Edwards, K.; Soslow, J.H.; Dendy, J.M.; Schlaudecker, E.; et al. Myocarditis Cases Reported After mRNA-Based COVID-19 Vaccination in the US From December 2020 to August 2021. JAMA 2022, 327, 331–340. [Google Scholar] [CrossRef]
- Heymans, S.; Cooper, L.T. Myocarditis after COVID-19 mRNA vaccination: Clinical observations and potential mechanisms. Nat. Rev. Cardiol. 2021, 19, 75–77. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef]
- Rijkers, G.T.; Weterings, N.; Obregon-Henao, A.; Lepolder, M.; Dutt, T.S.; van Overveld, F.J.; Henao-Tamayo, M. Antigen Presentation of mRNA-Based and Virus-Vectored SARS-CoV-2 Vaccines. Vaccines 2021, 9, 848. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.W.; Lagniton, P.N.P.; Liu, Y.; Xu, R.-H. mRNA vaccines for COVID-19: What, why and how. Int. J. Biol. Sci. 2021, 17, 1446–1460. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthukumar, A.; Narasimhan, M.; Li, Q.-Z.; Mahimainathan, L.; Hitto, I.; Fuda, F.; Batra, K.; Jiang, X.; Zhu, C.; Schoggins, J.; et al. In-Depth Evaluation of a Case of Presumed Myocarditis After the Second Dose of COVID-19 mRNA Vaccine. Circulation 2021, 144, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Parra-Lucares, A.; Toro, L.; Weitz-Muñoz, S.; Ramos, C. Cardiomyopathy Associated with Anti-SARS-CoV-2 Vaccination: What Do We Know? Viruses 2021, 13, 2493. [Google Scholar] [CrossRef]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 2020, 217, 108480. [Google Scholar] [CrossRef]
- Mascaro-Blanco, A.; Alvarez, K.; Yu, X.; Lindenfeld, J.; Olansky, L.; Lyons, T.; Duvall, D.; Heuser, J.S.; Gosmanova, A.; Rubenstein, C.J.; et al. Consequences of unlocking the cardiac myosin molecule in human myocarditis and cardiomyopathies. Autoimmunity 2008, 41, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Heuser, J.S.; Cunningham, L.C.; Kosanke, S.D.; Cunningham, M.W. Mimicry and Antibody-Mediated Cell Signaling in Autoimmune Myocarditis. J. Immunol. 2006, 177, 8234–8240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockman, H.A.; Koch, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, Y.; Zhao, Y.; Lung, D.C.; Ye, Z.; Song, W.; Liu, F.-F.; Cai, J.-P.; Wong, W.-M.; Yip, C.C.-Y.; et al. ntravenous injection of COVID-19 mRNA vaccine can induce acute myopericarditis in mouse model. Clin. Infect. Dis. 2021, 73, 2372–2373. [Google Scholar] [CrossRef]
- Larson, K.F.; Ammirati, E.; Adler, E.D.; Cooper, L.T.; Hong, K.N.; Saponara, G.; Couri, D.; Cereda, A.; Procopio, A.; Cavalotti, C.; et al. After BNT162b2 and mRNA-1273 Vaccination. Circulation 2021, 144, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Segal, Y.; Shoenfeld, Y. Vaccine-induced autoimmunity: The role of molecular mimicry and immune crossreaction. Cell. Mol. Immunol. 2018, 15, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrama, D.; Mahita, J.; Sette, A.; Peters, B. Lack of evidence of significant homology of SARS-CoV-2 spike sequences to myocarditis-associated antigens. EBioMedicine 2022, 75, 103807. [Google Scholar] [CrossRef] [PubMed]
- Di Florio, D.N.; Sin, J.; Coronado, M.J.; Atwal, P.S.; Fairweather, D. Sex differences in inflammation, redox biology, mitochondria and autoimmunity. Redox Biol. 2020, 31, 101482. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Won, J.-H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.-W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisancho-Kiss, S.; Davis, S.E.; Nyland, J.F.; Frisancho, J.A.; Cihakova, D.; Barrett, M.A.; Rose, N.R.; Fairweather, D. Cutting Edge: Cross-Regulation by TLR4 and T cell Ig Mucin-3 Determines Sex Differences in Inflammatory Heart Disease. J. Immunol. 2007, 178, 6710–6714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Qin, J.; Bu, P. Pathways Involved in Interleukin-1β–Mediated Murine Cardiomyocyte Apoptosis. Tex. Heart Inst. J. 2015, 42, 109–116. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, L.C.; Mezzaroma, E.; Van Tassell, B.W.; Marchetti, C.; Carbone, S.; Abbate, A.; Toldo, S. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol. Med. 2014, 20, 221–229. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pannucci, P.; Jefferson, S.R.; Hampshire, J.; Cooper, S.L.; Hill, S.J.; Woolard, J. COVID-19-Induced Myocarditis: Pathophysiological Roles of ACE2 and Toll-like Receptors. Int. J. Mol. Sci. 2023, 24, 5374. https://doi.org/10.3390/ijms24065374
Pannucci P, Jefferson SR, Hampshire J, Cooper SL, Hill SJ, Woolard J. COVID-19-Induced Myocarditis: Pathophysiological Roles of ACE2 and Toll-like Receptors. International Journal of Molecular Sciences. 2023; 24(6):5374. https://doi.org/10.3390/ijms24065374
Chicago/Turabian StylePannucci, Patrizia, Sophie R. Jefferson, Jonathan Hampshire, Samantha L. Cooper, Stephen J. Hill, and Jeanette Woolard. 2023. "COVID-19-Induced Myocarditis: Pathophysiological Roles of ACE2 and Toll-like Receptors" International Journal of Molecular Sciences 24, no. 6: 5374. https://doi.org/10.3390/ijms24065374
APA StylePannucci, P., Jefferson, S. R., Hampshire, J., Cooper, S. L., Hill, S. J., & Woolard, J. (2023). COVID-19-Induced Myocarditis: Pathophysiological Roles of ACE2 and Toll-like Receptors. International Journal of Molecular Sciences, 24(6), 5374. https://doi.org/10.3390/ijms24065374