
A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing

Abstract
:1. Introduction
2. Results
2.1. Sequencing Results and Data Assembly
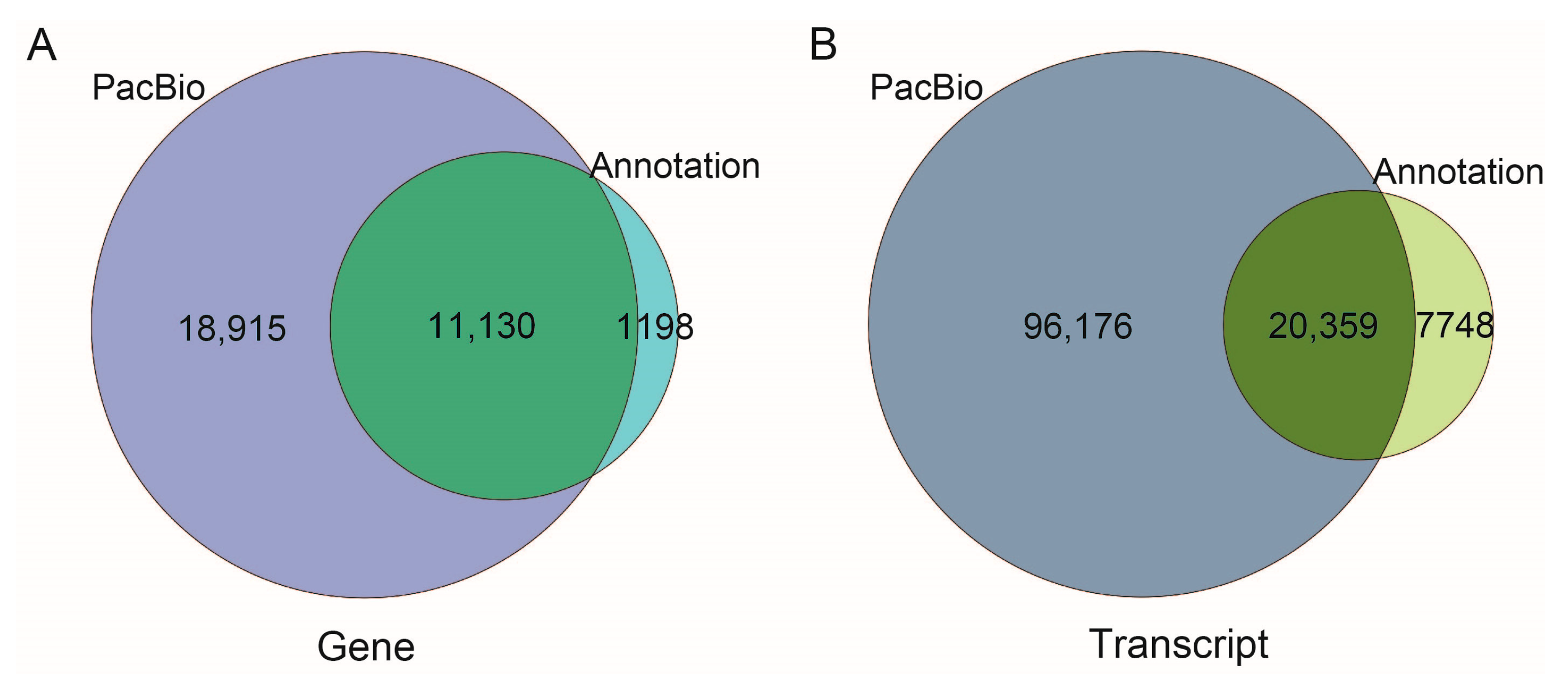
2.2. Functional Annotation of Transcripts
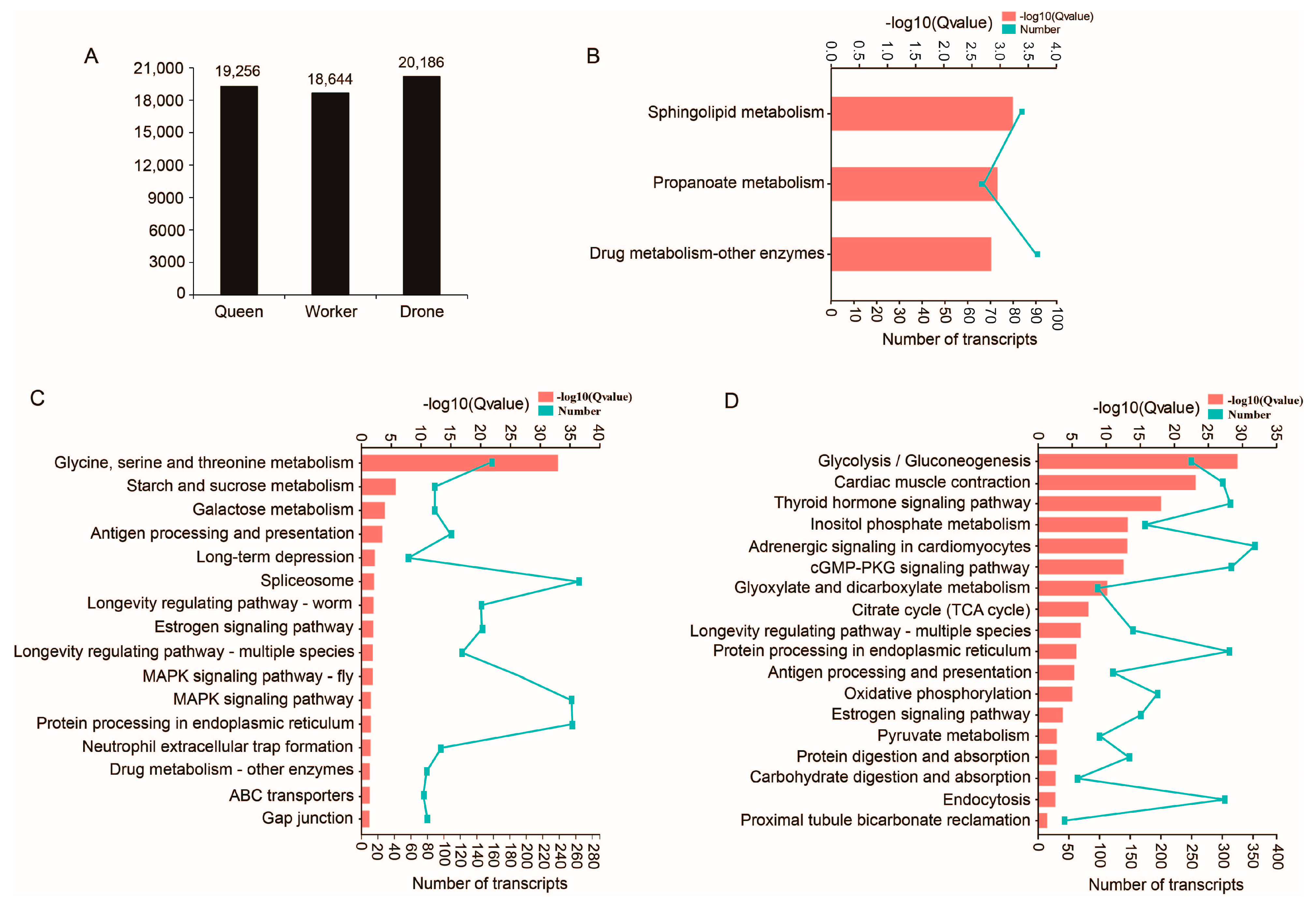
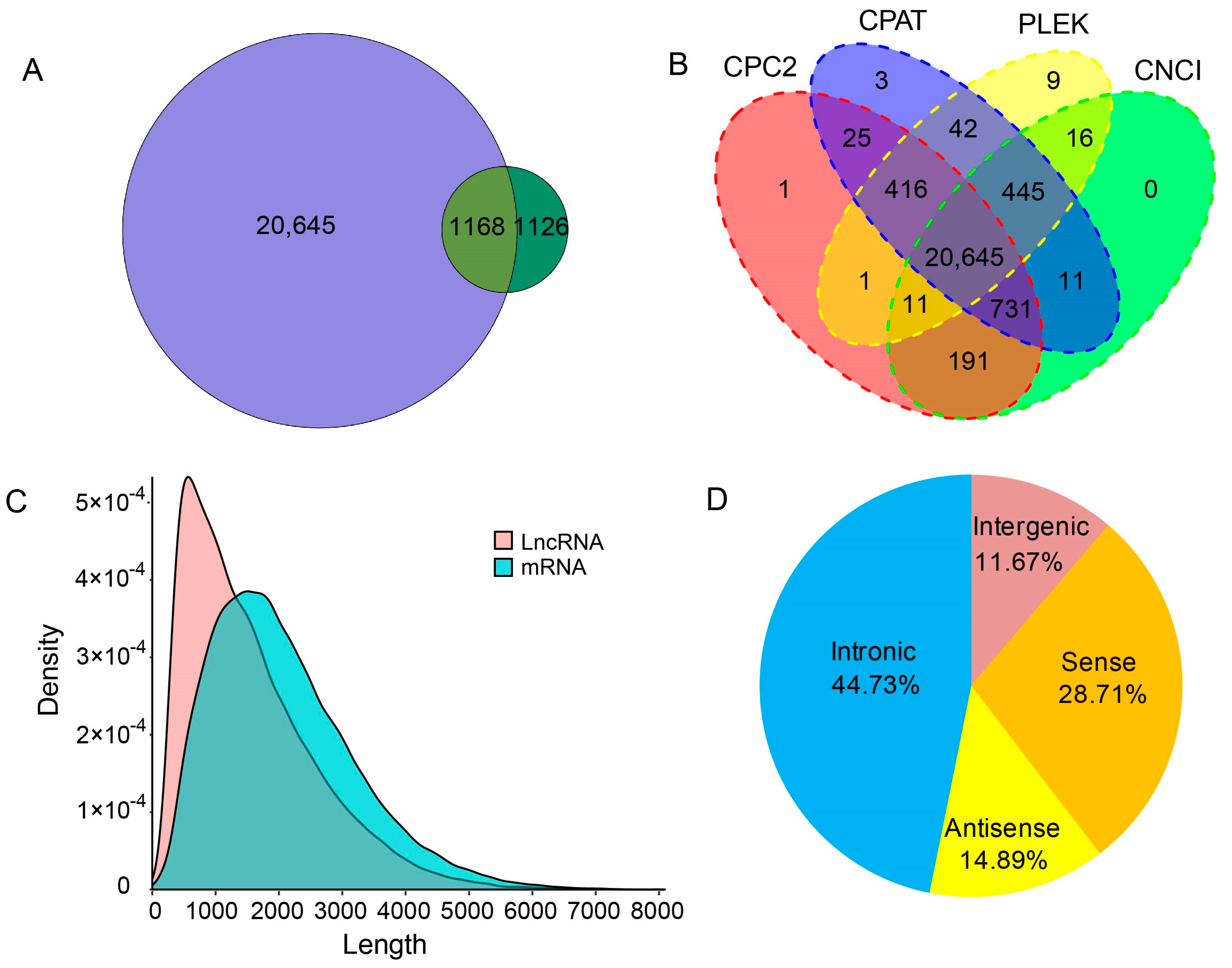
2.3. Unique Transcripts from Queen, Worker and Drone Data
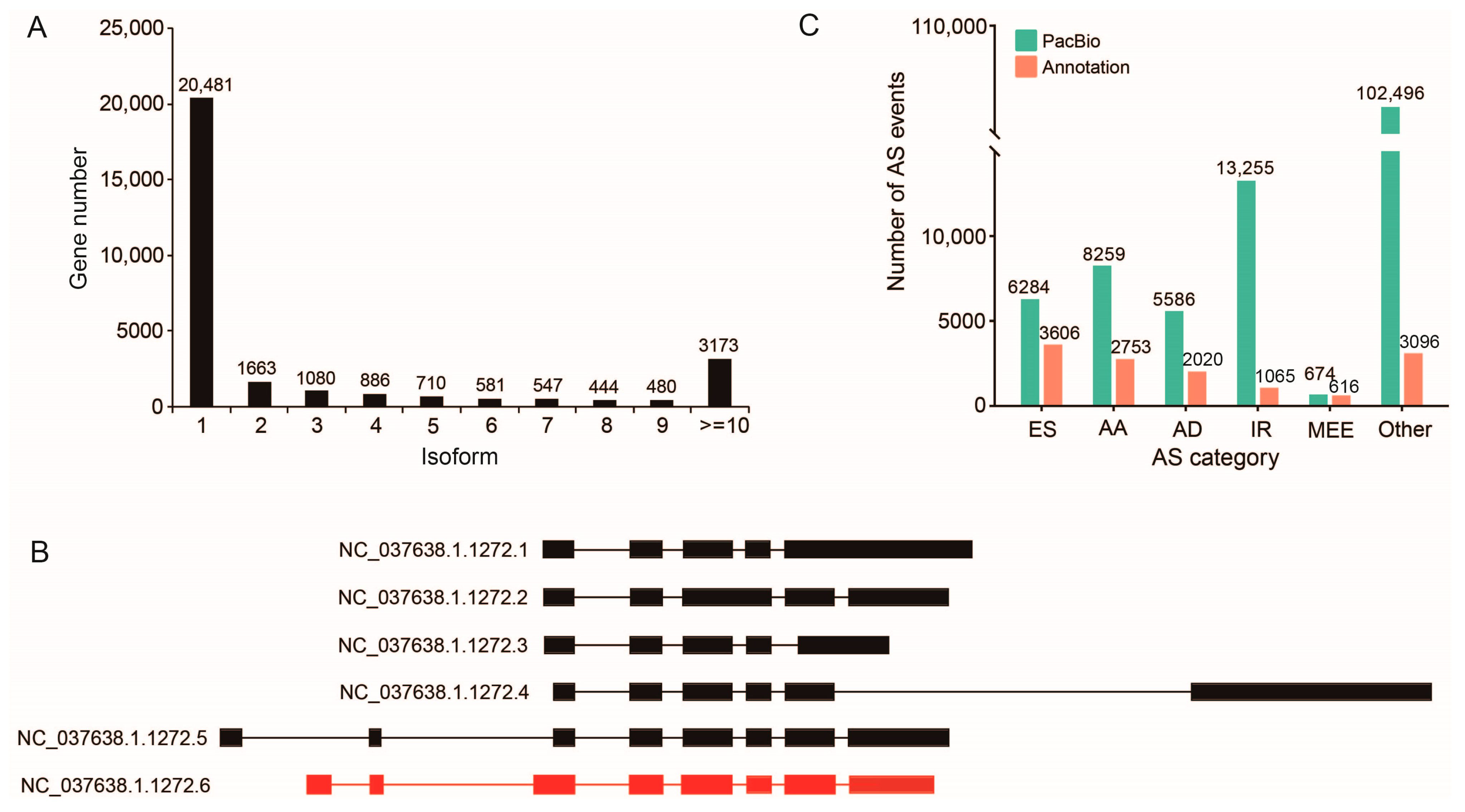
2.4. Alternative Splicing
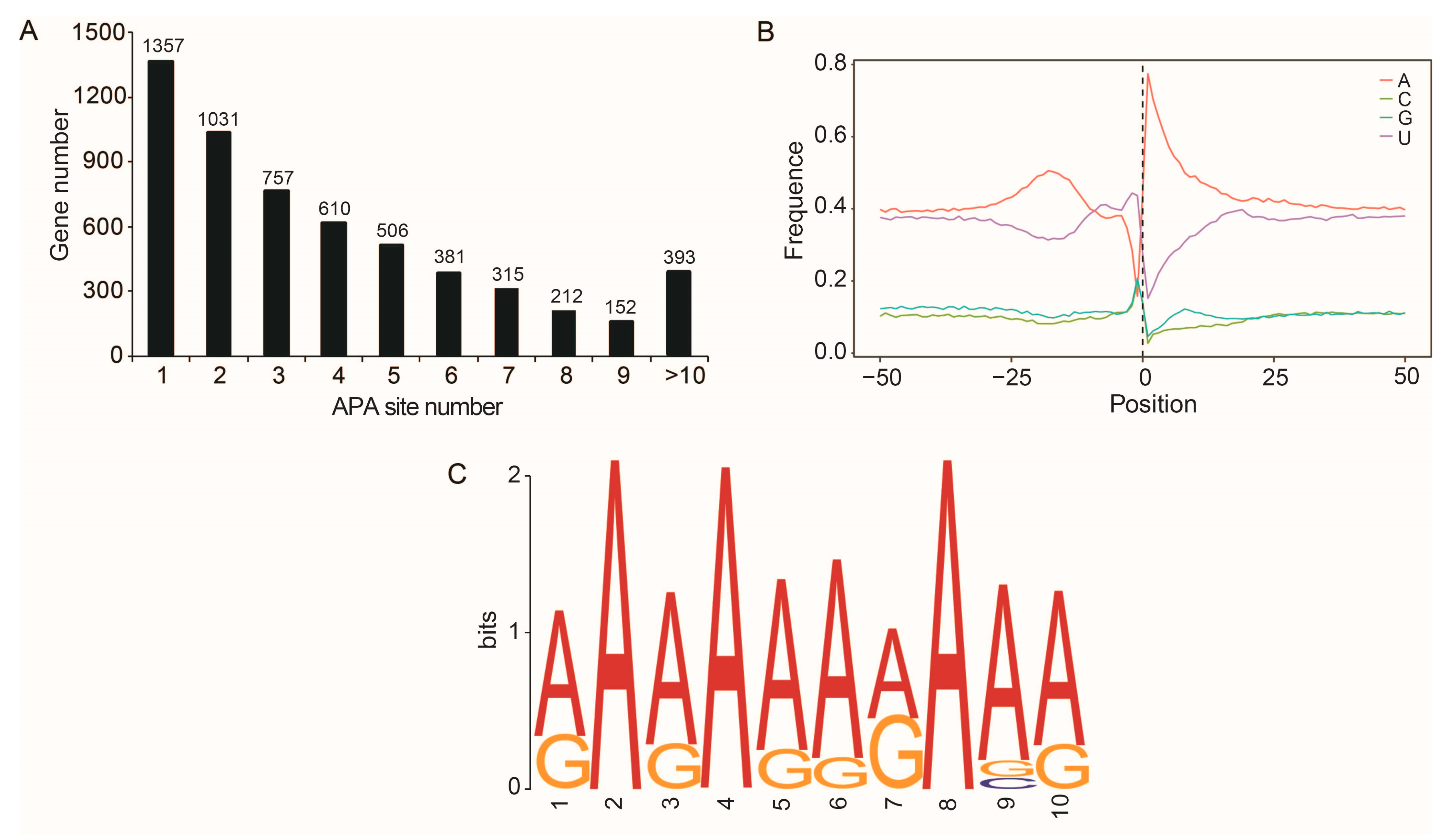
2.5. APA Sites
2.6. Long Non-Coding RNAs
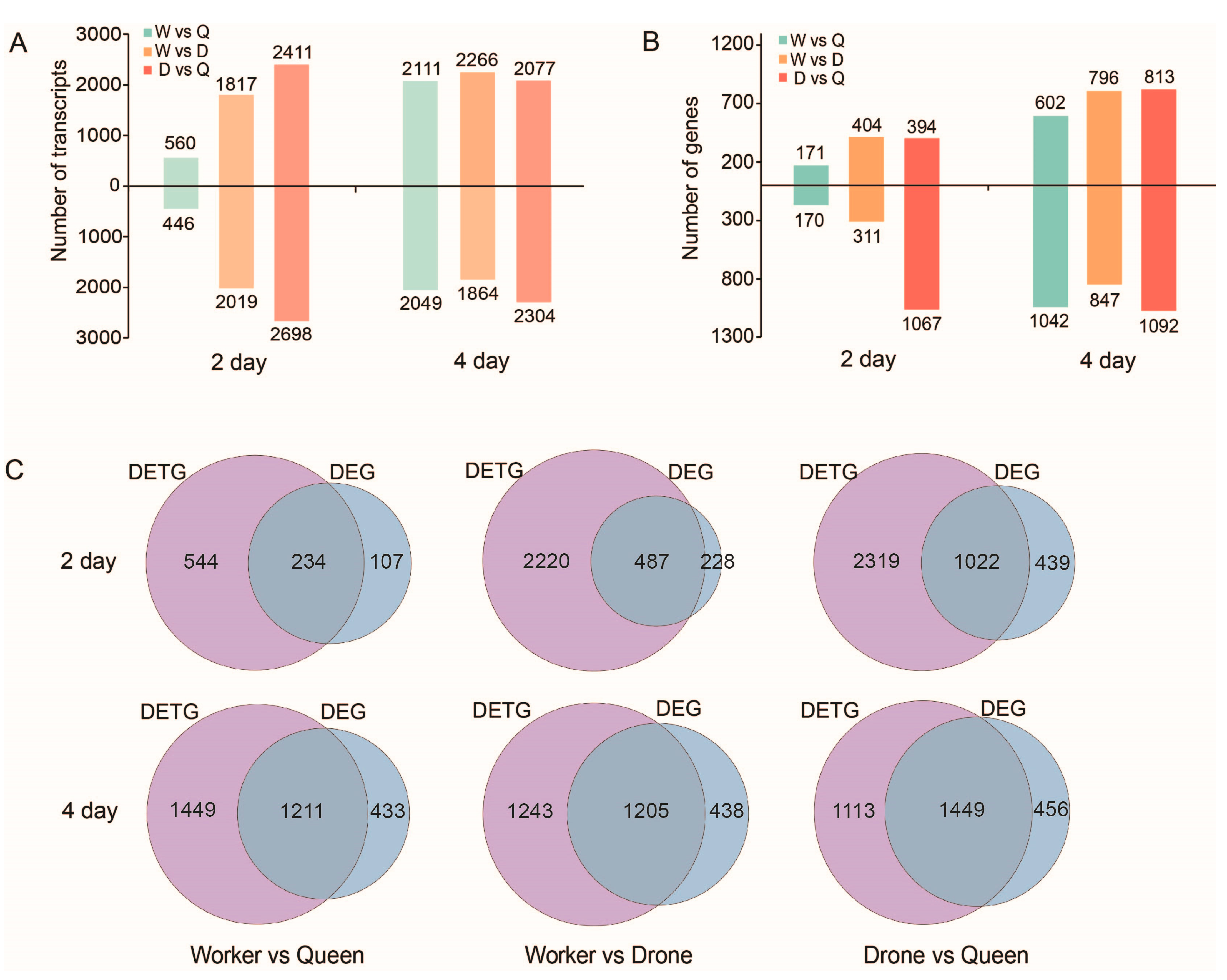
2.7. DETs and DEGs between the Three Honey Bee Castes
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Library Construction and Sequencing
4.3. Analysis of the Raw Data
4.4. Identification of Genes and Transcripts
4.5. Identification of Alternative Splicing Events, lncRNAs and APA Sites
4.6. Identification of DETs and DEGs
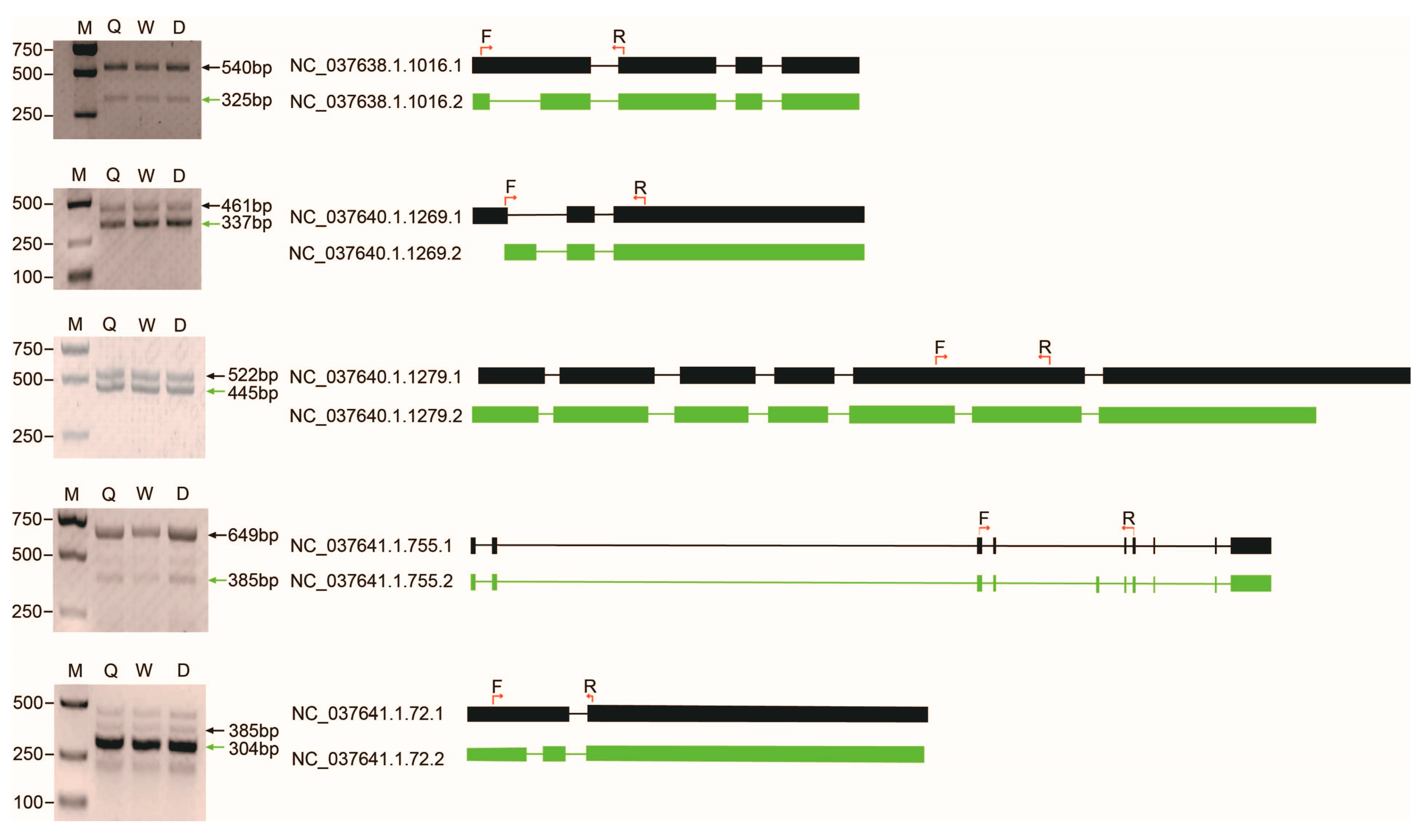
4.7. RT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, S.L. Honey bee germplasm resources. In The Apicultural Science in China, 1st ed.; Liu, B.H., Ed.; Chinese Agricultural Press: Beijing, China, 2001; pp. 16–24. [Google Scholar]
- Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.-B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, K.; Wakamiya, T.; Bono, H. Meta-Analysis of the Public RNA-Seq Data of the Western Honeybee Apis mellifera to Construct Reference Transcriptome Data. Insects 2022, 13, 931. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, S.T.; Döke, M.A.; Giray, T.; Grozinger, C.M. Tissue-specific transcriptional patterns underlie seasonal phenotypes in honey bees (Apis mellifera). Mol. Ecol. 2022, 31, 174–184. [Google Scholar] [CrossRef]
- Hunt, G.J.; Page, R.E., Jr. Linkage map of the honey bee, Apis mellifera, based on RAPD markers. Genetics 1995, 139, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Solignac, M.; Vautrin, D.; Baudry, E.; Mougel, F.; Loiseau, A.; Cornuet, J.-M. A Microsatellite-Based Linkage Map of the Honeybee, Apis mellifera L. Genetics 2004, 167, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Solignac, M.; Mougel, F.; Vautrin, D.; Monnerot, M.; Cornuet, J.-M. A third-generation microsatellite-based linkage map of the honey bee, Apis mellifera, and its comparison with the sequence-based physical map. Genome Biol. 2007, 8, R66. [Google Scholar] [CrossRef] [Green Version]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Staiger, D.; Brown, J.W. Alternative Splicing at the Intersection of Biological Timing, Development, and Stress Responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary Dynamics of Gene and Isoform Regulation in Mammalian Tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.-B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbazuk, W.B.; Fu, Y.; McGinnis, K.M. Genome-wide analyses of alternative splicing in plants: Opportunities and challenges. Genome Res. 2008, 18, 1381–1392. [Google Scholar] [CrossRef] [Green Version]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA -Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High Throughput Sequencing: An Overview of Sequencing Chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.; Puiu, D.; Wegrzyn, J.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. Gigascience 2017, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Chen, H.-F.; Yin, Z.-Y.; Chen, W.-L.; Lu, L.-T. The genetic adaptations of Toxoptera aurantii facilitated its rapid multiple plant hosts dispersal and invasion. Genomics 2022, 114, 110472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Liu, H.; Pan, X.; Xu, M.; Zhang, G.; He, M. PacBio single molecule long-read sequencing provides insight into the complexity and diversity of the Pinctada fucata martensii transcriptome. BMC Genom. 2020, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Jiang, W.; Zhou, M.; Barron, A.B.; Zeng, Z. A comparison of honeybee (Apis mellifera) queen, worker and drone larvae by RNA-Seq. Insect Sci. 2019, 26, 499–509. [Google Scholar] [CrossRef]
- Gibilisco, L.; Zhou, Q.; Mahajan, S.; Bachtrog, D. Alternative Splicing within and between Drosophila Species, Sexes, Tissues, and Developmental Stages. PLoS Genet. 2016, 12, e1006464. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Zhao, Q.-Y.; Wang, X.-Y.; Xu, X.-Y.; Tang, Q.; Li, M.; Li, X.; Xu, Y.-Z. Alternative splicing and trans-splicing events revealed by analysis of the Bombyx mori transcriptome. Rna 2012, 18, 1395–1407. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhong, W.; He, W.; Li, Y.; Li, Y.; Li, T.; Vasseur, L.; You, M. Genome-wide profiling of the alternative splicing provides insights into development in Plutella xylostella. BMC Genom. 2019, 20, 463. [Google Scholar] [CrossRef] [Green Version]
- Smibert, P.; Miura, P.; Westholm, J.O.; Shenker, S.; May, G.; Duff, M.O.; Zhang, D.; Eads, B.D.; Carlson, J.; Brown, J.B.; et al. Global Patterns of Tissue-Specific Alternative Polyadenylation in Drosophila. Cell Rep. 2012, 1, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, C.H.; Friedman, R.C.; Ruby, J.G.; Bartel, D.P. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 2011, 469, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Subtelny, A.O.; Koppstein, D.; Bell, G.W.; Sive, H.; Bartel, D.P. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012, 22, 2054–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Venu, R.; Nobuta, K.; Wu, X.; Notibala, V.; Demirci, C.; Meyers, B.C.; Wang, G.-L.; Ji, G.; Li, Q.Q. Transcriptome dynamics through alternative polyadenylation in developmental and environmental responses in plants revealed by deep sequencing. Genome Res. 2011, 21, 1478–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Castro, I.; Moreira, A. On the function and relevance of alternative 3′- UTRs in gene expression regulation. Wiley Interdiscip. Rev. RNA 2021, 12, e1653. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cheng, T.; Liu, C.; Liu, D.; Zhang, Q.; Long, R.; Zhao, P.; Xia, Q. Systematic Identification and Characterization of Long Non-Coding RNAs in the Silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-J.; Song, Y.-J.; Han, H.-L.; Xu, H.-Q.; Wei, D.; Smagghe, G.; Wang, J.-J. Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly, Zeugodacus cucurbitae (Coquillett). BMC Genom. 2020, 21, 600. [Google Scholar] [CrossRef]
- Azlan, A.; Obeidat, S.M.; Das, K.T.; Yunus, M.A.; Azzam, G. Genome-wide identification of Aedes albopictus long noncoding RNAs and their association with dengue and Zika virus infection. PLoS Negl. Trop. Dis. 2021, 15, e0008351. [Google Scholar] [CrossRef]
- Meng, L.; Yuan, G.; Chen, M.; Dou, W.; Jing, T.; Zheng, L.; Peng, M.; Bai, W.; Wang, J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with malathion resistance in Bactrocera dorsalis. Pest Manag. Sci. 2021, 77, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Leeb, M.; Steffen, P.A.; Wutz, A. X chromosome inactivation sparked by non-coding RNAs. RNA Biol. 2009, 6, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Hierholzer, A.; Chureau, C.; Liverziani, A.; Ruiz, N.B.; Cattanach, B.M.; Young, A.N.; Kumar, M.; Cerase, A.; Avner, P. A long noncoding RNA influences the choice of the X chromosome to be inactivated. Proc. Natl. Acad. Sci. USA 2022, 119, e2118182119. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA Exploits Three-Dimensional Genome Architecture to Spread Across the X Chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [Green Version]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.-H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Zhang, H.; Zeitz, M.J.; Wang, H.; Niu, B.; Ge, S.; Li, W.; Cui, J.; Wang, G.; Qian, G.; Higgins, M.J.; et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J. Cell Biol. 2014, 204, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Attaway, M.; Chwat-Edelstein, T.; Vuong, B.Q. Regulatory Non-Coding RNAs Modulate Transcriptional Activation During B Cell Development. Front. Genet. 2021, 12, 678084. [Google Scholar] [CrossRef]
- Li, X.L.; Subramanian, M.; Jones, M.F.; Chaudhary, R.; Singh, D.K.; Zong, X.; Gryder, B.; Sindri, S.; Mo, M.; Schetter, A.; et al. Long Noncoding RNA PURPL Suppresses Basal p53 Levels and Promotes Tumorigenicity in Colorectal Cancer. Cell Rep. 2017, 20, 2408–2423. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Sammeth, M.; Foissac, S.; Guigó, R. A general definition and nomenclature for alternative splicing events. PLoS Comput. Biol. 2008, 4, e1000147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37 (Suppl. S2), w202–w208. [Google Scholar] [CrossRef]
- Langmead, B. Aligning Short Sequencing Reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Bases (Gbp) | Number of Polymerase Reads | Number of Subreads | Number of CCSs | Number of FLNCs | Average Length of FLNCs (bp) |
|---|---|---|---|---|---|---|
| Queen | 64.80 | 1,100,885 | 43,491,193 | 795,183 | 556,221 | 1504 |
| Worker | 59.74 | 1,042,133 | 40,055,924 | 735,061 | 514,591 | 1511 |
| Drone | 65.49 | 1,126,347 | 40,663,357 | 792,515 | 546,907 | 1651 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.-Y.; Pan, L.-X.; Cheng, F.-P.; Jin, M.-J.; Wang, Z.-L. A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing. Int. J. Mol. Sci. 2023, 24, 5827. https://doi.org/10.3390/ijms24065827
Zheng S-Y, Pan L-X, Cheng F-P, Jin M-J, Wang Z-L. A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing. International Journal of Molecular Sciences. 2023; 24(6):5827. https://doi.org/10.3390/ijms24065827
Chicago/Turabian StyleZheng, Shuang-Yan, Lu-Xia Pan, Fu-Ping Cheng, Meng-Jie Jin, and Zi-Long Wang. 2023. "A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing" International Journal of Molecular Sciences 24, no. 6: 5827. https://doi.org/10.3390/ijms24065827
APA StyleZheng, S. -Y., Pan, L. -X., Cheng, F. -P., Jin, M. -J., & Wang, Z. -L. (2023). A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing. International Journal of Molecular Sciences, 24(6), 5827. https://doi.org/10.3390/ijms24065827