
New Drugs and Therapies in Pulmonary Arterial Hypertension



Abstract
:1. Introduction
2. Pathophysiology of Pulmonary Arterial Hypertension
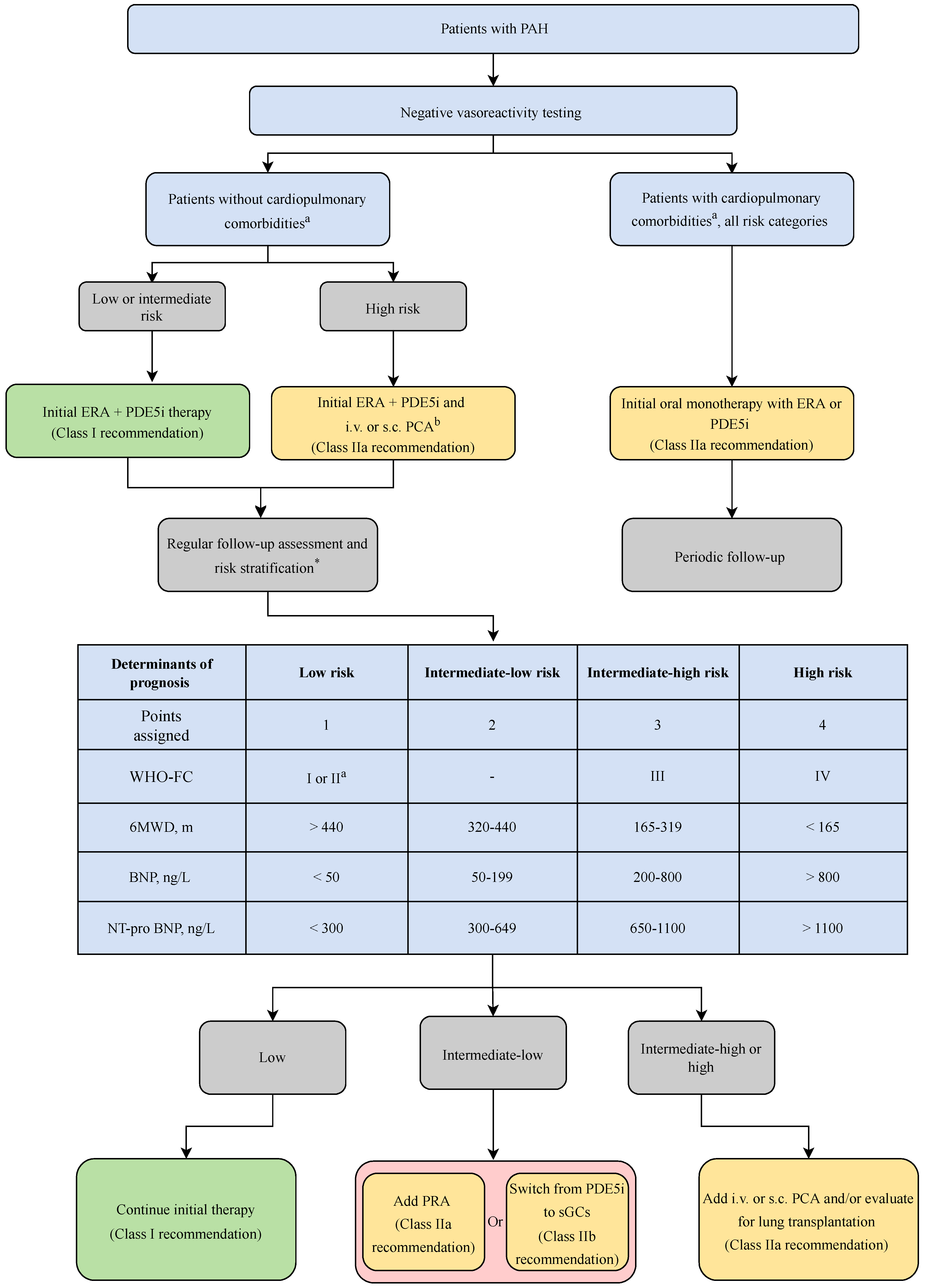
3. Current FDA Approved Therapies for Pulmonary Arterial Hypertension
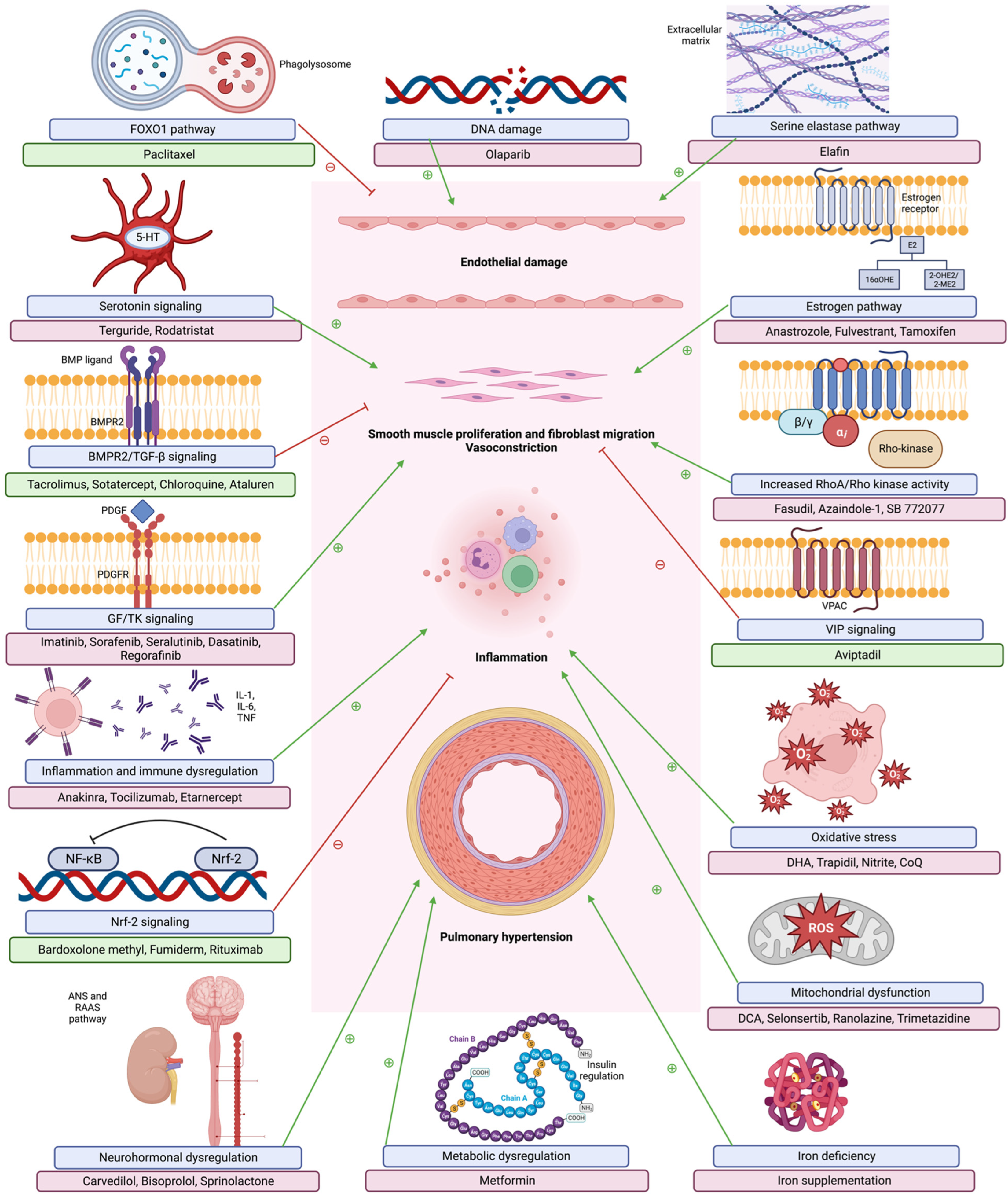
4. Novel Therapies
4.1. BMPR2 Pathway
4.1.1. BMP Ligands
4.1.2. Other BMPR2 Modulating Therapies
4.2. Inflammation and Immunity
4.2.1. Interleukin-6 (IL-6)
4.2.2. Interleukin-1 (IL-1)
4.2.3. Tumor Necrosis Factor-α (ΤΝF-α)
4.2.4. Nuclear Factor κβ
4.3. GF/TK Signaling Pathway
4.4. RhoA/Rho Kinase Pathway
4.5. Mitochondrial Dysfunction
4.6. Oxidative Stress
4.7. Extracellular Matrix
4.8. Metabolic Pathway
4.9. Neurohormonal Pathways
4.9.1. Beta-Blockers
4.9.2. Alternate RAAS Pathway
4.9.3. Mineralocorticoid Receptor Antagonists
4.9.4. Vasoactive Intestinal Peptide
4.10. Serotonin Signaling
4.11. Estrogen Pathway
4.12. Iron Deficiency
4.13. Deoxyribonucleic Acid (DNA) Damage
4.14. FoxO1 Pathway
5. Interventional Modalities
5.1. Balloon Pulmonary Angioplasty
5.2. Pulmonary Artery Denervation
5.3. Atrial Septostomy
6. Newer Emerging Therapies
6.1. Immunotherapy
6.2. Micro RNAs (miR)
6.3. Gene Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valerio, C.J.; Schreiber, B.E.; Handler, C.E.; Denton, C.P.; Coghlan, J.G. Borderline Mean Pulmonary Artery Pressure in Patients With Systemic Sclerosis: Transpulmonary Gradient Predicts Risk of Developing Pulmonary Hypertension. Arthritis Rheum. 2013, 65, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.B.; Mazur, W.; Kalra, D.K. Pulmonary hypertension: Diagnosis, imaging techniques, and novel therapies. Cardiovasc. Diagn. Ther. 2017, 7, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Mayer, E.; Simonneau, G.; Rubin, L.J. Chronic Thromboembolic Pulmonary Hypertension. Circulation 2006, 113, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Berghold, A.; Scheidl, S.; Olschewski, H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur. Respir. J. 2009, 34, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.; Douschan, P.; Maron, B.A.; Condliffe, R.; Olschewski, H. Mildly increased pulmonary arterial pressure: A new disease entity or just a marker of poor prognosis? Eur. J. Heart Fail. 2019, 21, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Saggar, R.; Bolster, M.B.; Chung, L.; Csuka, M.E.; Derk, C.; Domsic, R.; Fischer, A.; Frech, T.; Goldberg, A.; et al. Baseline characteristics and follow-up in patients with normal haemodynamics versus borderline mean pulmonary arterial pressure in systemic sclerosis: Results from the PHAROS registry. Ann. Rheum. Dis. 2012, 71, 1335–1342. [Google Scholar] [CrossRef]
- Maron, B.A.; Hess, E.; Maddox, T.M.; Opotowsky, A.R.; Tedford, R.J.; Lahm, T.; Joynt, K.E.; Kass, D.J.; Stephens, T.; Stanislawski, M.; et al. Association of Borderline Pulmonary Hypertension With Mortality and Hospitalization in a Large Patient Cohort: Insights From the Veterans Affairs Clinical Assessment, Reporting, and Tracking Program. Circulation 2016, 133, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.J.; Vorla, M.; Kalra, D.K. Molecular Pathways in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2022, 23, 10001. [Google Scholar] [CrossRef]
- Zolty, R. Novel Experimental Therapies for Treatment of Pulmonary Arterial Hypertension. J. Exp. Pharmacol. 2021, 13, 817–857. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef]
- Vetter, N. Editor’s Choice. Br. Med. Bull. 2010, 94, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Huber, L.; Bye, H.; Brock, M. The pathogenesis of pulmonary hypertension—An update. Swiss Med. Wkly. 2015, 145, w14202. [Google Scholar] [CrossRef] [Green Version]
- Farber, H.W.; Loscalzo, J. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and Immunity in the Pathogenesis of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef]
- Maron, B.A. Pulmonary arterial hypertension: Cellular and molecular changes in the lung. Glob. Cardiol. Sci. Pract. 2020, 2020, e202003. [Google Scholar] [CrossRef]
- Wilkins, M.R. Pulmonary hypertension: The science behind the disease spectrum. Eur. Respir. Rev. 2012, 21, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.; Rochefort, G.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Bonnet, S.N.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423. [Google Scholar] [CrossRef] [Green Version]
- Connolly, M.J.; Aaronson, P.I. Key role of the RhoA/Rho kinase system in pulmonary hypertension. Pulm. Pharmacol. Ther. 2011, 24, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Do, E.Z.; Fukumoto, Y.; Takaki, A.; Tawara, S.; Ohashi, J.; Nakano, M.; Tada, T.; Saji, K.; Sugimura, K.; Fujita, H.; et al. Evidence for Rho-Kinase Activation in Patients with Pulmonary Arterial Hypertension. Circ. J. 2009, 73, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Mair, K.M.; Yang, X.D.; Long, L.; White, K.; Wallace, E.; Ewart, M.-A.; Docherty, C.K.; Morrell, N.W.; MacLean, M.R. Sex Affects Bone Morphogenetic Protein Type II Receptor Signaling in Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Crit. Care Med. 2015, 191, 693–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelkel, N.F.; Quaife, R.A.; Leinwand, L.A.; Barst, R.J.; McGoon, M.D.; Meldrum, D.R.; Dupuis, J.; Long, C.S.; Rubin, L.J.; Smart, F.W.; et al. Right Ventricular Function and Failure. Circulation 2006, 114, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Barst, R.J.; McGoon, M.; Torbicki, A.; Sitbon, O.; Krowka, M.J.; Olschewski, H.; Gaine, S. Diagnosis and differential assessment of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S40–S47. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Humbert, M.; Jais, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-Term Response to Calcium Channel Blockers in Idiopathic Pulmonary Arterial Hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- Rich, S.; Kaufmann, E.; Levy, P.S. The Effect of High Doses of Calcium-Channel Blockers on Survival in Primary Pulmonary Hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef]
- Zolty, R. Pulmonary arterial hypertension specific therapy: The old and the new. Pharmacol. Ther. 2020, 214, 107576. [Google Scholar] [CrossRef]
- Lavelle, A.; Sugrue, R.; Lawler, G.; Mulligan, N.; Kelleher, B.; Murphy, D.M.; Gaine, S. Sitaxentan-induced hepatic failure in two patients with pulmonary arterial hypertension. Eur. Respir. J. 2009, 34, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-T.N.; Kirkham, N.; Johnson, M.K.; Lordan, J.L.; Fisher, A.J.; Peacock, A.J. Sitaxentan-related acute liver failure in a patient with pulmonary arterial hypertension. Eur. Respir. J. 2011, 37, 472–474. [Google Scholar] [CrossRef] [Green Version]
- Bin-Nun, A.; Schreiber, M.D. Role of iNO in the modulation of pulmonary vascular resistance. J. Perinatol. 2008, 28, S84–S92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triposkiadis, F.; Xanthopoulos, A.; Skoularigis, J.; Starling, R.C. Therapeutic augmentation of NO-sGC-cGMP signalling: Lessons learned from pulmonary arterial hypertension and heart failure. Heart Fail. Rev. 2022, 27, 1991–2003. [Google Scholar] [CrossRef]
- Stasch, J.-P.; Pacher, P.; Evgenov, O.V. Soluble Guanylate Cyclase as an Emerging Therapeutic Target in Cardiopulmonary Disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimminger, F.; Weimann, G.; Frey, R.; Voswinckel, R.; Thamm, M.; Bolkow, D.; Weissmann, N.; Muck, W.; Unger, S.; Wensing, G.; et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur. Respir. J. 2009, 33, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Ruopp, N.F.; Cockrill, B.A. Diagnosis and Treatment of Pulmonary Arterial Hypertension. JAMA 2022, 327, 1379–1391. [Google Scholar] [CrossRef]
- Barst, R. How has epoprostenol changed the outcome for patients with pulmonary arterial hypertension? Int. J. Clin. Pract. 2010, 64, 23–32. [Google Scholar] [CrossRef]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharmacol. 2021, 178, 6–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.A. Clarifying the Pulmonary Arterial Hypertension Molecular Landscape Using Functional Genetics. Am. J. Respir. Crit. Care Med. 2020, 202, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J.P.; Loyd, J.E.; Austin, E.D. The Genetics of Pulmonary Arterial Hypertension in the Post-BMPR2 Era. Pulm. Circ. 2011, 1, 305–319. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Happé, C.; Kurakula, K.; Sun, X.-Q.; Bos, D.D.S.G.; Rol, N.; Guignabert, C.; Tu, L.; Schalij, I.; Wiesmeijer, K.C.; Tura-Ceide, O.; et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasim, T.; Ogo, T.; Chowdhury, H.M.; Zhao, L.; Chen, C.-N.; Rhodes, C.; Trembath, R. BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGFβ-TAK1-MAPK pathways in PAH. Hum. Mol. Genet. 2012, 21, 2548–2558. [Google Scholar] [CrossRef] [Green Version]
- Dewachter, L.; Adnot, S.; Guignabert, C.; Tu, L.; Marcos, E.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; et al. Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. Eur. Respir. J. 2009, 34, 1100–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.K.; Ichimura, K.; Spiekerkoetter, E. Promising therapeutic approaches in pulmonary arterial hypertension. Curr. Opin. Pharmacol. 2021, 59, 127–139. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602449. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Feng, J.-A.; Li, P.; Xing, D.; Zhang, Y.; Serra, R.; Ambalavanan, N.; Majid-Hassan, E.; Oparil, S. Dominant negative mutation of the TGF-β receptor blocks hypoxia-induced pulmonary vascular remodeling. J. Appl. Physiol. 2006, 100, 564–571. [Google Scholar] [CrossRef]
- Tielemans, B.; Delcroix, M.; Belge, C.; Quarck, R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov. Today 2019, 24, 703–716. [Google Scholar] [CrossRef]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.; Dunmore, B.J.; Morrell, N. Chloroquine Prevents Progression of Experimental Pulmonary Hypertension via Inhibition of Autophagy and Lysosomal Bone Morphogenetic Protein Type II Receptor Degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef]
- Drake, K.M.; Dunmore, B.J.; McNelly, L.N.; Morrell, N.W.; Aldred, M.A. Correction of Nonsense BMPR2 and SMAD9 Mutations by Ataluren in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, N.; Sonenberg, N. Proposing a mechanism of action for ataluren. Proc. Natl. Acad. Sci. USA 2016, 113, 12353–12355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouthon, L.; Guillevin, L.; Humbert, M. Pulmonary arterial hypertension: An autoimmune disease? Eur. Respir. J. 2005, 26, 986–988. [Google Scholar] [CrossRef] [Green Version]
- Mukerjee, D.; George, D.S.; Coleiro, B.; Knight, C.; Denton, C.P.; Davar, J.; Black, C.M.; Coghlan, J.G. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: Application of a registry approach. Ann. Rheum. Dis. 2003, 62, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolls, M.R.; Taraseviciene-Stewart, L.; Rai, P.R.; Voelkel, N.F.; Badesch, D.B. Autoimmunity and pulmonary hypertension: A perspective. Eur. Respir. J. 2005, 26, 1110–1118. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern Age Pathology of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Molossi, S.; Clausell, N.; Rabinovitch, M. Reciprocal induction of tumor necrosis factor-? and interleukin-? activity mediates fibronectin synthesis in coronary artery smooth muscle cells. J. Cell. Physiol. 1995, 163, 19–29. [Google Scholar] [CrossRef]
- Jones, P.L.; Cowan, K.N.; Rabinovitch, M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am. J. Pathol. 1997, 150, 1349–1360. [Google Scholar]
- Terrier, B.; Tamby, M.C.; Camoin, L.; Guilpain, P.; Broussard, C.; Bussone, G.; Yaïci, A.; Hotellier, F.; Simonneau, G.; Guillevin, L.; et al. Identification of Target Antigens of Antifibroblast Antibodies in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 1128–1134. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.J.; Holmes, A.M.; Deighton, J.; Long, L.; Yang, X.; Barker, L.; Walker, C.; Budd, D.C.; Upton, P.D.; Morrell, N.W. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: Role of proinflammatory cytokines. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L604–L615. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, S.Y.; Jiang, X.; Tamosiuniene, R.; Kim, D.; Guan, T.; Arsalane, S.; Pasupneti, S.; Voelkel, N.F.; Tang, Q.; et al. The Role of Regulatory T Cells in Pulmonary Arterial Hypertension. Front. Immunol. 2021, 12, 684657. [Google Scholar] [CrossRef] [PubMed]
- Golembeski, S.M.; West, J.; Tada, Y.; Fagan, K.A. Interleukin-6 Causes Mild Pulmonary Hypertension and Augments Hypoxia-Induced Pulmonary Hypertension in Mice. Chest 2005, 128, 572S–573S. [Google Scholar] [CrossRef]
- Savale, L.; Tu, L.; Rideau, D.; Izziki, M.; Maitre, B.; Adnot, S.; Eddahibi, S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 2009, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Arita, Y.; Sakata, Y.; Sudo, T.; Maeda, T.; Matsuoka, K.; Tamai, K.; Higuchi, K.; Shioyama, W.; Nakaoka, Y.; Kanakura, Y.; et al. The efficacy of tocilizumab in a patient with pulmonary arterial hypertension associated with Castleman’s disease. Heart Vessel. 2010, 25, 444–447. [Google Scholar] [CrossRef]
- Taniguchi, K.; Shimazaki, C.; Fujimoto, Y.; Shimura, K.; Uchiyama, H.; Matsumoto, Y.; Kuroda, J.; Horiike, S.; Taniwaki, M. Tocilizumab is effective for pulmonary hypertension associated with multicentric Castleman’s disease. Int. J. Hematol. 2009, 90, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, S.; Zapantis, E.; Zolty, R.; Efthimiou, P. A novel therapeutic approach in pulmonary arterial hypertension as a complication of adult-onset Still’s disease: Targeting IL-6. Int. J. Rheum. Dis. 2014, 17, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Toshner, M.; Church, C.; Harbaum, L.; Rhodes, C.; Moreschi, S.S.V.; Liley, J.; Jones, R.; Arora, A.; Batai, K.; Desai, A.A.; et al. Mendelian randomisation and experimental medicine approaches to interleukin-6 as a drug target in pulmonary arterial hypertension. Eur. Respir. J. 2022, 59, 2002463. [Google Scholar] [CrossRef]
- Lawrie, A.; Waterman, E.; Southwood, M.; Evans, D.; Suntharalingam, J.; Francis, S.; Crossman, D.; Croucher, P.; Morrell, N.; Newman, C. Evidence of a Role for Osteoprotegerin in the Pathogenesis of Pulmonary Arterial Hypertension. Am. J. Pathol. 2008, 172, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Tuder, R. Interleukin-1 Receptor Antagonist Inhibits Pulmonary Hypertension Induced by Inflammationa. Ann. N. Y. Acad. Sci. 1994, 725, 104–109. [Google Scholar] [CrossRef]
- Campos, M.; Schiopu, E. Pulmonary Arterial Hypertension in Adult-Onset Still’s Disease: Rapid Response to Anakinra. Case Rep. Rheumatol. 2012, 2012, 537613. [Google Scholar] [CrossRef] [Green Version]
- Trankle, C.R.; Canada, J.M.; Kadariya, D.; Markley, R.; De Chazal, H.M.; Pinson, J.; Fox, A.; Van Tassell, B.W.; Abbate, A.; Grinnan, D. IL-1 Blockade Reduces Inflammation in Pulmonary Arterial Hypertension and Right Ventricular Failure: A Single-Arm, Open-Label, Phase IB/II Pilot Study. Am. J. Respir. Crit. Care Med. 2019, 199, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Shannon, J.M.; Irvin, C.G.; Fagan, K.A.; Cool, C.; Augustin, A.; Mason, R.J.; Eurlings, I.M.J.; Dentener, M.A.; Mercken, E.M.; et al. Overexpression of tumor necrosis factor-α produces an increase in lung volumes and pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L39–L49. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Itoh, A.; Nishihira, J.; Makita, H.; Miyamoto, K.; Yamaguchi, E.; Nishimura, M. Effects of IL-1beta, TNF-alpha, and macrophage migration inhibitory factor on prostacyclin synthesis in rat pulmonary artery smooth muscle cells. Respirology 2003, 8, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W. TNF potentiates PAF-induced pulmonary vasoconstriction in the rat: Role of neutrophils and thromboxane A2. J. Appl. Physiol. 1994, 77, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zuo, X.-R.; Wang, Y.-Y.; Xie, W.-P.; Wang, H.; Zhang, M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-α antagonists via the suppression of TNF-α expression and NF-κB pathway in rats. Vasc. Pharmacol. 2013, 58, 71–77. [Google Scholar] [CrossRef]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef] [Green Version]
- Mutschler, D.; Wikström, G.; Lind, L.; Larsson, A.; Lagrange, A.; Eriksson, M. Etanercept Reduces Late Endotoxin-Induced Pulmonary Hypertension in the Pig. J. Interf. Cytokine Res. 2006, 26, 661–667. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Lu, J.; Li, M.-T.; Wang, Q.; Zeng, X.-F. Preventive and remedial application of etanercept attenuate monocrotaline-induced pulmonary arterial hypertension. Int. J. Rheum. Dis. 2019, 19, 192–198. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Sawada, H.; Mitani, Y.; Maruyama, J.; Jiang, B.H.; Ikeyama, Y.; Dida, F.A.; Yamamoto, H.; Imanaka-Yoshida, K.; Shimpo, H.; Mizoguchi, A.; et al. A Nuclear Factor-κB Inhibitor Pyrrolidine Dithiocarbamate Ameliorates Pulmonary Hypertension in Rats. Chest 2007, 132, 1265–1274. [Google Scholar] [CrossRef]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear Factor κ-B Is Activated in the Pulmonary Vessels of Patients with End-Stage Idiopathic Pulmonary Arterial Hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me Blocks the NF-κB Pathway by Direct Inhibition of IKKβ on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.H.; Huang, W.C.; Chang, W.T. Future Perspectives of Pulmonary Hypertension Treatment. Acta Cardiol. Sin. 2022, 38, 435–442. [Google Scholar] [CrossRef]
- Grzegorzewska, A.P.; Seta, F.; Han, R.; Czajka, C.A.; Makino, K.; Stawski, L.; Isenberg, J.S.; Browning, J.L.; Trojanowska, M. Dimethyl Fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci. Rep. 2017, 7, 41605. [Google Scholar] [CrossRef] [Green Version]
- Kong, K.; Koontz, D.; Morse, C.; Roth, E.; Domsic, R.T.; Simon, M.A.; Stratton, E.; Buchholz, C.; Tobin-Finch, K.; Simms, R.; et al. A pilot study of dimethyl fumarate in pulmonary arterial hypertension associated with systemic sclerosis. J. Scleroderma Relat. Disord. 2021, 6, 242–246. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Badesch, D.; Chung, L.; Domsic, R.T.; Medsger, T.; Pinckney, A.; Keyes-Elstein, L.; D’Aveta, C.; Spychala, M.; White, R.J.; et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis–associated Pulmonary Arterial Hypertension: A Multicenter, Double-Blind, Randomized, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 209–221. [Google Scholar] [CrossRef]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc. Med. 2020, 21, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Frantz, R.P.; Benza, R.L.; Channick, R.N.; Chin, K.; Howard, L.S.; McLaughlin, V.V.; Sitbon, O.; Zamanian, R.T.; Hemnes, A.R.; Cravets, M.; et al. TORREY, a Phase 2 study to evaluate the efficacy and safety of inhaled seralutinib for the treatment of pulmonary arterial hypertension. Pulm. Circ. 2021, 11, 20458940211057071. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Schermuly, R.T.; Ellinghaus, P.; Milting, H.; Riedl, B.; Nikolova, S.; Pullamsetti, S.S.; Weissmann, N.; Dony, E.; Savai, R.; et al. Combined Tyrosine and Serine/Threonine Kinase Inhibition by Sorafenib Prevents Progression of Experimental Pulmonary Hypertension and Myocardial Remodeling. Circulation 2008, 118, 2081–2090. [Google Scholar] [CrossRef]
- Kimura, G.; Kataoka, M.; Inami, T.; Fukuda, K.; Yoshino, H.; Satoh, T. Sorafenib as a potential strategy for refractory pulmonary arterial hypertension. Pulm. Pharmacol. Ther. 2017, 44, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Dhoble, S.; Patravale, V.; Weaver, E.; Lamprou, D.A.; Patravale, T. Comprehensive review on novel targets and emerging therapeutic modalities for pulmonary arterial Hypertension. Int. J. Pharm. 2022, 621, 121792. [Google Scholar] [CrossRef] [PubMed]
- Veeroju, S.; Kojonazarov, B.; Weiss, A.; Ghofrani, H.A.; Weissmann, N.; Grimminger, F.; Seeger, W.; Novoyatleva, T.; Schermuly, R.T. Therapeutic Potential of Regorafenib—A Multikinase Inhibitor in Pulmonary Hypertension. Int. J. Mol. Sci. 2021, 22, 1502. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Wang, Z.; Liu, W.; Deng, N.; Ding, Y.; Tang, L.; Hla, T.; Zeng, R.; Li, L.; et al. Regulation of PTEN by Rho small GTPases. Nature 2005, 7, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-Y.; Li, C.-Y.; Chen, J.; Pan, L.; Saito, S.; Terashita, T.; Saito, K.; Miyawaki, K.; Shigemoto, K.; Mominoki, K.; et al. Rho-ROCK Signal Pathway Regulates Microtubule-Based Process Formation of Cultured Podocytes—Inhibition of ROCK Promoted Process Elongation. Nephron Exp. Nephrol. 2004, 97, e49–e61. [Google Scholar] [CrossRef] [PubMed]
- Kosako, H.; Yoshida, T.; Matsumura, F.; Ishizaki, T.; Narumiya, S.; Inagaki, M. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene 2000, 19, 6059–6064. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Amano, M.; Nagata, K.-I.; Inagaki, N.; Nakamura, H.; Saya, H.; Kaibuchi, K.; Inagaki, M. Roles of Rho-associated Kinase in Cytokinesis; Mutations in Rho-associated Kinase Phosphorylation Sites Impair Cytokinetic Segregation of Glial Filaments. J. Cell Biol. 1998, 143, 1249–1258. [Google Scholar] [CrossRef] [Green Version]
- Riento, K.; Ridley, A.J. ROCKs: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Guilluy, C.; Eddahibi, S.; Agard, C.; Guignabert, C.; Izikki, M.; Tu, L.; Savale, L.; Humbert, M.; Fadel, E.; Adnot, S.; et al. RhoA and Rho Kinase Activation in Human Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2009, 179, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wu, S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm. Pharmacol. Ther. 2017, 46, 54–63. [Google Scholar] [CrossRef]
- Otani, N.; Tomoe, T.; Kawabe, A.; Sugiyama, T.; Horie, Y.; Sugimura, H.; Yasu, T.; Nakamoto, T. Recent Advances in the Treatment of Pulmonary Arterial Hypertension. Pharmaceuticals 2022, 15, 1277. [Google Scholar] [CrossRef]
- Fagan, K.A.; Oka, M.; Bauer, N.R.; Gebb, S.A.; Ivy, D.D.; Morris, K.G.; McMurtry, I.F. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am. J. Physiol. Cell. Mol. Physiol. 2004, 287, L656–L664. [Google Scholar] [CrossRef]
- Dahal, B.K.; Kosanovic, D.; Pamarthi, P.K.; Sydykov, A.; Lai, Y.-J.; Kast, R.; Schirok, H.; Stasch, J.-P.; Ghofrani, H.A.; Weissmann, N.; et al. Therapeutic efficacy of azaindole-1 in experimental pulmonary hypertension. Eur. Respir. J. 2010, 36, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, J.S.; Badejo, A.M.; Casey, D.B.; Murthy, S.N.; Kadowitz, P.J. Analysis of Pulmonary Vasodilator Responses to SB-772077-B [4-(7-((3-Amino-1-pyrrolidinyl)carbonyl)-1-ethyl-1H-imidazo(4,5-c)pyridin-2-yl)-1,2,5-oxadiazol-3-amine], a Novel Aminofurazan-Based Rho Kinase Inhibitor. Experiment 2009, 330, 334–341. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Matoba, T.; Ito, A.; Takata-Tanaka, H.; Kishi, T.; Hayashidani, S.; Abe, K.; Takeshita, A.; Shimokawa, H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 2005, 91, 391–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikura, K.; Yamada, N.; Ito, M.; Ota, S.; Nakamura, M.; Isaka, N.; Nakano, T. Beneficial Acute Effects of Rho-Kinase Inhibitor in Patients With Pulmonary Arterial Hypertension. Circ. J. 2006, 70, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Fukumoto, Y.; Saji, K.; Sugimura, K.; Demachi, J.; Nawata, J.; Shimokawa, H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessel. 2010, 25, 144–149. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Y.-F.; Zhao, Q.-H.; Jiang, R.; Wu, Y.; Peng, F.-H.; Xu, X.-Q.; Wang, L.; He, J.; Jing, Z.-C. Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: A randomized, controlled, crossover study. Int. J. Cardiol. 2014, 177, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Yamada, N.; Matsubara, H.; Mizoguchi, M.; Uchino, K.; Yao, A.; Kihara, Y.; Kawano, M.; Watanabe, H.; Takeda, Y.; et al. Double-Blind, Placebo-Controlled Clinical Trial With a Rho-Kinase Inhibitor in Pulmonary Arterial Hypertension. Circ. J. 2013, 77, 2619–2625. [Google Scholar] [CrossRef] [Green Version]
- Ranchoux, B.; Meloche, J.; Paulin, R.; Boucherat, O.; Provencher, S.; Bonnet, S. DNA Damage and Pulmonary Hypertension. Int. J. Mol. Sci. 2016, 17, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, L.; Sidhu, V.K.; Fang, Y.-H.; Ryan, J.J.; Parikh, K.S.; Hong, Z.; Toth, P.; Morrow, E.; Kutty, S.; Lopaschuk, G.D.; et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: Therapeutic benefits of dichloroacetate. J. Mol. Med. 2013, 91, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.-C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a Metabolic Modulator, Prevents and Reverses Chronic Hypoxic Pulmonary Hypertension in Rats. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.Y.; Hindmarch, C.C.T.; et al. Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [Green Version]
- Budas, G.R.; Boehm, M.; Kojonazarov, B.; Viswanathan, G.; Tian, X.; Veeroju, S.; Novoyatleva, T.; Grimminger, F.; Hinojosa-Kirschenbaum, F.; Ghofrani, H.A.; et al. ASK1 Inhibition Halts Disease Progression in Preclinical Models of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 373–385. [Google Scholar] [CrossRef]
- Boucherat, O.; Provencher, S.; Bonnet, S. Therapeutic Value of ASK1 Inhibition in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Feldman, J.; McLaughlin, V.V.; Rischard, F.; Lange, T.J.; White, R.J.; Peacock, A.J.; Gerhardt, F.; Ebrahimi, R.; Brooks, G.; et al. Selonsertib in adults with pulmonary arterial hypertension (ARROW): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2022, 10, 35–46. [Google Scholar] [CrossRef]
- Gong, M.; Fragakis, N.; Zhang, C.; Zhang, Z.; Li, G.; Liu, T. Ranolazine as a novel therapy for pulmonary arterial hypertension. Int. J. Cardiol. 2016, 223, 860–862. [Google Scholar] [CrossRef]
- Rocchetti, M.; Sala, L.; Rizzetto, R.; Staszewsky, L.I.; Alemanni, M.; Zambelli, V.; Russo, I.; Barile, L.; Cornaghi, L.; Altomare, C.; et al. Ranolazine prevents INaL enhancement and blunts myocardial remodelling in a model of pulmonary hypertension. Cardiovasc. Res. 2014, 104, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Liles, J.T.; Hoyer, K.; Oliver, J.; Chi, L.; Dhalla, A.K.; Belardinelli, L. Ranolazine Reduces Remodeling of the Right Ventricle and Provoked Arrhythmias in Rats with Pulmonary Hypertension. Experiment 2015, 353, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Tocchetti, C.G.; Carpi, A.; Coppola, C.; Quintavalle, C.; Rea, D.; Campesan, M.; Arcari, A.; Piscopo, G.; Cipresso, C.; Monti, M.G.; et al. Ranolazine protects from doxorubicin-induced oxidative stress and cardiac dysfunction. Eur. J. Heart Fail. 2014, 16, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Schilz, R.; Mediratta, A.; Addetia, K.; Coslet, S.; Thomeas, V.; Gillies, H.; Oudiz, R.J. Phase I Safety Study of Ranolazine in Pulmonary Arterial Hypertension. Pulm. Circ. 2015, 5, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.S.; Cuttica, M.J.; Beussink-Nelson, L.; Kozyleva, A.; Sanchez, C.; Mkrdichian, H.; Selvaraj, S.; Dematte, J.E.; Lee, D.C.; Shah, S.J. Effects of Ranolazine on Exercise Capacity, Right Ventricular Indices, and Hemodynamic Characteristics in Pulmonary Arterial Hypertension: A Pilot Study. Pulm. Circ. 2015, 5, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Forfia, P.R.; Vaidya, A.; Mazurek, J.A.; Park, M.H.; Ramani, G.; Chan, S.Y.; Waxman, A.B. Rationale and design of the ranolazine PH–RV study: A multicentred randomised and placebo-controlled study of ranolazine to improve RV function in patients with non-group 2 pulmonary hypertension. Open Heart 2018, 5, e000736. [Google Scholar] [CrossRef] [Green Version]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.B.; Michelakis, E.D. Fatty Acid Oxidation and Malonyl-CoA Decarboxylase in the Vascular Remodeling of Pulmonary Hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef]
- Leopold, J.A.; Maron, B.A. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2016, 17, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Aldred, M.A. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes 2020, 11, 1224. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, Y.-H.; Gou, X.; Li, F.-Y.; Yang, X.-Y.; Li, Y.-M.; Chen, F. Oxidative Stress and Antioxidative Therapy in Pulmonary Arterial Hypertension. Molecules 2022, 27, 3724. [Google Scholar] [CrossRef]
- Xue, C.; Senchanthisai, S.; Sowden, M.; Pang, J.; White, R.J.; Berk, B.C. Endothelial-to-Mesenchymal Transition and Inflammation Play Key Roles in Cyclophilin A–Induced Pulmonary Arterial Hypertension. Hypertension 2020, 76, 1113–1123. [Google Scholar] [CrossRef]
- Xu, T.; Shao, L.; Wang, A.; Liang, R.; Lin, Y.; Wang, G.; Zhao, Y.; Hu, J.; Liu, S. Retracted: CD248 as a novel therapeutic target in pulmonary arterial hypertension. Clin. Transl. Med. 2020, 10, e175. [Google Scholar] [CrossRef] [PubMed]
- Weise-Cross, L.; Resta, T.C.; Jernigan, N.L. Redox Regulation of Ion Channels and Receptors in Pulmonary Hypertension. Antioxid. Redox Signal. 2019, 31, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Wang, R.; Feng, P.; Dong, Q.; Chen, W.; Zhao, Y.; Li, A.; Li, H.; Chen, J.; Huang, W. Dihydroartemisinin Attenuates Pulmonary Hypertension Through Inhibition of Pulmonary Vascular Remodeling in Rats. J. Cardiovasc. Pharmacol. 2020, 76, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, J.; Dong, Y.; Xu, M.; Xu, L.; Guan, H.; Xia, X.; Wang, L. Anti-hypoxic effect of dihydroartemisinin on pulmonary artery endothelial cells. Biochem. Biophys. Res. Commun. 2018, 506, 840–846. [Google Scholar] [CrossRef]
- Li, Y.; Cai, H.; Wei, J.; Zhu, L.; Yao, Y.; Xie, M.; Song, L.; Zhang, C.; Huang, X.; Wang, L. Dihydroartemisinin Attenuates Hypoxic Pulmonary Hypertension via the Downregulation of miR-335 Targeting Vangl2. DNA Cell Biol. 2022, 41, 750–767. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yamaguchi, K.; Shimada, S.; Kitamura, Y.; Ohnishi, H. Antithrombotic activity and the mechanism of action of trapidil (Rocornal®). Prostaglandins Leukot. Med. 1982, 9, 685–695. [Google Scholar] [CrossRef]
- Türck, P.; Lacerda, D.S.; Carraro, C.C.; de Lima-Seolin, B.G.; Teixeira, R.B.; Bonetto, J.H.P.; Colombo, R.; Schenkel, P.C.; Belló-Klein, A.; Araujo, A.S.D.R. Trapidil improves hemodynamic, echocardiographic and redox state parameters of right ventricle in monocrotaline-induced pulmonary arterial hypertension model. Biomed. Pharmacother. 2018, 103, 182–190. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; George, P.; Gladwin, M.T. Nitrite in pulmonary arterial hypertension: Therapeutic avenues in the setting of dysregulated arginine/nitric oxide synthase signalling. Cardiovasc. Res. 2011, 89, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.A.; Vanderpool, R.R.; Nouraie, M.; Bachman, T.N.; White, P.M.; Sugahara, M.; Gorcsan, J.; Parsley, E.L.; Gladwin, M.T. Acute hemodynamic effects of inhaled sodium nitrite in pulmonary hypertension associated with heart failure with preserved ejection fraction. J. Clin. Investig. 2016, 1, e89620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, J.; Farha, S.; Park, M.M.; Comhair, S.A.; Lundgrin, E.L.; Tang, W.W.; Bongard, R.D.; Merker, M.P.; Erzurum, S.C. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol. 2014, 2, 884–891. [Google Scholar] [CrossRef] [Green Version]
- Hoshikawa, Y.; Ono, S.; Suzuki, S.; Tanita, T.; Chida, M.; Song, C.; Noda, M.; Tabata, T.; Voelkel, N.F.; Fujimura, S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Physiol. 2001, 90, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Voelkel, M.A.; Wynne, K.M.; Badesch, D.B.; Groves, B.M.; Voelkel, N.F. Hyperuricemia in Severe Pulmonary Hypertension. Chest 2000, 117, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Bendayan, D.; Shitrit, D.; Ygla, M.; Huerta, M.; Fink, G.; Kramer, M. Hyperuricemia as a prognostic factor in pulmonary arterial hypertension. Respir. Med. 2003, 97, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Spiekermann, S.; Schenk, K.; Hoeper, M.M. Increased xanthine oxidase activity in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2009, 34, 276. [Google Scholar] [CrossRef]
- Tan, W.; Madhavan, K.; Hunter, K.S.; Park, D.; Stenmark, K.R. Vascular Stiffening in Pulmonary Hypertension: Cause or Consequence? (2013 Grover Conference Series). Pulm. Circ. 2014, 4, 560–580. [Google Scholar] [CrossRef] [Green Version]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Poiani, G.J.; Tozzi, C.A.; Yohn, S.E.; Pierce, R.A.; Belsky, S.A.; Berg, R.A.; Yu, S.Y.; Deak, S.B.; Riley, D.J. Collagen and elastin metabolism in hypertensive pulmonary arteries of rats. Circ. Res. 1990, 66, 968–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.L.; Rabinovitch, M. Tenascin-C Is Induced With Progressive Pulmonary Vascular Disease in Rats and Is Functionally Related to Increased Smooth Muscle Cell Proliferation. Circ. Res. 1996, 79, 1131–1142. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Haghighat, L.; Spiekerkoetter, E.; Sawada, H.; Alvira, C.M.; Wang, L.; Acharya, S.; Rodriguez-Colon, G.; Orton, A.; Zhao, M.; et al. Neutrophil Elastase Is Produced by Pulmonary Artery Smooth Muscle Cells and Is Linked to Neointimal Lesions. Am. J. Pathol. 2011, 179, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Rabinovitch, M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular matrix-bound basic fibroblast growth factor. J. Cell. Physiol. 1996, 166, 495–505. [Google Scholar] [CrossRef]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.-Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [Green Version]
- Vieillard-Baron, A.; Frisdal, E.; Raffestin, B.; Baker, A.H.; Eddahibi, S.; Adnot, S.; D’Ortho, M.-P. Inhibition of Matrix Metalloproteinases by Lung TIMP-1 Gene Transfer Limits Monocrotaline-Induced Pulmonary Vascular Remodeling in Rats. Hum. Gene Ther. 2003, 14, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Mawatari, E.; Hongo, M.; Sakai, A.; Terasawa, F.; Takahashi, M.; Yazaki, Y.; Kinoshita, O.; Ikeda, U. Amlodipine prevents monocrotaline-induced pulmonary arterial hypertension and prolongs survival in rats independent of blood pressure lowering. Clin. Exp. Pharmacol. Physiol. 2007, 34, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.S.; Kim, K.C.; Hong, Y.M. Gene Expressions of Nitric Oxide Synthase and Matrix Metalloproteinase-2 in Monocrotaline-Induced Pulmonary Hypertension in Rats After Bosentan Treatment. Korean Circ. J. 2011, 41, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.T. Matrix Metalloproteinase Inhibitor Development and the Remodeling of Drug Discovery. Heart Fail. Rev. 2004, 9, 63–79. [Google Scholar] [CrossRef]
- Cowan, K.; Heilbut, A.; Humpl, T.; Lam, C.; Ito, S.; Rabinovitch, M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat. Med. 2000, 6, 698–702. [Google Scholar] [CrossRef]
- Cowan, K.N.; Jones, P.L.; Rabinovitch, M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J. Clin. Investig. 2000, 105, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Ilkiw, R.; Todorovich-Hunter, L.; Maruyama, K.; Shin, J.; Rabinovitch, M. SC-39026, a serine elastase inhibitor, prevents muscularization of peripheral arteries, suggesting a mechanism of monocrotaline-induced pulmonary hypertension in rats. Circ. Res. 1989, 64, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Molteni, A.; Ward, W.F.; Ts’Ao, C.-H.; Hinz, J.M. Monocrotaline-induced cardiopulmonary injury in rats. Biochem. Pharmacol. 1989, 38, 2411–2419. [Google Scholar] [CrossRef]
- Zaidi, S.H.; You, X.-M.; Ciura, S.; Husain, M.; Rabinovitch, M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 2002, 105, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Nickel, N.P.; Spiekerkoetter, E.; Gu, M.; Li, C.G.; Li, H.; Kaschwich, M.; Diebold, I.; Hennigs, J.K.; Kim, K.-Y.; Miyagawa, K.; et al. Elafin Reverses Pulmonary Hypertension via Caveolin-1–Dependent Bone Morphogenetic Protein Signaling. Am. J. Respir. Crit. Care Med. 2015, 191, 1273–1286. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, A.J.; Miyagawa, K.; Rhodes, C.J.; Taylor, S.; Del Rosario, P.A.; Hsi, A.; Haddad, F.; Spiekerkoetter, E.; Bental-Roof, M.; Bland, R.D.; et al. Severe Pulmonary Arterial Hypertension Is Characterized by Increased Neutrophil Elastase and Relative Elafin Deficiency. Chest 2021, 160, 1442–1458. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Niswender, K.D.; Johnson, J.A.; Pugh, M.E.; Gleaves, L.; Fessel, J.P.; Hemnes, A.R. A potential role for insulin resistance in experimental pulmonary hypertension. Eur. Respir. J. 2013, 41, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2008, 33, 318–324. [Google Scholar] [CrossRef]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.-H. Activation of the AMP-Activated Kinase by Antidiabetes Drug Metformin Stimulates Nitric Oxide Synthesis In Vivo by Promoting the Association of Heat Shock Protein 90 and Endothelial Nitric Oxide Synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Awad, K.S.; West, J.D.; Perez, V.; MacLean, M. Novel Signaling Pathways in Pulmonary Arterial Hypertension (2015 Grover Conference Series). Pulm. Circ. 2016, 6, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Agard, C.; Rolli-Derkinderen, M.; Dumas-De-La-Roque, E.; Rio, M.; Sagan, C.; Savineau, J.; Loirand, G.; Pacaud, P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br. J. Pharmacol. 2009, 158, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Brittain, E.L.; Niswender, K.; Agrawal, V.; Chen, X.; Fan, R.; Pugh, M.E.; Rice, T.W.; Robbins, I.M.; Song, H.; Thompson, C.; et al. Mechanistic Phase II Clinical Trial of Metformin in Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2020, 9, e018349. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Sutliff, R.L.; Kang, B.-Y.; Hart, C.M. PPARγ as a potential therapeutic target in pulmonary hypertension. Ther. Adv. Respir. Dis. 2010, 4, 143–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Boudina, S.; Banke, N.H.; Sambandam, N.; Han, X.; Young, D.M.; Leone, T.C.; Gross, R.W.; Lewandowski, E.D.; Abel, E.D.; et al. The transcriptional coactivator PGC-1α is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am. J. Physiol. Circ. Physiol. 2008, 295, H185–H196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, G.; Han, D.; Zhang, Y.; Xu, J.; Lu, J.; Li, S.; Xie, X.; Liu, L.; Dong, L.; et al. Activation of PPAR-γ ameliorates pulmonary arterial hypertension via inducing heme oxygenase-1 and p21WAF1: An in vivo study in rats. Life Sci. 2014, 98, 39–43. [Google Scholar] [CrossRef]
- Xie, X.; Wang, G.; Zhang, D.; Zhang, Y.; Zhu, Y.; Li, F.; Li, S.; Li, M. Activation of peroxisome proliferator-activated receptor γ ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. Biomed. Rep. 2015, 3, 537–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, E.L.; Bogaard, H.J.; Noordegraaf, A.V.; de Man, F.S. Neurohormonal modulation in pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2004633. [Google Scholar] [CrossRef]
- Maron, B.A.; Leopold, J.A.; Hemnes, A.R. Metabolic syndrome, neurohumoral modulation, and pulmonary arterial hypertension. Br. J. Pharmacol. 2020, 177, 1457–1471. [Google Scholar] [CrossRef] [Green Version]
- Bristow, M.R.; Minobe, W.; Rasmussen, R.; Larrabee, P.; Skerl, L.; Klein, J.W.; Anderson, F.L.; Murray, J.; Mestroni, L.; Karwande, S.V. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J. Clin. Investig. 1992, 89, 803–815. [Google Scholar] [CrossRef]
- Rain, S.; Andersen, S.; Najafi, A.; Schultz, J.G.; Bós, D.D.S.G.; Handoko, M.L.; Bogaard, H.-J.; Vonk-Noordegraaf, A.; Andersen, A.; van der Velden, J.; et al. Right Ventricular Myocardial Stiffness in Experimental Pulmonary Arterial Hypertension. Circ. Heart Fail. 2016, 9, e002636. [Google Scholar] [CrossRef]
- Maron, B.A.; Leopold, J.A. Emerging Concepts in the Molecular Basis of Pulmonary Arterial Hypertension. Circulation 2015, 131, 2079–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Lunar, I.; Pereda, D.; Ibanez, B.; García-Álvarez, A. Neurohormonal Modulation as a Therapeutic Target in Pulmonary Hypertension. Cells 2020, 9, 2521. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638. [Google Scholar] [CrossRef]
- Sandoval, J.; Del Valle-Mondragón, L.; Masso, F.; Zayas, N.; Pulido, T.; Teijeiro, R.; Gonzalez-Pacheco, H.; Olmedo-Ocampo, R.; Sisniega, C.; Paez-Arenas, A.; et al. Angiotensin converting enzyme 2 and angiotensin (1–7) axis in pulmonary arterial hypertension. Eur. Respir. J. 2020, 56, 1902416. [Google Scholar] [CrossRef]
- Perros, F.; Man, F.H.-D.; Bogaard, H.J.; Antigny, F.; Simonneau, G.; Bonnet, S.; Provencher, S.; Galiè, N.; Humbert, M. Use of β-Blockers in Pulmonary Hypertension. Circ. Heart Fail. 2017, 10, e003703. [Google Scholar] [CrossRef]
- Badagliacca, R.; Mercurio, V.; Romeo, E.; Correale, M.; Masarone, D.; Papa, S.; Tocchetti, C.; Agostoni, P. Beta-blockers in pulmonary arterial hypertension: Time for a second thought? Vasc. Pharmacol. 2022, 144, 106974. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Bajaj, N.S.; Zein, J.; Minai, O.A.; Dweik, R.A. Outcomes of β-blocker use in pulmonary arterial hypertension: A propensity-matched analysis. Eur. Respir. J. 2015, 46, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Grinnan, D.; Bogaard, H.-J.; Grizzard, J.; Van Tassell, B.; Abbate, A.; DeWilde, C.; Priday, A.; Voelkel, N.F. Treatment of Group I Pulmonary Arterial Hypertension with Carvedilol Is Safe. Am. J. Respir. Crit. Care Med. 2014, 189, 1562–1564. [Google Scholar] [CrossRef] [PubMed]
- De Man, F.S.; Handoko, M.L.; van Ballegoij, J.J.; Schalij, I.; Bogaards, S.J.; Postmus, P.E.; van der Velden, J.; Westerhof, N.; Paulus, W.J.; Vonk-Noordegraaf, A. Bisoprolol Delays Progression Towards Right Heart Failure in Experimental Pulmonary Hypertension. Circ. Heart Fail. 2012, 5, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Van Campen, J.S.; de Boer, K.; van de Veerdonk, M.C.; van der Bruggen, C.E.; Allaart, C.P.; Raijmakers, P.G.; Heymans, M.W.; Marcus, J.T.; Harms, H.J.; Handoko, M.L.; et al. Bisoprolol in idiopathic pulmonary arterial hypertension: An explorative study. Eur. Respir. J. 2016, 48, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.A.; Hanrott, K.; Budd, D.C.; Torres, F.; Grünig, E.; Escribano-Subias, P.; Meseguer, M.L.; Halank, M.; Opitz, C.; Hall, D.A.; et al. An open-label, dose-escalation study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of GSK2586881 in participants with pulmonary arterial hypertension. Pulm. Circ. 2022, 12, 816–825. [Google Scholar] [CrossRef]
- Maron, B.A.; Leopold, J.A. The Role of the Renin-Angiotensin-Aldosterone System in the Pathobiology of Pulmonary Arterial Hypertension (2013 Grover Conference Series). Pulm. Circ. 2014, 4, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, T.; Shelley, P.; Patel, D.; Vorla, M.; Kalra, D.K. Strategizing Drug Therapies in Pulmonary Hypertension for Improved Outcomes. Pharmaceuticals 2022, 15, 1242. [Google Scholar] [CrossRef] [PubMed]
- Safdar, Z.; Cho, E. Effect of spironolactone use in pulmonary arterial hypertension—Analysis from pivotal trial databases. Pulm. Circ. 2021, 11, 20458940211045618. [Google Scholar] [CrossRef] [PubMed]
- Favre, J.; Gao, J.; Di Zhang, A.; Remy-Jouet, I.; Ouvrard-Pascaud, A.; Dautreaux, B.; Escoubet, B.; Thuillez, C.; Jaisser, F.; Richard, V. Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. Am. J. Physiol. Circ. Physiol. 2011, 300, H2035–H2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, I.R.; Sagliani, K.D.; Warburton, R.R.; Hill, N.S.; Fanburg, B.L.; Jaffe, I.Z.; Penumatsa, K.C.; Toksoz, D.; Kharnaf, M.; Kapur, N.K.; et al. Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L678–L688. [Google Scholar] [CrossRef] [Green Version]
- Soderman, C.; Eriksson, L.S.; Juhlin-Dannfelt, A.; Lundberg, J.M.; Broman, L.; Holmgren, A. Effect of vasoactive intestinal polypeptide (VIP) on pulmonary ventilation-perfusion relationships and central haemodynamics in healthy subjects. Clin. Physiol. Funct. Imaging 1993, 13, 677–685. [Google Scholar] [CrossRef]
- Busto, R.; Prieto, J.C.; Bodega, G.; Zapatero, J.; Carrero, I. Immunohistochemical localization and distribution of VIP/PACAP receptors in human lung☆. Peptides 2000, 21, 265–269. [Google Scholar] [CrossRef]
- Petkov, V.; Mosgoeller, W.; Ziesche, R.; Raderer, M.; Stiebellehner, L.; Vonbank, K.; Funk, G.-C.; Hamilton, G.; Novotny, C.; Burian, B.; et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J. Clin. Investig. 2003, 111, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuchte, H.H.; Baezner, C.; Baumgartner, R.A.; Bevec, D.; Bacher, G.; Neurohr, C.; Behr, J. Inhalation of vasoactive intestinal peptide in pulmonary hypertension. Eur. Respir. J. 2008, 32, 1289–1294. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, H.M.; Denning, J.; Wring, S.; Palacios, M.; Hoffman, S.; Crizer, K.; Kamau-Kelley, W.; Symonds, W.; Feldman, J. A trial design to maximize knowledge of the effects of rodatristat ethyl in the treatment of pulmonary arterial hypertension (ELEVATE 2). Pulm. Circ. 2022, 12, e12088. [Google Scholar] [CrossRef]
- Lazarus, H.; Denning, J.; Kamau-Kelley, W.; Wring, S.; Palacios, M.; Humbert, M. ELEVATE 2: A multicenter study of rodatristat ethyl in patients with WHO Group 1 pulmonary arterial hypertension (PAH). Eur. Respir. J. 2021, 58, PA1919. [Google Scholar] [CrossRef]
- Lahm, T.; Crisostomo, P.R.; Markel, T.A.; Wang, M.; Wang, Y.; Weil, B.; Meldrum, D.R. Exogenous estrogen rapidly attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction. Shock 2008, 30, 660–667. [Google Scholar] [CrossRef]
- Liu, A.; Philip, J.; Vinnakota, K.C.; Bergh, F.V.D.; Tabima, D.M.; Hacker, T.; Beard, D.A.; Chesler, N.C. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol. Rep. 2017, 5, e13157. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Baird, G.L.; Barr, R.G.; Bluemke, D.A.; Fritz, J.S.; Hill, N.S.; Klinger, J.R.; Lima, J.A.C.; Ouyang, P.; Palevsky, H.I.; et al. Higher Estradiol and Lower Dehydroepiandrosterone-Sulfate Levels Are Associated with Pulmonary Arterial Hypertension in Men. Am. J. Respir. Crit. Care Med. 2016, 193, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lin, F.; Xiao, Z.; Sun, B.; Wei, Z.; Liu, B.; Xue, L.; Xiong, C. Investigational pharmacotherapy and immunotherapy of pulmonary arterial hypertension: An update. Biomed. Pharmacother. 2020, 129, 110355. [Google Scholar] [CrossRef]
- Kawut, S.M.; Archer-Chicko, C.L.; DeMichele, A.; Fritz, J.S.; Klinger, J.R.; Ky, B.; Palevsky, H.I.; Palmisciano, A.J.; Patel, M.; Pinder, D.; et al. Anastrozole in Pulmonary Arterial Hypertension. A Randomized, Double-Blind, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2016, 195, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Austin, E.D.; Talati, M.; Fessel, J.P.; Farber-Eger, E.H.; Brittain, E.L.; Hemnes, A.R.; Loyd, J.E.; West, J. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur. Respir. J. 2017, 50, 1602337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Roque, E.D.; Savineau, J.-P.; Metivier, A.-C.; Billes, M.-A.; Kraemer, J.-P.; Doutreleau, S.; Jougon, J.; Marthan, R.; Moore, N.; Fayon, M.; et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): A pilot study. Ann. D’Endocrinol. 2012, 73, 20–25. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.R.; Wharton, J.; Wilkins, M.R. Iron Deficiency and Raised Hepcidin in Idiopathic Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.G.; Balanos, G.M.; Croft, Q.P.P.; Talbot, N.P.; Dorrington, K.L.; Ratcliffe, P.J.; Robbins, P.A. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J. Physiol. 2008, 586, 5999–6005. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Dorrington, K.L.; Maxwell, P.; Robbins, P. Effects of desferrioxamine on serum erythropoietin and ventilatory sensitivity to hypoxia in humans. J. Appl. Physiol. 2000, 89, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron Homeostasis and Pulmonary Hypertension. Circ. Res. 2015, 116, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Wunderer, F.; Traeger, L.; Sigurslid, H.H.; Meybohm, P.; Bloch, D.B.; Malhotra, R. The role of hepcidin and iron homeostasis in atherosclerosis. Pharmacol. Res. 2020, 153, 104664. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L.; Pedersen, S.L.; Toe, Q.K.; Quinlan, G.J.; Wort, S.J. Pulmonary Arterial Hypertension: Iron Matters. Front. Physiol. 2018, 9, 641. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.-X.; Qiu, B.-Q.; Lai, S.-Q.; Zhou, X.-L.; Gong, C.-W.; Wang, L.-J.; Yuan, M.-M.; He, A.-D.; Liu, J.-C.; Huang, H. Iron Metabolism and Idiopathic Pulmonary Arterial Hypertension: New Insights from Bioinformatic Analysis. BioMed Res. Int. 2021, 2021, 5669412. [Google Scholar] [CrossRef] [PubMed]
- Howard, L.S.G.E.; He, J.; Watson, G.M.J.; Huang, L.; Wharton, J.; Luo, Q.; Kiely, D.G.; Condliffe, R.; Pepke-Zaba, J.; Morrell, N.W.; et al. Supplementation with Iron in Pulmonary Arterial Hypertension. Two Randomized Crossover Trials. Ann. Am. Thorac. Soc. 2021, 18, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Radisavljevic, Z.; Shroyer, K.R.; Polak, J.M.; Voelkel, N.F. Monoclonal Endothelial Cells in Appetite Suppressant–associated Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 1999–2001. [Google Scholar] [CrossRef]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, È.; et al. Role for DNA Damage Signaling in Pulmonary Arterial Hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Drake, K.M.; Rigelsky, C.M.; McNelly, L.N.; Meade, S.L.; Comhair, S.A.A.; Erzurum, S.C.; Aldred, M.A. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Perez, V.A.D.J. In Defense of the Nucleus: NUDT1 and Oxidative DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2021, 203, 541–542. [Google Scholar] [CrossRef]
- Feng, W.; Wang, J.; Yan, X.; Zhai, C.; Shi, W.; Wang, Q.; Zhang, Q.; Li, M. Paclitaxel alleviates monocrotaline-induced pulmonary arterial hypertension via inhibition of FoxO1-mediated autophagy. Naunyn-Schmiedebergs Arch. Pharmacol. 2019, 392, 605–613. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- Voorburg, J.A.; Cats, V.M.; Buis, B.; Bruschke, A.V. Balloon Angioplasty in the Treatment of Pulmonary Hypertension Caused by Pulmonary Embolism. Chest 1988, 94, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, J.; Goldhaber, S.Z.; Lock, J.E.; Ferndandes, S.M.; Landzberg, M.J. Balloon Pulmonary Angioplasty for Treatment of Chronic Thromboembolic Pulmonary Hypertension. Circulation 2001, 103, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Zhao, Z.-H.; Luo, Q.; Zhao, Q.; Yan, L.; Zhang, Y.; Li, X.; Yang, T.; Zeng, Q.-X.; Xiong, C.-M.; et al. Balloon pulmonary angioplasty for chronic thromboembolic pulmonary hypertension: State of the art. World J. Clin. Cases 2020, 8, 2679–2702. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Miyagawa, K.; Nakayama, K.; Kinutani, H.; Shinke, T.; Okada, K.; Okita, Y.; Hirata, K.-I.; Emoto, N. Balloon pulmonary angioplasty: An additional treatment option to improve the prognosis of patients with chronic thromboembolic pulmonary hypertension. Eurointervention 2014, 10, 518–525. [Google Scholar] [CrossRef]
- Brenot, P.; Jaïs, X.; Taniguchi, Y.; Alonso, C.G.; Gerardin, B.; Mussot, S.; Mercier, O.; Fabre, D.; Parent, F.; Jevnikar, M.; et al. French experience of balloon pulmonary angioplasty for chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1802095. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, A.; Satoh, T.; Fukuda, T.; Sugimura, K.; Fukumoto, Y.; Emoto, N.; Yamada, N.; Yao, A.; Ando, M.; Ogino, H.; et al. Balloon Pulmonary Angioplasty for Chronic Thromboembolic Pulmonary Hypertension. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e004029. [Google Scholar] [CrossRef]
- Magon, W.; Stepniewski, J.; Jonas, K.; Waligora, M.; Podolec, P.; Kopec, G. P4679Changes in systemic inflammation and endothelial dysfunction after balloon pulmonary angioplasty. Eur. Heart J. 2019, 40 (Suppl. S1), ehz745-1061. [Google Scholar] [CrossRef]
- Cannon, J.E.; Su, L.; Kiely, D.G.; Page, K.; Toshner, M.; Swietlik, E.; Treacy, C.; Ponnaberanam, A.; Condliffe, R.; Sheares, K.; et al. Dynamic Risk Stratification of Patient Long-Term Outcome After Pulmonary Endarterectomy. Circulation 2016, 133, 1761–1771. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Ogawa, A.; Miyaji, K.; Mizoguchi, H.; Shimokawahara, H.; Naito, T.; Oka, T.; Yunoki, K.; Munemasa, M.; Matsubara, H. Novel Angiographic Classification of Each Vascular Lesion in Chronic Thromboembolic Pulmonary Hypertension Based on Selective Angiogram and Results of Balloon Pulmonary Angioplasty. Circ. Cardiovasc. Interv. 2016, 9, e003318. [Google Scholar] [CrossRef]
- Martín, M.V.; Melón, N.M.; González-Trevilla, A.A.; Cebada, F.S.; Nieto, S.H.; Cruz-Utrilla, A.; Hinojosa, W.; López-Gude, M.J.; Charterina, S.A.; Ostolaza, Y.R.; et al. Balloon pulmonary angioplasty can be an effective and safe therapeutic option in non-surgical elderly patients. Front. Cardiovasc. Med. 2022, 9, 1001518. [Google Scholar] [CrossRef]
- Ciarka, A.; Doan, V.; Velez-Roa, S.; Naeije, R.; van de Borne, P. Prognostic Significance of Sympathetic Nervous System Activation in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2010, 181, 1269–1275. [Google Scholar] [CrossRef]
- Wensel, R.; Jilek, C.; Dorr, M.; Francis, D.P.; Stadler, H.; Lange, T.; Blumberg, F.; Opitz, C.; Pfeifer, M.; Ewert, R. Impaired cardiac autonomic control relates to disease severity in pulmonary hypertension. Eur. Respir. J. 2009, 34, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Velez-Roa, S.; Ciarka, A.; Najem, B.; Vachiery, J.-L.; Naeije, R.; van de Borne, P. Increased Sympathetic Nerve Activity in Pulmonary Artery Hypertension. Circulation 2004, 110, 1308–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perros, F.; Ranchoux, B.; Izikki, M.; Bentebbal, S.; Happé, C.; Antigny, F.; Jourdon, P.; Dorfmüller, P.; Lecerf, F.; Fadel, E.; et al. Nebivolol for Improving Endothelial Dysfunction, Pulmonary Vascular Remodeling, and Right Heart Function in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2015, 65, 668–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhang, J.; Jiang, X.-M.; Xie, D.-J.; Wang, J.-S.; Li, L.; Li, B.; Wang, Z.-M.; Rothman, A.M.; Lawrie, A.; et al. Pulmonary Artery Denervation Attenuates Pulmonary Arterial Remodeling in Dogs With Pulmonary Arterial Hypertension Induced by Dehydrogenized Monocrotaline. JACC Cardiovasc. Interv. 2015, 8, 2013–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-L.; Zhang, Y.-J.; Zhou, L.; Xie, D.-J.; Zhang, F.-F.; Du-Jiang, X.; Wong, S.S.; Kwan, T.W. Percutaneous pulmonary artery denervation completely abolishes experimental pulmonary arterial hypertension in vivo. Eurointervention 2013, 9, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Rothman, A.M.; Arnold, N.D.; Chang, W.; Watson, O.; Swift, A.J.; Condliffe, R.; Elliot, C.A.; Kiely, D.G.; Suvarna, S.K.; Gunn, J.; et al. Pulmonary Artery Denervation Reduces Pulmonary Artery Pressure and Induces Histological Changes in an Acute Porcine Model of Pulmonary Hypertension. Circ. Cardiovasc. Interv. 2015, 8, e002569. [Google Scholar] [CrossRef]
- Chen, S.-L.; Zhang, F.-F.; Xu, J.; Xie, D.-J.; Zhou, L.; Nguyen, T.; Stone, G.W. Pulmonary Artery Denervation to Treat Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2013, 62, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, J.; Chen, M.; Xie, D.-J.; Kan, J.; Yu, W.; Li, X.-B.; Xu, T.; Gu, Y.; Dong, J.; et al. Pulmonary Artery Denervation Significantly Increases 6-Min Walk Distance for Patients with Combined Pre- and Post-Capillary Pulmonary Hypertension Associated with Left Heart Failure. JACC Cardiovasc. Interv. 2019, 12, 274–284. [Google Scholar] [CrossRef]
- Rubin, L.J. Pulmonary Artery Denervation for Pulmonary Artery Hypertension. JACC Cardiovasc. Interv. 2015, 8, 2024–2025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wei, Y.; Zhang, C.; Yang, Z.; Kan, J.; Gu, H.; Fan, F.; Gu, H.; Wang, Q.; Xie, D.; et al. Pulmonary Artery Denervation for Pulmonary Arterial Hypertension: A Sham-Controlled Randomized Trial. JACC Cardiovasc. Interv. 2022, 15, 2412–2423. [Google Scholar] [CrossRef]
- Kurzyna, M.; Dąbrowski, M.; Bielecki, D.; Fijalkowska, A.; Pruszczyk, P.; Opolski, G.; Burakowski, J.; Florczyk, M.; Tomkowski, W.Z.; Wawrzyńska, L.; et al. Atrial Septostomy in Treatment of End-Stage Right Heart Failure in Patients with Pulmonary Hypertension. Chest 2007, 131, 977–983. [Google Scholar] [CrossRef]
- Rich, S.; Lam, W. Atrial septostomy as palliative therapy for refractory primary pulmonary hypertension. Am. J. Cardiol. 1983, 51, 1560–1561. [Google Scholar] [CrossRef]
- Sandoval, J.; Gaspar, J.; Pena, H.; Santos, L.E.; Cordova, J.; Del Valle, K.; Rodriguez, A.; Pulido, T. Effect of atrial septostomy on the survival of patients with severe pulmonary arterial hypertension. Eur. Respir. J. 2011, 38, 1343–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Wan, L.; Li, H.; Wang, C.; Guo, T.; Niu, H.; Li, S.; Yundan, P.; Wang, L.; Fang, W. First in-human modified atrial septostomy combining radiofrequency ablation and balloon dilation. Heart 2022, 108, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Chen, X.; Song, X.; Chen, X.; Ma, W.; Lin, J.; Wu, H.; Hu, X.; Zhou, Y.; Zhang, H.; et al. Immunotherapy of Endothelin-1 Receptor Type A for Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2019, 73, 2567–2580. [Google Scholar] [CrossRef]
- Bockmeyer, C.L.; Maegel, L.; Janciauskiene, S.; Rische, J.; Lehmann, U.; Maus, U.A.; Nickel, N.; Haverich, A.; Hoeper, M.; Golpon, H.A.; et al. Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J. Heart Lung Transplant. 2012, 31, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.J.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L1182–L1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Drugs | Animals (Model) | Biological Indicators | Administration | Observed Effect |
|---|---|---|---|---|
| Alginate oligosaccharide | Sprague-Dawley rats (MCT, intraperitoneal) | p47-phox, p67-phox, and gp91-phox, subunits of NADPH oxidase, MDA | intraperitoneal | Down-regulate the expressions of malondialdehyde and NADPH by inhibiting the TGF 1/p-Smad2 signaling pathway to prevent the pulmonary vascular remodeling induced by MCT |
| Vardenafil | Sprague-Dawley rats (MCT, intraperitoneal) | 8-iso-prostaglandin-F2a, 3-nitrotyrosine, eNOS, NO, MDA, SOD, Nox2, Nox4 | intragluteal | Suppress proliferation of enhanced apoptosis of pulmonary artery smooth muscle cells, attenuating small pulmonary artery remodeling, and right ventricular hypertrophy |
| Pentaerythritol Tetranitrate (PETN) | Wistar rats (MCT, intravenous) | HO-1, ICAM-1 | intragluteal | PETN therapy improved endothelium-dependent relaxation in pulmonary arteries and reduced oxidative stress |
| Sulforaphane | Male mice (SU5416 and 10% hypoxia, SuHx) | Nrf2, NQO1, NLRP3 | intragluteal | Reduce SuHx-induced pulmonary vascular remodeling, inflammation, and fibrosis |
| Crocin | Sprague-Dawley rats (Hypoxia) | OXR1, P21, Nrf2 | intraperitoneal | Crocin co-treatment significantly improved the hemodynamic, oxidative stress biomarkers and histological data of the PAH rats |
| Melatonin | Newborn sheep (Chronic hypobaric hypoxia, Putre, 3600 m) | SOD2, CAT, GPx1, VDAC, p47-phox, Xanthine Oxidase, 8-isoprostanes, 4HNE, and NT | intragluteal | Reduced major sources of pro-oxidative ROS at the cellular level, reduced oxidative stress and enhanced antioxidant status at the pulmonary level of neonatal PAH |
| Resveratrol | Sprague-Dawley rats (MCT, hypoxia) | Nrf2, HIF-1 | intragluteal | Exert antiproliferation, antioxidant, and anti-inflammation effects |
| Ellagic acid | Male Sprague-Dawley rats (Porcine pancreatic elastase, intra-tracheal) | SOD, CAT, and glutathione | intragluteal | Reduce oxidative stress and prevent PAH |
| 18-Glycrrhetinic Acid | Male Sprague-Dawley rats (MCT, intraperitoneal) | Nox2, Nox4 | intragluteal | Reduce the changes in oxidative stress biomarkers and inhibit Nox2 and Nox4 expression |
| Celastramycin | Wild-type mice; SD rats (3 wks. of hypoxic exposure; SU5416, subcutaneous) | ROS, Nrf2, Nox, GSH/GSSG, SOD2 | Osmotic pump; intraperitoneal | Increase protein levels of Nrf2 and improve pulmonary hypertension |
| Celastrol | cROCK1-/- and cROCK2-/- mice (TAC) | CyPA, Bsg, Nox2, Nox4 | intraperitoneal | Inhibit CyPA/Bsg-NF-B axis and enhance ROS production |
| Hybridization of Isosorbide 5 Mononitrate and Bardoxolone Methyl | Male Sprague-Dawley rats (MCT, intracerebral) | NO, Nox4 | intratracheal | By inactivating Nox4, excessive proliferation of vascular pericytes was inhibited, macrophage infiltration and oxidative stress were reduced, cardiac hypertrophy and fibrosis were significantly reduced in rats with pulmonary hypertension |
| Combination of DCA/ATO | Male Sprague-Dawley rats (MCT, intracerebral) | CHOP, Bcl2 | intragluteal | The combined treatment of DCA/ATO significantly reduces the right ventricular systolic blood pressure accompanied by a decrease in right heart hypertrophy and reduces vascular remodeling, thereby inhibiting excessive PASMC proliferation |
| Baicalein | Male Sprague-Dawley rats (MCT, intracerebral) | MDA, SOD, GSH-Px, Bax, Bcl-2 | intragluteal | Inhibit oxidative stress and alleviated pulmonary vascular remodeling in MCT-induced PAH |
| 17-β estradiol | Male Sprague-Dawley rats (MCT, intracerebral) | T-AOC, MDA, Nox4 | intraperitoneal | Inhibit Nox4-mediated oxidative stress and alleviated MCT-induced right ventricular remodeling of PAH rats |
| Copaiba Oil | Male Wistar rats (MCT, intraperitoneal) | eNOS | intragluteal | Reduce oxidative stress and apoptosis signaling in RV of rats with PAH |
| Dimethyl Fumarate | Male C57BL/6 mice (Hypoxic chamber) | HO-1, NOX4 | intraperitoneal | Mitigate oxidative stress damage and inflammation in lung |
| Bucindolol | Male Wistar rats (MCT, intraperitoneal) | eNOS, SOD-1 | intraperitoneal | Decrease (21%) PVR and increase RV workload, thereby improving the vascular remodeling of the pulmonary artery |
| Rosuvastatin | Male Ren2 and Sprague-Dawley rats (Transgenic (mRen2) 27 rats) | 3-NT, NO(x), Nox, and endothelial NO synthase expression | intraperitoneal | Improve cardiovascular outcomes/risk by restoring endothelial and SMC function, inhibiting SMC proliferation, reducing oxidative stress and inflammation in the vascular wall |
| Carvacrol | Male Wistar rats (Hypoxia) | SOD, GSH, MDA, caspase 3 | intraperitoneal | Attenuate the pulmonary vascular remodeling and promotes PASMC apoptosis |
| Trapidil | Male Wistar rats (MCT, intraperitoneal) | NADPH oxidases, glutathiones/total glutathiones | intraperitoneal | Improve hemodynamic, echocardiographic, and redox state parameters of right ventricle |
| Tetrandrine | Male Sprague-Dawley rats (MCT, intraperitoneal) | cGMP, PKG-1, iNOS | intraperitoneal | Alleviate MCT-induced PAH through regulation of NO signaling pathway and antioxidant and antiproliferation effects |
| Trimethoxystilbene | Male Sprague-Dawley rats (hypoxic chamber) | Nox2, Nox4, VPO1 | intragluteal | Attenuate hypoxia-induced pulmonary vascular remodeling and right ventricle hypertrophy accompanied by downregulation of Nox2, Nox4 and VOP1 expression |
| Hydrogen | Male Sprague-Dawley rats (MCT, intracerebral) | STAT3, NFAT | Housed ad libitumto hydrogen- saturated water | Ameliorate MCT-induced PAH in rats by suppressing macrophage accumulation, reducing oxidative stress, and modulating the STAT3/NFAT axis |
| Blueberry extract | Male Wistar rats (MCT, intragluteal) | NADPH, SOD, GPx, ETA/ETB | intragluteal | Decrease the mean pulmonary artery pressure and total reactive species concentration and lipid oxidation |
| Ocimum Sanctum (Linn) | Male Wistar rats (MCT, intracerebral) | Thiobarbituric Acid Reactive Substances (TBARS); GSH; Catalase; SOD; Nox1 | intragluteal | Decrease Nox-1 expression and increase expression of Bcl2/Bax ratio caused by MCT |
| Honokiol | Male Sprague-Dawley rats (MCT, intraperitoneal) | CyPA | intragluteal | Alleviate autophagy and PAH regulated by CyPA in PAECs |
| GS-444217/Selonsertib | SD rats (MCT (intracerebral)/Sugen/hypoxia) | phosphorylation of p38 and JNK | intragluteal | Reduce pulmonary arterial pressure and RV hypertrophy in PAH models associated with reduced ASK1 phosphorylation, reduced muscularization of the pulmonary arteries, and reduced fibrotic gene expression in the RV |
| SGI-1776, TP-3654 | Male SD rats (MCT, intracerebral/Fawn-Hooded Rats) | Repair of DNA damage | intragluteal | Improve significantly pulmonary hemodynamics (right heart catheterization) and vascular remodeling (Elastica van Gieson) |
| MicroRNA Expression in PAH; Human Model; Animal Model | Effect |
|---|---|
| miR-17-92 NA; Mouse-Hypoxia, Rat-monocrotaline, hypoxia | Increased PASMC proliferation, induced by IL-6; overexpression downregulates BMPR-II |
| miR-21 Pulmonary arteries, plexiform lesions; Mouse-hypoxia, Sugen5416/hypoxia, VHL null Interleukin-6 transgenic, Rat-monocrotaline | Decreased NOS expression in hypoxic PAECs, increased PASMC proliferation; miR-21 deletion enhances PH in mice |
| miR-126 Right ventricle; Rat-monocrotaline | Inhibition of VEGF pathway and decrease in RV vascular density |
| miR-145 Lung tissue, plexiform lesions; Mouse-hypoxia, BMPR2 mutation | Decrease in miR-145 is protective against hypoxia-induced PAH |
| miR-150 Plasma; NA | Associated with poor survival |
| miR-204 Lung, pulmonary arteries; Rat-monocrotaline, Sugen5416/hypoxia, Mouse-hypoxia | Increased NFAT, PASMC proliferation; miR-204 mimics prevent PH in monocrotaline model |
| miR-210 ; Pulmonary artery; Mouse-Sugen5416/hypoxia | Inhibits PASMC apoptosis by suppressing E2F3 transcription factor expression |
| miR-214 ; NA; Mouse-hypoxia, Sugen5416/hypoxia, Rat-monocrotaline, Sugen5416/hypoxia | Increased right ventricular hypertrophy in hypoxia models |
| miR-130/301 NA; Mouse-hypoxia, Sugen5416/hypoxia, VHL null, Interleukin-6 transgenic, BMPR2X transgenic, Schistosoma mansoni-infected, Rat-monocrotaline, Juvenile lamb-pulmonary artery-aorta shunt | Increased PAEC proliferation and PASMC contraction via PPAR-γ mediated pathways |
| miRNA-21 and miRNA-27a PAECs and PASMCs; NA | Suppress PAEC and PASMC proliferation |
| miR-26a PAH patient plasma; Rats-monocrotaline | Inhibition of miR-26a promotes apoptosis of rat cardiomyocytes and pathological right ventricular hypertrophy in PAH |
| miR-124 ; Pulmonary artery smooth muscle cells; Mouse-chronic hypoxia | Suppression of NFAT pathway, antiproliferative |
| miR-138 and miR-25 Pulmonary artery smooth muscle cells; Rats-monocrotaline | Downregulation of MCU, increased PASMC proliferation, apoptosis resistance; inhibition of miRs prevent PH in monocrotaline model |
| miR-140-5p ; NA; Rat-monocrotaline, Sugen-hypoxia | Inhibition of miR 140-5p promotes smooth muscle cell proliferation |
| miR-424/503 ; Pulmonary artery endothelial cells; APLN knock-out mice | Reduced endothelial proliferation, decreased expression of FGF-2 and FGF receptor-1; restoration of miRs prevents monocrotaline and Sugen-hypoxia induced PH. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.J.; Beckmann, T.; Vorla, M.; Kalra, D.K. New Drugs and Therapies in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2023, 24, 5850. https://doi.org/10.3390/ijms24065850
Shah AJ, Beckmann T, Vorla M, Kalra DK. New Drugs and Therapies in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2023; 24(6):5850. https://doi.org/10.3390/ijms24065850
Chicago/Turabian StyleShah, Aangi J., Taylor Beckmann, Mounica Vorla, and Dinesh K. Kalra. 2023. "New Drugs and Therapies in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 24, no. 6: 5850. https://doi.org/10.3390/ijms24065850
APA StyleShah, A. J., Beckmann, T., Vorla, M., & Kalra, D. K. (2023). New Drugs and Therapies in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 24(6), 5850. https://doi.org/10.3390/ijms24065850