
The Major Components of Cerebrospinal Fluid Dictate the Characteristics of Inhibitors against Amyloid-Beta Aggregation

 , ,
, , 
Abstract
:1. Introduction
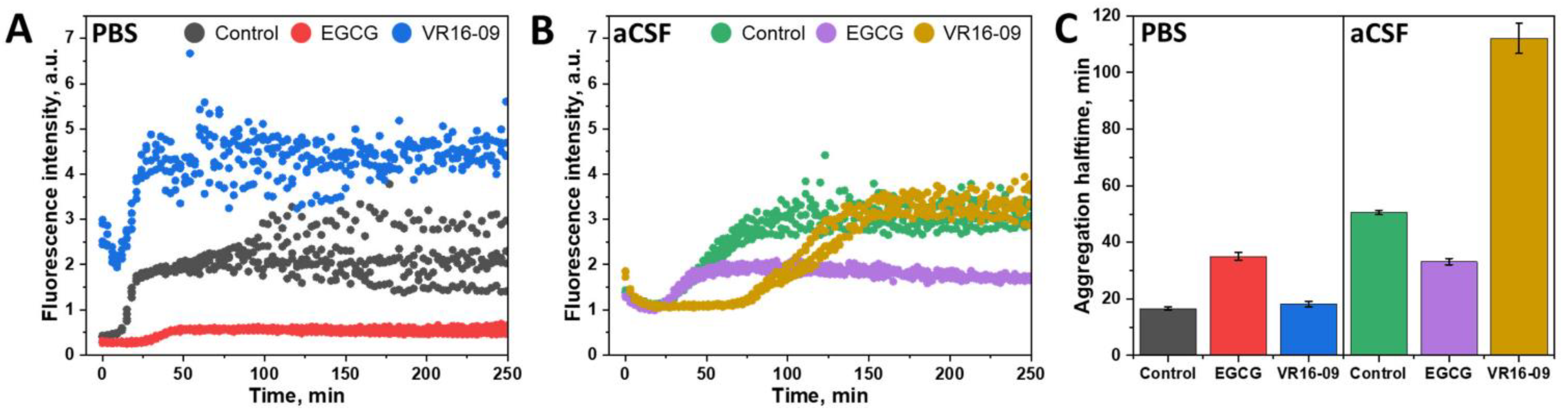
2. Results
3. Discussion
4. Materials and Methods
4.1. Preparation of aCSF Reaction Mixture
4.2. Preparation of Epigallocatechin-3-Gallate (EGCG)
4.3. Aβ42 Aggregation Experiments
4.4. Atomic Force Microscopy (AFM)
4.5. Cell Culturing
4.6. MTT Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- De, S.; Whiten, D.R.; Ruggeri, F.S.; Hughes, C.; Rodrigues, M.; Sideris, D.I.; Taylor, C.G.; Aprile, F.A.; Muyldermans, S.; Knowles, T.P.J.; et al. Soluble aggregates present in cerebrospinal fluid change in size and mechanism of toxicity during Alzheimer’s disease progression. Acta Neuropathol. Commun. 2019, 7, 120. [Google Scholar] [CrossRef] [Green Version]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investig. 2019, 99, 958–970. [Google Scholar] [CrossRef]
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [Green Version]
- Athar, T.; Al Balushi, K.; Khan, S.A. Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 5629–5645. [Google Scholar] [CrossRef]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef]
- Benoit, S.L.; Maier, R.J. The nickel-chelator dimethylglyoxime inhibits human amyloid beta peptide in vitro aggregation. Sci. Rep. 2021, 11, 6622. [Google Scholar] [CrossRef] [PubMed]
- Finder, V.H.; Vodopivec, I.; Nitsch, R.M.; Glockshuber, R. The Recombinant Amyloid-β Peptide Aβ1-42 Aggregates Faster and Is More Neurotoxic than Synthetic Aβ1-42. J. Mol. Biol. 2010, 396, 9–18. [Google Scholar] [CrossRef]
- Kodali, R.; Williams, A.D.; Chemuru, S.; Wetzel, R. Aβ(1–40) Forms Five Distinct Amyloid Structures whose β-Sheet Contents and Fibril Stabilities Are Correlated. J. Mol. Biol. 2010, 401, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Foderà, V.; Groenning, M.; Vetri, V.; Librizzi, F.; Spagnolo, S.; Cornett, C.; Olsen, L.; van de Weert, M.; Leone, M. Thioflavin T Hydroxylation at Basic pH and Its Effect on Amyloid Fibril Detection. J. Phys. Chem. B 2008, 112, 15174–15181. [Google Scholar] [CrossRef]
- Di Terlizzi, R.; Platt, S. The function, composition and analysis of cerebrospinal fluid in companion animals: Part I—Function and composition. Vet. J. 2006, 172, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Ashok, K.S.; Gabriele, Z. The Interstitial System of the Brain in Health and Disease. Aging Dis. 2020, 11, 200. [Google Scholar] [CrossRef] [Green Version]
- Vernau, W.; Vernau, K.A.; Bailey, C.S. Cerebrospinal Fluid. In Clinical Biochemistry of Domestic Animals; Elsevier: Amsterdam, The Netherlands, 2008; pp. 769–819. [Google Scholar]
- Padayachee, E.R.; Zetterberg, H.; Portelius, E.; Borén, J.; Molinuevo, J.L.; Andreasen, N.; Cukalevski, R.; Linse, S.; Blennow, K.; Andreasson, U. Cerebrospinal fluid-induced retardation of amyloid β aggregation correlates with Alzheimer’s disease and the APOE ε4 allele. Brain Res. 2016, 1651, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Frankel, R.; Törnquist, M.; Meisl, G.; Hansson, O.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Frohm, B.; Cedervall, T.; Knowles, T.P.J.; et al. Autocatalytic amplification of Alzheimer-associated Aβ42 peptide aggregation in human cerebrospinal fluid. Commun. Biol. 2019, 2, 365. [Google Scholar] [CrossRef] [Green Version]
- Milojevic, J.; Raditsis, A.; Melacini, G. Human Serum Albumin Inhibits Aβ Fibrillization through a “Monomer-Competitor” Mechanism. Biophys. J. 2009, 97, 2585–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Wärmländer, S.K.T.S.; Gräslund, A.; Abrahams, J.P. Non-chaperone Proteins Can Inhibit Aggregation and Cytotoxicity of Alzheimer Amyloid β Peptide. J. Biol. Chem. 2014, 289, 27766–27775. [Google Scholar] [CrossRef] [Green Version]
- Baram, M.; Miller, Y. Inhibitory Activity of Insulin on Aβ Aggregation Is Restricted Due to Binding Selectivity and Specificity to Polymorphic Aβ States. ACS Chem. Neurosci. 2020, 11, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Ban, T.; Aritake, K.; Huang, Z.-L.; Qu, W.-M.; Okazaki, I.; Mohri, I.; Murayama, S.; Ozono, K.; Taniike, M.; et al. Lipocalin-type prostaglandin D synthase/β-trace is a major amyloid β-chaperone in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2007, 104, 6412–6417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilde, L.M.; Kösters, S.; Steinbach, S.; Schork, K.; Eisenacher, M.; Galozzi, S.; Turewicz, M.; Barkovits, K.; Mollenhauer, B.; Marcus, K.; et al. Protein variability in cerebrospinal fluid and its possible implications for neurological protein biomarker research. PLoS ONE 2018, 13, e0206478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanyon, H.F.; Viles, J.H. Human Serum Albumin Can Regulate Amyloid-β Peptide Fiber Growth in the Brain Interstitium. J. Biol. Chem. 2012, 287, 28163–28168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaret, D.L.; Morrison, N.; Gulbranson, R.; Keren, D.F. Immunofixation to quantify beta 2-transferrin in cerebrospinal fluid to detect leakage of cerebrospinal fluid from skull injury. Clin. Chem. 1992, 38, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Rainesalo, S.; Keränen, T.; Palmio, J.; Peltola, J.; Oja, S.S.; Saransaari, P. Plasma and Cerebrospinal Fluid Amino Acids in Epileptic Patients. Neurochem. Res. 2004, 29, 319–324. [Google Scholar] [CrossRef]
- Itkin, A.; Dupres, V.; Dufrêne, Y.F.; Bechinger, B.; Ruysschaert, J.-M.; Raussens, V. Calcium Ions Promote Formation of Amyloid β-Peptide (1–40) Oligomers Causally Implicated in Neuronal Toxicity of Alzheimer’s Disease. PLoS ONE 2011, 6, e18250. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.M.; Locatelli, L.; Fedele, G.; Cazzaniga, A.; Mazur, A. Magnesium and the Brain: A Focus on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 24, 223. [Google Scholar] [CrossRef]
- Hadi Ali Janvand, S.; Ladefoged, L.K.; Zubrienė, A.; Sakalauskas, A.; Christiansen, G.; Dudutienė, V.; Schiøtt, B.; Matulis, D.; Smirnovas, V.; Otzen, D.E. Inhibitory effects of fluorinated benzenesulfonamides on insulin fibrillation. Int. J. Biol. Macromol. 2023, 227, 590–600. [Google Scholar] [CrossRef]
- Sneideris, T.; Sakalauskas, A.; Sternke-Hoffmann, R.; Peduzzo, A.; Ziaunys, M.; Buell, A.K.; Smirnovas, V. The Environment Is a Key Factor in Determining the Anti-Amyloid Efficacy of EGCG. Biomolecules 2019, 9, 855. [Google Scholar] [CrossRef] [Green Version]
- Ziaunys, M.; Mikalauskaite, K.; Sakalauskas, A.; Smirnovas, V. Interplay between epigallocatechin-3-gallate and ionic strength during amyloid aggregation. PeerJ 2021, 9, e12381. [Google Scholar] [CrossRef] [PubMed]
- Sakalauskas, A.; Ziaunys, M.; Snieckute, R.; Smirnovas, V. Autoxidation Enhances Anti-Amyloid Potential of Flavone Derivatives. Antioxidants 2021, 10, 1428. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.A.; Ecroyd, H.; Kee, T.W.; Carver, J.A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009, 276, 5960–5972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef] [Green Version]
- Sakalauskas, A.; Ziaunys, M.; Smirnovas, V. Gallic acid oxidation products alter the formation pathway of insulin amyloid fibrils. Sci. Rep. 2020, 10, 14466. [Google Scholar] [CrossRef]
- Griner, S.L.; Seidler, P.; Bowler, J.; Murray, K.A.; Yang, T.P.; Sahay, S.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Philipp, S.; et al. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. eLife 2019, 8, e46924. [Google Scholar] [CrossRef]
- Bode, D.C.; Stanyon, H.F.; Hirani, T.; Baker, M.D.; Nield, J.; Viles, J.H. Serum Albumin’s Protective Inhibition of Amyloid-β Fiber Formation Is Suppressed by Cholesterol, Fatty Acids and Warfarin. J. Mol. Biol. 2018, 430, 919–934. [Google Scholar] [CrossRef]
- Sakalauskas, A.; Janoniene, A.; Zvinys, G.; Mikalauskaite, K.; Ziaunys, M.; Smirnovas, V. Exploring the Formation of Polymers with Anti-Amyloid Properties within the 2′3′-Dihydroxyflavone Autoxidation. Process. Antioxid. 2022, 11, 1711. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.I.; Masuda, Y.; Takegoshi, K.; Irie, K. Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.-L.; Tsai, M.-Y.; Schafer, N.P.; Wolynes, P.G. Modeling Protein Aggregation Kinetics: The Method of Second Stochasticization. J. Phys. Chem. B 2021, 125, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Ziaunys, M.; Mikalauskaite, K.; Smirnovas, V. Amyloidophilic Molecule Interactions on the Surface of Insulin Fibrils: Cooperative Binding and Fluorescence Quenching. Sci. Rep. 2019, 9, 20303. [Google Scholar] [CrossRef] [Green Version]
- Plum, F. The physiology and pathophysiology of the cerebrospinal fluid by Hugh Davson, Keasley Welch, and Malcolm B. Segal New York. Livingstone, I987 1013 pp. illustrated, $198.00. Ann. Neurol. 1988, 24, 106. [Google Scholar] [CrossRef]
- Johnson, K.S.; Sexton, D.J. Cerebrospinal Fluid: Physiology and Utility of an Examination in Disease States; UpToDate: Waltham, MA, USA, 2022; pp. 1–14. [Google Scholar]
- Elbohouty, M.; Wilson, M.T.; Voss, L.J.; Steyn-Ross, D.A.; Hunt, L.A. In vitro electrical conductivity of seizing and non-seizing mouse brain slices at 10 kHz. Phys. Med. Biol. 2013, 58, 3599–3613. [Google Scholar] [CrossRef] [PubMed]
- Kozak, L.R.; Bango, M.; Szabo, M.; Rudas, G.; Vidnyanszky, Z.; Nagy, Z. Using diffusion MRI for measuring the temperature of cerebrospinal fluid within the lateral ventricles. Acta Paediatr. Int. J. Paediatr. 2010, 99, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Šneideris, T.; Baranauskiene, L.; Cannon, J.G.; Rutkiene, R.; Meškys, R.; Smirnovas, V. Looking for a generic inhibitor of amyloid-like fibril formation among flavone derivatives. PeerJ 2015, 3, e1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int. J. Mol. Sci. 2021, 22, 12382. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition of Concentrated aCSF (in 3 Separate Parts) | |||
|---|---|---|---|
| Amount in Pt 1, mM | Amount in Pt 2, mM | Amount in Pt 3, mM | |
| NaCl | 1270 | - | 127 |
| KCl | 18 | - | 1.8 |
| KH2PO4 | 12 | - | 1.2 |
| Na2HPO4 | 78.1 | - | 7.81 |
| NaH2PO4 | 31.9 | - | 3.19 |
| D-glucose | - | 40 | - |
| CaCl2 | - | 14 | - |
| MgCl2 | - | 13 | - |
| Urea | - | 6.5 | - |
| HSA | - | - | 0.0615 |
| Cholesterol | - | - | 0.052 |
| Sodium lactate | - | - | 24 |
| Glutamine | - | - | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakalauskas, A.; Ziaunys, M.; Snieckute, R.; Janoniene, A.; Veiveris, D.; Zvirblis, M.; Dudutiene, V.; Smirnovas, V. The Major Components of Cerebrospinal Fluid Dictate the Characteristics of Inhibitors against Amyloid-Beta Aggregation. Int. J. Mol. Sci. 2023, 24, 5991. https://doi.org/10.3390/ijms24065991
Sakalauskas A, Ziaunys M, Snieckute R, Janoniene A, Veiveris D, Zvirblis M, Dudutiene V, Smirnovas V. The Major Components of Cerebrospinal Fluid Dictate the Characteristics of Inhibitors against Amyloid-Beta Aggregation. International Journal of Molecular Sciences. 2023; 24(6):5991. https://doi.org/10.3390/ijms24065991
Chicago/Turabian StyleSakalauskas, Andrius, Mantas Ziaunys, Ruta Snieckute, Agne Janoniene, Dominykas Veiveris, Mantas Zvirblis, Virginija Dudutiene, and Vytautas Smirnovas. 2023. "The Major Components of Cerebrospinal Fluid Dictate the Characteristics of Inhibitors against Amyloid-Beta Aggregation" International Journal of Molecular Sciences 24, no. 6: 5991. https://doi.org/10.3390/ijms24065991
APA StyleSakalauskas, A., Ziaunys, M., Snieckute, R., Janoniene, A., Veiveris, D., Zvirblis, M., Dudutiene, V., & Smirnovas, V. (2023). The Major Components of Cerebrospinal Fluid Dictate the Characteristics of Inhibitors against Amyloid-Beta Aggregation. International Journal of Molecular Sciences, 24(6), 5991. https://doi.org/10.3390/ijms24065991