
Genome-Wide Association Mapping of QTL Underlying Groat Protein Content of a Diverse Panel of Oat Accessions


Abstract
:1. Introduction
2. Results
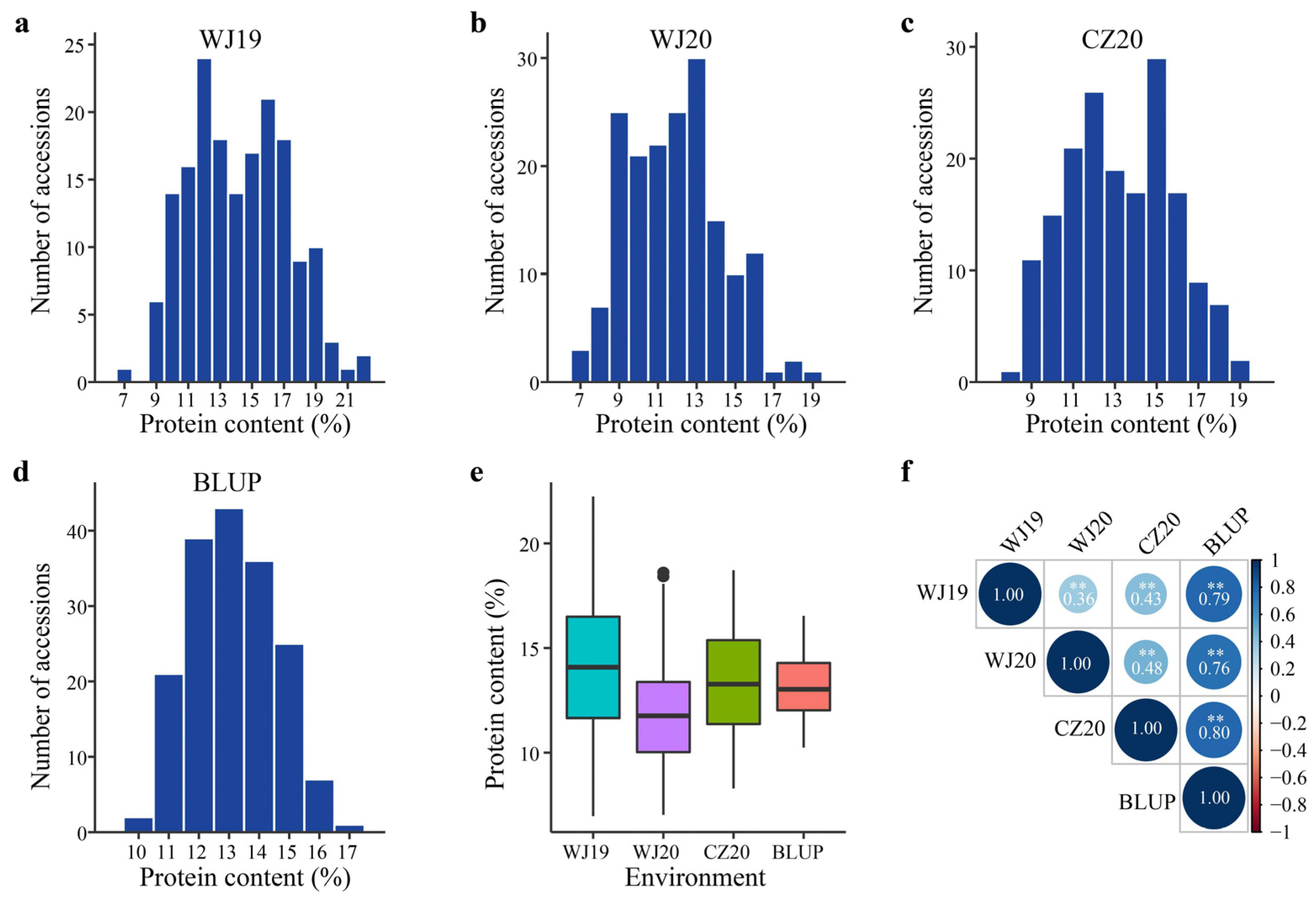
2.1. Variation in Groat Protein Content among Diverse Oat Accessions
2.2. Marker-Trait Associations for Protein Content
2.3. Comparison to Previous QTL Studies
2.4. Favorable Haplotype Analyses
2.5. Putative Candidate Genes and Annotations
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Field Trials and Measurement of GPC
4.3. Statistical Analysis
4.4. Genomic Data Analysis
4.5. Marker-Trait Association and Gene Annotations
4.6. Comparative Mapping
4.7. Estimate of Haplotye Effects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, H.; Yu, K.; Xu, Y.; Zhou, P.; Zhao, J.; Li, Y.; Liu, X.; Ren, C.; Peng, Y. Position validation of the dwarfing gene Dw6 in oat (Avena sativa L.) and its correlated effects on agronomic traits. Front. Plant Sci. 2021, 12, 668847. [Google Scholar] [CrossRef]
- Kamal, N.; Tsardakas Renhuldt, N.; Bentzer, J.; Gundlach, H.; Haberer, G.; Juhász, A.; Lux, T.; Bose, U.; Tye-Din, J.A.; Lang, D.; et al. The mosaic oat genome gives insights into a uniquely healthy cereal crop. Nature 2022, 606, 113–119. [Google Scholar] [CrossRef]
- Zimmer, C.M.; McNish, I.G.; Klos, K.E.; Oro, T.; Arruda, K.M.A.; Gutkoski, L.C.; Pacheco, M.T.; Smith, K.P.; Federizzi, L.C. Genome-wide association for β-glucan content, population structure, and linkage disequilibrium in elite oat germplasm adapted to subtropical environments. Mol. Breed. 2020, 40, 103. [Google Scholar] [CrossRef]
- Gulvady, A.A.; Brown, R.C.; Bell, J.A. Nutritional comparison of oats and other commonly consumed whole grains. In Oats Nutrition and Technology; Chu, Y., Ed.; Academic Press: New York, NY, USA; John Wiley and Sons: New York, NY, USA, 2014; pp. 71–93. [Google Scholar] [CrossRef]
- Tinker, N.A.; Wight, C.P.; Bekele, W.A.; Yan, W.; Jellen, E.N.; Renhuldt, N.T.; Sirijovski, N.; Lux, T.; Spannagl, M.; Mascher, M. Genome analysis in Avena sativa reveals hidden breeding barriers and opportunities for oat improvement. Commun. Biol. 2022, 5, 474. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Sehrawat, R.; Kong, Y. Oat proteins: A perspective on functional properties. LWT 2021, 152, 112307. [Google Scholar] [CrossRef]
- Brown, C.M.; Alexander, D.E.; Carmer, S.G. Variation in oil content and its relation to other characters in oats (Avena sativa L.)1. Crop Sci. 1966, 6, 190–191. [Google Scholar] [CrossRef]
- Mäkinen, O.E.; Sozer, N.; Ercili-Cura, D.; Poutanen, K. Protein from oat: Structure, processes, functionality, and nutrition. In Sustainable Protein Sources; Nadathur, S.R., Wanasundara, J.P.D., Scanlin, L., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 105–119. [Google Scholar]
- Robert, L.S.; Nozzolillo, C.; Cudjoe, A.; Altosaar, I. Total solubilization of groat proteins in high protein oat (Avena sativa L. cv. Hinoat): Evidence that glutelins are a minor component. Can. Inst. Food Sci. Technol. J. 1983, 16, 196–200. [Google Scholar] [CrossRef]
- Fric, P.; Gabrovska, D.; Nevoral, J. Celiac disease, gluten-free diet, and oats. Nutr. Rev. 2011, 69, 107–115. [Google Scholar] [CrossRef]
- Ma, C.Y.; Harwalkar, V.R. Chemical characterization and functionality assessment of oat protein fractions. J. Agric. Food Chem. 1984, 32, 144–149. [Google Scholar] [CrossRef]
- Welch, R.W.; Yong, Y.Y. The effects of variety and nitrogen fertiliser on protein production in oats. J. Sci. Food Agric. 1980, 31, 541–548. [Google Scholar] [CrossRef]
- Humphreys, D.G.; Smith, D.L.; Mather, D.E. Nitrogen fertilizer and seeding date induced changes in protein, oil and β-glucan contents of four oat cultivars. J. Cereal Sci. 1994, 20, 283–290. [Google Scholar] [CrossRef]
- Diederichsen, A. Assessments of genetic diversity within a world collection of cultivated hexaploid oat (Avena sativa L.) based on qualitative morphological characters. Genet. Resour. Crop. Evol. 2008, 55, 419–440. [Google Scholar] [CrossRef]
- Bityutskii, N.P.; Loskutov, I.; Yakkonen, K.; Konarev, A.; Shelenga, T.; Khoreva, V.; Blinova, E.; Ryumin, A. Screening of Avena sativa cultivars for iron, zinc, manganese, protein and oil content and fatty acid composition in whole grains. Cereal Res. Commun. 2020, 48, 87–94. [Google Scholar] [CrossRef]
- Zhu, S.; Rossnagel, B.G.; Kaeppler, H.F. Genetic analysis of quantitative trait loci for groat protein and oil content in oat. Crop Sci. 2004, 44, 254–260. [Google Scholar] [CrossRef]
- De Koeyer, D.L.; Tinker, N.A.; Wight, C.P.; Deyl, J.; Burrows, V.D.; O’Donoughue, L.S.; Lybaert, A.; Molnar, S.J.; Armstrong, K.C.; Fedak, G.; et al. A molecular linkage map with associated QTLs from a hulless × covered spring oat population. Theor. Appl. Genet. 2004, 108, 1285–1298. [Google Scholar] [CrossRef]
- Tanhuanpää, P.; Manninen, O.; Beattie, A.; Eckstein, P.; Scoles, G.; Rossnagel, B.; Kiviharju, E. An updated doubled haploid oat linkage map and QTL mapping of agronomic and grain quality traits from Canadian field trials. Genome 2012, 55, 289–301. [Google Scholar] [CrossRef]
- Hizbai, B.T.; Gardner, K.M.; Wight, C.P.; Dhanda, R.K.; Molnar, S.J.; Johnson, D.; Frégeau-Reid, J.; Yan, W.; Rossnagel, B.G.; Holland, J.B.; et al. Quantitative trait loci affecting oil content, oil composition, and other agronomically important traits in oat. Plant Genome 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, M.H.; Yu, J.; Beuch, S.; Weber, W.E. Quantitative trait loci for quality and agronomic traits in two advanced backcross populations in oat (Avena sativa L.). Plant Breed. 2014, 133, 588–601. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, S.; Shah, T.; Xie, C.; Hao, Z.; Li, X.; Farkhari, M.; Ribaut, J.-M.; Cao, M.; Rong, T.; et al. Joint linkage-linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize. Proc. Natl. Acad. Sci. USA 2010, 107, 19585–19590. [Google Scholar] [CrossRef] [Green Version]
- Asoro, F.G.; Newell, M.A.; Scott, M.P.; Beavis, W.D.; Jannink, J.-L. Genome-wide association study for beta-glucan concentration in elite North American oat. Crop Sci. 2013, 53, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Newell, M.A.; Asoro, F.G.; Scott, M.P.; White, P.J.; Beavis, W.D.; Jannink, J.-L. Genome-wide association study for oat (Avena sativa L.) beta-glucan concentration using germplasm of worldwide origin. Theor. Appl. Genet. 2012, 125, 1687–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klos, K.E.; Huang, Y.-F.; Bekele, W.A.; Obert, D.E.; Babiker, E.; Beattie, A.D.; Bjørnstad, Å.; Bonman, J.M.; Carson, M.L.; Chao, S.; et al. Population genomics related to adaptation in elite oat germplasm. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Klos, K.E.; Yimer, B.A.; Babiker, E.M.; Beattie, A.D.; Bonman, J.M.; Carson, M.L.; Chong, J.; Harrison, S.A.; Ibrahim, A.M.H.; Kolb, F.L.; et al. Genome-wide association mapping of crown rust resistance in oat elite germplasm. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-T.; Klos, K.E.; Huang, Y.-F. Genome-wide association study reveals the genetic architecture of seed vigor in oats. G3 Genes Genomes Genet. 2020, 10, 4489–4503. [Google Scholar] [CrossRef]
- Yan, H.; Zhou, P.; Peng, Y.; Bekele, W.A.; Ren, C.; Tinker, N.A.; Peng, Y. Genetic diversity and genome-wide association analysis in Chinese hulless oat germplasm. Theor. Appl. Genet. 2020, 133, 3365–3380. [Google Scholar] [CrossRef]
- Peng, Y.; Yan, H.; Guo, L.; Deng, C.; Wang, C.; Wang, Y.; Kang, L.; Zhou, P.; Yu, K.; Dong, X.; et al. Reference genome assemblies reveal the origin and evolution of allohexaploid oat. Nat. Genet. 2022, 54, 1248–1258. [Google Scholar] [CrossRef]
- Biel, W.; Bobko, K.; Maciorowski, R. Chemical composition and nutritive value of husked and naked oats grain. J. Cereal Sci. 2009, 49, 413–418. [Google Scholar] [CrossRef]
- Givens, D.I.; Davies, T.W.; Laverick, R.M. Effect of variety, nitrogen fertiliser and various agronomic factors on the nutritive value of husked and naked oats grain. Anim. Feed. Sci. Technol. 2004, 113, 169–181. [Google Scholar] [CrossRef]
- Ronald, P.S.; Brown, P.D.; Penner, G.A.; Brûlé-Babel, A.; Kibite, S. Heritability of hull percentage in oat. Crop Sci. 1999, 39, 52–57. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, J.; Wang, J.; Ding, S. Cultivar and environmental effects on (1→3,1→4)-β-D-glucan and protein content in malting barley. J. Cereal Sci. 2001, 34, 295–301. [Google Scholar] [CrossRef]
- Klindworth, D.L.; Hareland, G.A.; Elias, E.M.; Faris, J.D.; Chao, S.; Xu, S.S. Agronomic and quality characteristics of two new sets of Langdon durum–wild emmer wheat chromosome substitution lines. J. Cereal Sci. 2009, 50, 29–35. [Google Scholar] [CrossRef]
- Miller, S.; Wood, P.; Pietrzak, L.; Fulcher, R. Mixed linkage -glucan, protein content, and kernel weight in Avena Species. J. Cereal Chem. 1993, 70, 231. [Google Scholar]
- Welch, R.W.; Brown, J.C.W.; Leggett, J.M. Interspecific and intraspecific variation in grain and groat characteristics of wild oat (Avena) species: Very high groat (1→3),(1→4)-β- D -glucan in an Avena atlantica genotype. J. Cereal Sci. 2000, 31, 273–279. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, X.; He, K.; Liu, M.; Li, J.; Gao, Z.; Lin, Z.; Zhang, Y.; Wang, X.; Qiu, X.; et al. The MYB transcription factor superfamily of Arabidopsis: Expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 2006, 60, 107–124. [Google Scholar]
- Liu, J.; Huang, L.; Wang, C.; Liu, Y.; Yan, Z.; Wang, Z.; Xiang, L.; Zhong, X.; Gong, F.; Zheng, Y.; et al. Genome-wide association study reveals novel genomic regions associated with high grain protein content in wheat lines derived from wild emmer wheat. Front. Plant Sci. 2019, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Kibite, S.; Evans, L.E. Causes of negative correlations between grain yield and grain protein concentration in common wheat. Euphytica 1984, 33, 801–810. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.; Zhu, Y.; Huang, X.; Li, S.; Wu, X.; Zhao, Y.; Bao, Z.; Qin, L.; Jin, Y.; et al. THP9 enhances seed protein content and nitrogen-use efficiency in maize. Nature 2022, 612, 292–300. [Google Scholar] [CrossRef]
- Peng, B.; Kong, H.; Li, Y.; Wang, L.; Zhong, M.; Sun, L.; Gao, G.; Zhang, Q.; Luo, L.; Wang, G.; et al. OsAAP6 functions as an important regulator of grain protein content and nutritional quality in rice. Nat. Commun. 2014, 5, 4847. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Dong, S.-S.; He, W.-M.; Ji, J.-J.; Zhang, C.; Guo, Y.; Yang, T.-L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment * | Range (%) | Mean ± SD (%) | Coefficient of Variation (%) |
|---|---|---|---|
| WJ19 | 6.97–22.24 | 14.25 ± 3.06 | 21.48 |
| WJ20 | 7.03–18.62 | 11.90 ± 2.44 | 20.51 |
| CZ20 | 8.29–18.72 | 13.29 ± 2.55 | 19.16 |
| BLUP | 10.24–16.54 | 13.15 ± 1.43 | 10.86 |
| Source | DF | SS | MS | F-Value | p-Value |
|---|---|---|---|---|---|
| Accessions | 173 | 2298.10 | 13.28 | 3.11 | <0.001 |
| Environment | 2 | 486.33 | 243.16 | 56.99 | <0.001 |
| Error | 346 | 1476.23 | 4.28 |
| Accession a | Origin | Improvement | Grain Type | Group b | Protein Content (%) c |
|---|---|---|---|---|---|
| Dominik | Germany | Cultivar | Hulled | Low | 10.71 ± 2.40 |
| Bullion | United Kingdom | Cultivar | Hulless | Low | 8.88 ± 0.31 |
| CN 21952 | Ethiopia | Landrace | Hulled | Low | 9.65 ± 0.81 |
| CN 22244 | Ethiopia | Landrace | Hulled | Low | 9.33 ± 1.61 |
| CN 53617 | Tibet, China | Landrace | Hulless | Low | 9.48 ± 1.26 |
| CN 55126 | Bulgaria | Landrace | Hulled | Low | 10.25 ± 3.45 |
| CN 69522 | Ethiopia | Landrace | Hulled | Low | 8.93 ± 0.82 |
| CN 2841 | Czech Republic | Landrace | Hulled | Low | 9.77 ± 0.82 |
| ZY001500 | Hebei, China | Cultivar | Hulless | High | 17.09 ± 4.52 |
| ZY001746 | Inner Mongolia, China | Cultivar | Hulless | High | 17.22 ± 0.83 |
| ZY000054 | Hebei, China | Landrace | Hulless | High | 17.18 ± 2.32 |
| ZY001821 | Shanxi, China | Cultivar | Hulless | High | 17.44 ± 4.30 |
| ZY001960 | Shanxi, China | Landrace | Hulless | High | 16.25 ± 3.42 |
| ZY002019 | Shanxi, China | Landrace | Hulless | High | 18.13 ± 1.73 |
| ZY000100 | Inner Mongolia, China | Landrace | Hulless | High | 16.52 ± 3.42 |
| 20130090 | Shanxi, China | Unknown | Hulless | High | 15.90 ± 1.21 |
| ZY000264 | Inner Mongolia, China | Cultivar | Hulless | High | 15.66 ± 3.32 |
| ZY000517 | Shanxi, China | Landrace | Hulless | High | 15.95 ± 2.91 |
| ZY000584 | Gansu, China | Landrace | Hulless | High | 16.34 ± 1.83 |
| QTL | Chromosome | SNP | Gene ID | Dist. From SNP (in Bp) | Annotation |
|---|---|---|---|---|---|
| QTL1 | 1D | S1D_250255160 | A.satnudSFS1D01G005211 | +42,460 | - |
| QTL2 | 1D | S1D_442130558 | A.satnudSFS1D01G001001 | +4643 | Omega-amidase, chloroplastic |
| QTL3 | 2C | S2C_532984931 | A.satnudSFS2C01G004386 | +11,305 | BTB/POZ and MATH domain-containing protein 2-like |
| QTL4 | 2D | S2D_170852683 | A.satnudSFS2D01G003434 | +33,688 | Transcription factor MYB39-like |
| QTL5 | 2D | S2D_475816930 | A.satnudSFS2D01G006139 | +265,929 | - |
| S2D_476389956 | A.satnudSFS2D01G006142 | −112,580 | Dirigent protein 7-like | ||
| S2D_479293592 | A.satnudSFS2D01G006174 | +2123 | Gibberellin 2-beta-dioxygenase 8-like | ||
| S2D_481506595 | A.satnudSFS2D01G006200 | −78,257 | - | ||
| S2D_481506711 | A.satnudSFS2D01G006200 | −78,141 | - | ||
| S2D_483972532 | A.satnudSFS2D01G006225 | −272,059 | Flowering-promoting factor 1-like protein 4 | ||
| S2D_484596713 | A.satnudSFS2D01G006226 | −168,983 | Fasciclin-like arabinogalactan protein 14 | ||
| S2D_485481234 | A.satnudSFS2D01G006232 | −194,688 | Transcription factor 3C polypeptide 5-like | ||
| QTL6 | 3C | S3C_539312598 | A.satnudSFS3C01G003718 | +6886 | - |
| QTL7 | 3D | S3D_93982802 | A.satnudSFS3D01G001144 | +50,123 | - |
| QTL8 | 3D | S3D_323864116 | A.satnudSFS3D01G002332 | +23,877 | Zinc finger protein BRUTUS-like |
| S3D_329141631 | A.satnudSFS3D01G002420 | +22,075 | Ribosomal protein L36a | ||
| QTL9 | 3D | S3D_404716753 | A.satnudSFS3D01G003689 | −2211 | Ribosomal protein L18-2 |
| QTL10 | 4A | S4A_435900993 | A.satnudSFS4A01G006735 | +7552 | Axoneme-associated protein mst101(2)-like |
| QTL11 | 4D | S4D_422496061 | A.satnudSFS4D01G000876 | +41,624 | Probable leucine-rich repeat receptor-like protein kinase |
| S4D_425631870 | A.satnudSFS4D01G000779 | −3731 | - | ||
| S4D_425631876 | A.satnudSFS4D01G000779 | −3725 | - | ||
| S4D_425632011 | A.satnudSFS4D01G000779 | −3590 | - | ||
| QTL12 | 5C | S5C_42034228 | A.satnudSFS5C01G000409 | +28,493 | - |
| QTL13 | 6C | S6C_112862861 | A.satnudSFS6C01G002256 | +4234 | Putative disease resistance RPP13-like protein 3 |
| QTL14 | 6C | S6C_154091753 | A.satnudSFS6C01G003130 | −39,300 | - |
| QTL15 | 6C | S6C_238593165 | A.satnudSFS6C01G003976 | −3784 | - |
| QTL16 | 6C | S6C_285259972 | A.satnudSFS6C01G004265 | −9039 | - |
| QTL17 | 6C | S6C_516086587 | A.satnudSFS6C01G005207 | −58,400 | Polymerase 2-A |
| QTL18 | 6C | S6C_522114339 | A.satnudSFS6C01G005237 | +64,919 | Pop guanine nucleotide exchange factor 14 |
| QTL19 | 6C | S6C_528407767 | A.satnudSFS6C01G005283 | +68,520 | - |
| S6C_531364922 | A.satnudSFS6C01G005292 | −405,403 | Myb-related protein Zm1-like | ||
| QTL20 | 6C | S6C_540012059 | A.satnudSFS6C01G005352 | +34,825 | Cytosolic sulfotransferase 5 |
| QTL21 | 6C | S6C_583370024 | A.satnudSFS6C01G005704 | +94,938 | R2R3-MYB protein |
| QTL22 | 6D | S6D_26224217 | A.satnudSFS6D01G000303 | −2965 | - |
| S6D_26224354 | A.satnudSFS6D01G000303 | −2828 | - | ||
| QTL23 | 6D | S6D_196278046 | A.satnudSFS6D01G001314 | +64,283 | - |
| QTL24 | 7C | S7C_176236166 | A.satnudSFS7C01G002963 | +13,241 | Cytochrome P450 94B3-like |
| S7C_176236263 | A.satnudSFS7C01G002963 | +13,338 | Cytochrome P450 94B3-like | ||
| QTL25 | 7C | S7C_191492694 | A.satnudSFS7C01G003117 | +19,910 | - |
| QTL26 | 7C | S7C_413114334 | A.satnudSFS7C01G003963 | −636,910 | - |
| QTL27 | 7D | S7D_455264275 | A.satnudSFS7D01G004588 | +3682 | Hydrophobic protein RCI2A-like |
| QTL | Haplotype | Protein Content (%) | Hap Frequency (%) # | ||||
|---|---|---|---|---|---|---|---|
| WJ19 | WJ20 | CZ20 | BLUP | Hulless | Hulled | ||
| QTL16 | HAP-G | 16.30 | 13.67 | 15.09 | 14.43 | 86.67 | 10.53 |
| HAP-A | 13.04 | 10.72 | 12.11 | 12.34 | 10.00 | 62.28 | |
| QTL11 | HAP-CAA | 16.52 | 13.51 | 15.62 | 14.56 | 51.67 | 0.88 |
| HAP-TGC | 13.48 | 11.33 | 12.77 | 12.73 | 33.33 | 77.19 | |
| QTL16 + QTL11 | HAP-CAA + HAP-G | 16.81 | 13.85 | 15.84 | 14.75 | 43.33 | 0 |
| HAP-CAA + HAP-A | 15.73 | 11.41 | 15.08 | 13.78 | 5.00 | 0.89 | |
| HAP-TGC + HAP-G | 15.83 | 13.13 | 14.27 | 14.03 | 28.33 | 8.77 | |
| HAP-TGC + HAP-A | 12.52 | 10.57 | 12.16 | 12.20 | 5.00 | 46.49 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Zhang, H.; Zhou, P.; Ren, C.; Peng, Y. Genome-Wide Association Mapping of QTL Underlying Groat Protein Content of a Diverse Panel of Oat Accessions. Int. J. Mol. Sci. 2023, 24, 5581. https://doi.org/10.3390/ijms24065581
Yan H, Zhang H, Zhou P, Ren C, Peng Y. Genome-Wide Association Mapping of QTL Underlying Groat Protein Content of a Diverse Panel of Oat Accessions. International Journal of Molecular Sciences. 2023; 24(6):5581. https://doi.org/10.3390/ijms24065581
Chicago/Turabian StyleYan, Honghai, Haixu Zhang, Pingping Zhou, Changzhong Ren, and Yuanying Peng. 2023. "Genome-Wide Association Mapping of QTL Underlying Groat Protein Content of a Diverse Panel of Oat Accessions" International Journal of Molecular Sciences 24, no. 6: 5581. https://doi.org/10.3390/ijms24065581
APA StyleYan, H., Zhang, H., Zhou, P., Ren, C., & Peng, Y. (2023). Genome-Wide Association Mapping of QTL Underlying Groat Protein Content of a Diverse Panel of Oat Accessions. International Journal of Molecular Sciences, 24(6), 5581. https://doi.org/10.3390/ijms24065581