
Iron Limitation Restores Autophagy and Increases Lifespan in the Yeast Model of Niemann–Pick Type C1

 , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Characterization of the Vacuolar Membrane Proteome in the Yeast Model of NPC1
2.2. Cvt Pathway Is Induced and Autophagy Compromised in the Yeast Model of NPC1
2.3. Ncr1 Deficiency Induces the Iron Regulon and Aft1-Dependent Iron Overload
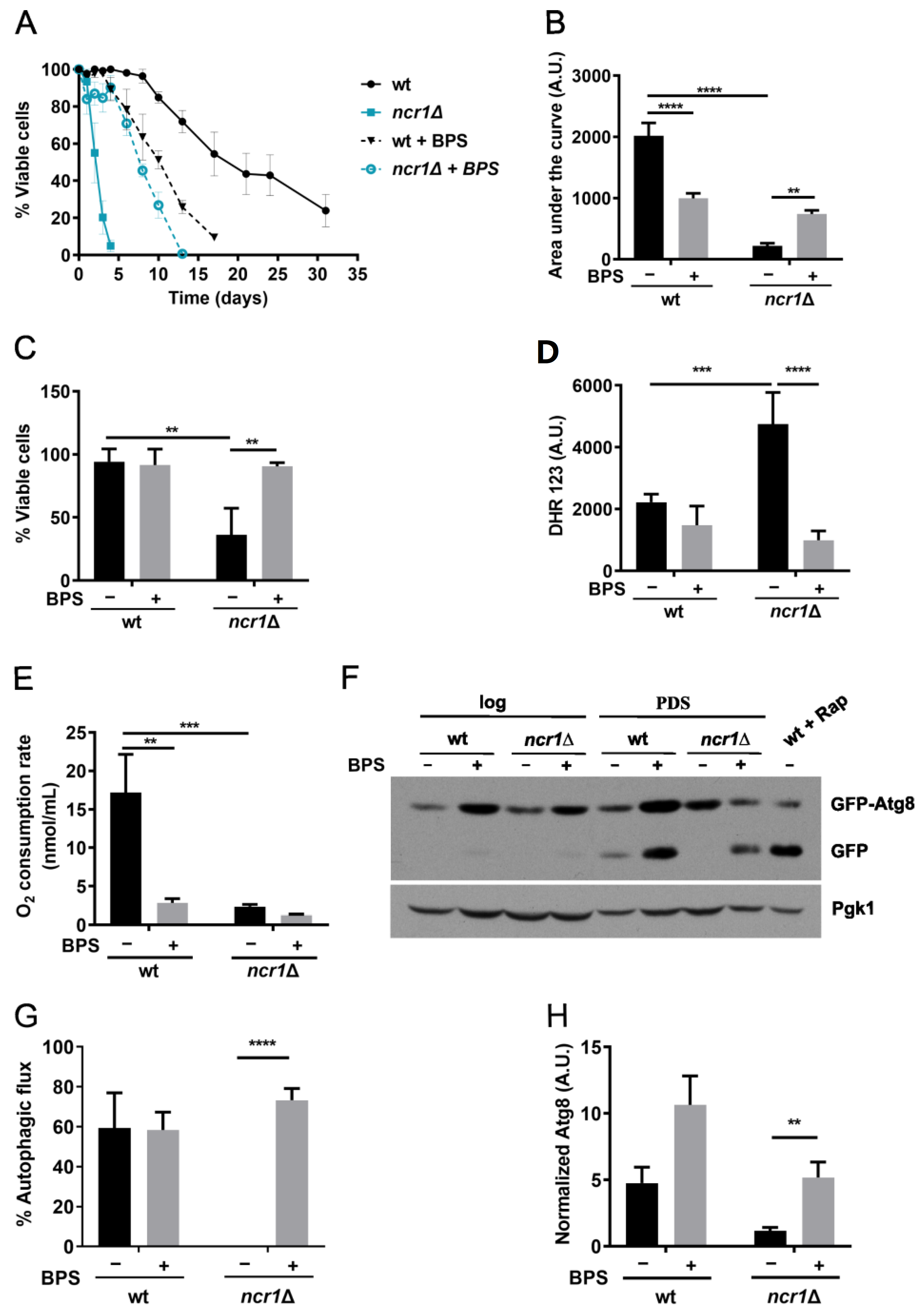
2.4. Iron Chelation with BPS Increases Lifespan, Oxidative Stress Resistance, and Restores Autophagic Flux in the Yeast Model of NPC1
3. Materials and Methods
3.1. Yeast Strains and Growth Conditions
3.2. Vacuolar Membranes Isolation
3.2.1. Spheroplasts Preparation and Lysis
3.2.2. Isolation of Intact Vacuoles
3.2.3. Isolation of Vacuolar Membranes
3.3. Proteomics
3.3.1. Sample Preparation and Data Acquisition
3.3.2. Data Analysis
3.4. Analysis of the AP-3 Pathway
3.5. Western Blotting
3.6. Iron Levels
3.7. β-Galactosidase Assay
3.8. Chronological Lifespan
3.9. Oxidative Stress Resistance
3.10. Intracellular Oxidation
3.11. Oxygen Consumption
3.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lawrence, R.E.; Zoncu, R. The Lysosome as a Cellular Centre for Signalling, Metabolism and Quality Control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-Y.; Zoncu, R. The Lysosome as a Command-and-Control Center for Cellular Metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrick, M.J.; Lieberman, A.P. Autophagic Dysfunction in a Lysosomal Storage Disorder Due to Impaired Proteolysis. Autophagy 2013, 9, 234–235. [Google Scholar] [CrossRef] [Green Version]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef] [Green Version]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the Second Gene of Niemann-Pick C Disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Maladie de Niemann-Pick Type C: Aspects Historiques et Actuels, Diagnostic Biochimique et Génétique. Arch. Pédiatrie 2010, 17, S41–S44. [Google Scholar] [CrossRef]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.A.; Scott, M.P. Cell-Autonomous Death of Cerebellar Purkinje Neurons with Autophagy in Niemann-Pick Type C Disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Mengel, E.; Klünemann, H.-H.; Lourenço, C.M.; Hendriksz, C.J.; Sedel, F.; Walterfang, M.; Kolb, S.A. Niemann-Pick Disease Type C Symptomatology: An Expert-Based Clinical Description. Orphanet J. Rare Dis. 2013, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.E.; Davies, J.P.; Chen, F.W.; Ioannou, Y.A. Niemann-Pick C1 Is a Late Endosome-Resident Protein That Transiently Associates with Lysosomes and the Trans-Golgi Network. Mol. Genet. Metab. 1999, 68, 1–13. [Google Scholar] [CrossRef]
- Ko, D.C.; Binkley, J.; Sidow, A.; Scott, M.P. The Integrity of a Cholesterol-Binding Pocket in Niemann–Pick C2 Protein Is Necessary to Control Lysosome Cholesterol Levels. Proc. Natl. Acad. Sci. USA 2003, 100, 2518–2525. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-Terminal Domain of NPC1 Reveals Distinct Subdomains for Binding and Transfer of Cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T. Complex Lipid Trafficking in Niemann-Pick Disease Type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick Disease Type C1 Is a Sphingosine Storage Disease That Causes Deregulation of Lysosomal Calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in Niemann-Pick C Disease Is Dependent upon Beclin-1 and Responsive to Lipid Trafficking Defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Rivera-Molina, F.; Xi, Z.; Toomre, D.; Schepartz, A. Endosome Motility Defects Revealed at Super-Resolution in Live Cells Using HIDE Probes. Nat. Chem. Biol. 2020, 16, 408–414. [Google Scholar] [CrossRef]
- Liedtke, M.; Völkner, C.; Hermann, A.; Frech, M.J. Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C. Cells 2022, 11, 507. [Google Scholar] [CrossRef]
- Roney, J.C.; Li, S.; Farfel-Becker, T.; Huang, N.; Sun, T.; Xie, Y.; Cheng, X.-T.; Lin, M.-Y.; Platt, F.M.; Sheng, Z.-H. Lipid-Mediated Motor-Adaptor Sequestration Impairs Axonal Lysosome Delivery Leading to Autophagic Stress and Dystrophy in Niemann-Pick Type C. Dev. Cell 2021, 56, 1452–1468.e8. [Google Scholar] [CrossRef]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.M.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired Autophagy in the Lipid-Storage Disorder Niemann-Pick Type C1 Disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Meske, V.; Priesnitz, T.; Albert, F.; Ohm, T.G. How to Reduce the Accumulation of Autophagic Vacuoles in NPC1-Deficient Neurons: A Comparison of Two Pharmacological Strategies. Neuropharmacology 2015, 89, 282–289. [Google Scholar] [CrossRef]
- Liao, G.; Cheung, S.; Galeano, J.; Ji, A.X.; Qin, Q.; Bi, X. Allopregnanolone Treatment Delays Cholesterol Accumulation and Reduces Autophagic/Lysosomal Dysfunction and Inflammation in Npc1−/− Mouse Brain. Brain Res. 2009, 1270, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Paulina Ordonez, M.; Roberts, E.A.; Kidwell, C.U.; Yuan, S.H.; Plaisted, W.C.; Goldstein, L.S.B. Disruption and Therapeutic Rescue of Autophagy in a Human Neuronal Model of Niemann Pick Type C1. Hum. Mol. Genet. 2012, 21, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Yui, N. β-Cyclodextrin-Threaded Biocleavable Polyrotaxanes Ameliorate Impaired Autophagic Flux in Niemann-Pick Type C Disease. J. Biol. Chem. 2015, 290, 9442–9454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soga, M.; Ishitsuka, Y.; Hamasaki, M.; Yoneda, K.; Furuya, H.; Matsuo, M.; Ihn, H.; Fusaki, N.; Nakamura, K.; Nakagata, N.; et al. HPGCD Outperforms HPBCD as a Potential Treatment for Niemann-Pick Disease Type C during Disease Modeling with IPS Cells. Stem Cells 2015, 33, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and Chemical Correction of Cholesterol Accumulation and Impaired Autophagy in Hepatic and Neural Cells Derived from Niemann-Pick Type C Patient-Specific IPS Cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharkeshwar, A.K.; Trekker, J.; Vermeire, W.; Pauwels, J.; Sannerud, R.; Priestman, D.A.; te Vruchte, D.; Vints, K.; Baatsen, P.; Decuypere, J.-P.; et al. A Novel Approach to Analyze Lysosomal Dysfunctions through Subcellular Proteomics and Lipidomics: The Case of NPC1 Deficiency. Sci. Rep. 2017, 7, 41408. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.; Yao, Y.; Liu, J.; Yu, Z.; Cheung, S.; Xie, A.; Liang, X.; Bi, X. Cholesterol Accumulation Is Associated with Lysosomal Dysfunction and Autophagic Stress in Npc1−/− Mouse Brain. Am. J. Pathol. 2007, 171, 962–975. [Google Scholar] [CrossRef] [Green Version]
- Balboa, E.; Marín, T.; Oyarzún, J.E.; Contreras, P.S.; Hardt, R.; van den Bosch, T.; Alvarez, A.R.; Rebolledo-Jaramillo, B.; Klein, A.D.; Winter, D.; et al. Proteomic Analysis of Niemann-Pick Type C Hepatocytes Reveals Potential Therapeutic Targets for Liver Damage. Cells 2021, 10, 2159. [Google Scholar] [CrossRef]
- Macías-Vidal, J.; Guerrero-Hernández, M.; Estanyol, J.M.; Aguado, C.; Knecht, E.; Coll, M.J.; Bachs, O. Identification of Lysosomal Npc1-Binding Proteins: Cathepsin D Activity Is Regulated by NPC1. Proteomics 2016, 16, 150–158. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal Cholesterol Activates mTORC1 via an SLC38A9–Niemann-Pick C1 Signaling Complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Davis, O.B.; Shin, H.R.; Lim, C.-Y.; Wu, E.Y.; Kukurugya, M.; Maher, C.F.; Perera, R.M.; Ordonez, M.P.; Zoncu, R. NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev. Cell 2021, 56, 260–276.e7. [Google Scholar] [CrossRef]
- Lim, C.Y.; Davis, O.B.; Shin, H.R.; Zhang, J.; Berdan, C.A.; Jiang, X.; Counihan, J.L.; Ory, D.S.; Nomura, D.K.; Zoncu, R. ER–Lysosome Contacts Enable Cholesterol Sensing by mTORC1 and Drive Aberrant Growth Signalling in Niemann–Pick Type C. Nat. Cell Biol. 2019, 21, 1206–1218. [Google Scholar] [CrossRef]
- Dai, S.; Dulcey, A.E.; Hu, X.; Wassif, C.A.; Porter, F.D.; Austin, C.P.; Ory, D.S.; Marugan, J.; Zheng, W. Methyl-β-Cyclodextrin Restores Impaired Autophagy Flux in Niemann-Pick C1-Deficient Cells through Activation of AMPK. Autophagy 2017, 13, 1435–1451. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Dang, Y.; Ren, Y.R.; Liu, J.O. Cholesterol Trafficking Is Required for mTOR Activation in Endothelial Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar] [CrossRef] [Green Version]
- Colaco, A.; Fernández-Suárez, M.E.; Shepherd, D.; Gal, L.; Bibi, C.; Chuartzman, S.; Diot, A.; Morten, K.; Eden, E.; Porter, F.D.; et al. Unbiased Yeast Screens Identify Cellular Pathways Affected in Niemann–Pick Disease Type C. Life Sci. Alliance 2020, 3, e201800253. [Google Scholar] [CrossRef]
- Yambire, K.F.; Fernandez-Mosquera, L.; Steinfeld, R.; Mühle, C.; Ikonen, E.; Milosevic, I.; Raimundo, N. Mitochondrial Biogenesis Is Transcriptionally Repressed in Lysosomal Lipid Storage Diseases. Elife 2019, 8, e39598. [Google Scholar] [CrossRef] [PubMed]
- Gläser, A.; Hammerl, F.; Gräler, M.H.; Coldewey, S.M.; Völkner, C.; Frech, M.J.; Yang, F.; Luo, J.; Tönnies, E.; Halbach, O. von B. und; et al. Identification of Brain-Specific Treatment Effects in NPC1 Disease by Focusing on Cellular and Molecular Changes of Sphingosine-1-Phosphate Metabolism. Int. J. Mol. Sci. 2020, 21, 4502. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, D.; Burgoyne, T.; Sanchez-Heras, E.; Hartwig, P.; Colaco, A.; Newton, J.; Futter, C.E.; Spiegel, S.; Platt, F.M.; Eden, E.R. NPC1 Regulates ER Contacts with Endocytic Organelles to Mediate Cholesterol Egress. Nat. Commun. 2019, 10, 4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid Ceramidase Improves Mitochondrial Function and Oxidative Stress in Niemann-Pick Type C Disease by Repressing STARD1 Expression and Mitochondrial Cholesterol Accumulation. Redox Biol. 2021, 45, 102052. [Google Scholar] [CrossRef]
- Tiscione, S.A.; Casas, M.; Horvath, J.D.; Lam, V.; Hino, K.; Ory, D.S.; Fernando Santana, L.; Simó, S.; Dixon, R.E.; Dickson, E.J. IP3R-Driven Increases in Mitochondrial Ca2+ Promote Neuronal Death in NPC Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2110629118. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Faux, N.G.; Killilea, D.W.; Yanjanin, N.; Firnkes, S.; Volitakis, I.; Ganio, G.; Walterfang, M.; Hastings, C.; Porter, F.D.; et al. Altered Transition Metal Homeostasis in Niemann-Pick Disease, Type C1. Metallomics 2014, 6, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, Y.H.; Lotan, A.; Yeshurun, S.; Schroeder, A.; Bush, A.I. Iron Chelation by Deferiprone Does Not Rescue the Niemann-Pick Disease Type C1 Mouse Model. BioMetals 2020, 33, 87–95. [Google Scholar] [CrossRef]
- Christomanou, H.; Vanier, M.T.; Santambrogio, P.; Arosio, P.; Kleijer, W.J.; Harzer, K. Deficient Ferritin Immunoreactivity in Tissues from Niemann–Pick Type C Patients: Extension of Findings to Fetal Tissues, H and L Ferritin Isoforms, but Also One Case of the Rare Niemann–Pick C2 Complementation Group. Mol. Genet. Metab. 2000, 70, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Argüello, G.; Martinez, P.; Peña, J.; Chen, O.; Platt, F.; Zanlungo, S.; González, M. Hepatic Metabolic Response to Restricted Copper Intake in a Niemann-Pick C Murine Model. Metallomics 2014, 6, 1527–1539. [Google Scholar] [CrossRef]
- Chen, O.C.W.; Siebel, S.; Colaco, A.; Nicoli, E.-R.; Platt, N.; Shepherd, D.; Newman, S.; Armitage, A.E.; Farhat, N.Y.; Seligmann, G.; et al. Defective Iron Homeostasis and Hematological Abnormalities in Niemann-Pick Disease Type C1. Wellcome Open Res. 2022, 7, 267. [Google Scholar] [CrossRef]
- Liang, L.; Wang, H.; Yao, J.; Wei, Q.; Lu, Y.; Wang, T.; Cao, X. NPC1 Deficiency Contributes to Autophagy-Dependent Ferritinophagy in HEI-OC1 Auditory Cells. Front. Mol. Biosci. 2022, 9, 952608. [Google Scholar] [CrossRef]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative Stress and Neurodegeneration: The Involvement of Iron. BioMetals 2018, 31, 715–735. [Google Scholar] [CrossRef]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative Stress: A Pathogenic Mechanism for Niemann-Pick Type C Disease. Oxid. Med. Cell. Longev. 2012, 2012, 205713. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.V.; Vilaça, R.; Santos, C.N.; Costa, V.; Menezes, R. Exploring the Power of Yeast to Model Aging and Age-Related Neurodegenerative Disorders. Biogerontology 2017, 18, 3–34. [Google Scholar] [CrossRef]
- Rajakumar, T.; Munkacsi, A.B.; Sturley, S.L. Exacerbating and Reversing Lysosomal Storage Diseases: From Yeast to Humans. Microb. Cell 2017, 4, 278–293. [Google Scholar] [CrossRef] [Green Version]
- Malathi, K.; Higaki, K.; Tinkelenberg, A.H.; Balderes, D.A.; Almanzar-Paramio, D.; Wilcox, L.J.; Erdeniz, N.; Redican, F.; Padamsee, M.; Liu, Y.; et al. Mutagenesis of the Putative Sterol-Sensing Domain of Yeast Niemann Pick C-Related Protein Reveals a Primordial Role in Subcellular Sphingolipid Distribution. J. Cell Biol. 2004, 164, 547–556. [Google Scholar] [CrossRef]
- Berger, A.C.; Vanderford, T.H.; Gernert, K.M.; Nichols, J.W.; Faundez, V.; Corbett, A.H. Saccharomyces Cerevisiae Npc2p Is a Functionally Conserved Homologue of the Human Niemann-Pick Disease Type C2 Protein, HNPC2. Eukaryot. Cell 2005, 4, 1851–1862. [Google Scholar] [CrossRef] [Green Version]
- Winkler, M.B.L.; Kidmose, R.T.; Szomek, M.; Thaysen, K.; Rawson, S.; Muench, S.P.; Wüstner, D.; Pedersen, B.P. Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 2019, 179, 485–497.e18. [Google Scholar] [CrossRef]
- Tsuji, T.; Fujimoto, M.; Tatematsu, T.; Cheng, J.; Orii, M.; Takatori, S.; Fujimoto, T. Niemann-Pick Type C Proteins Promote Microautophagy by Expanding Raft-like Membrane Domains in the Yeast Vacuole. eLife 2017, 6, e25960. [Google Scholar] [CrossRef] [PubMed]
- Vilaça, R.; Silva, E.; Nadais, A.; Teixeira, V.; Matmati, N.; Gaifem, J.; Hannun, Y.A.; Sá Miranda, M.C.; Costa, V. Sphingolipid Signalling Mediates Mitochondrial Dysfunctions and Reduced Chronological Lifespan in the Yeast Model of Niemann-Pick Type C1. Mol. Microbiol. 2014, 91, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Vilaça, R.; Barros, I.; Matmati, N.; Silva, E.; Martins, T.; Teixeira, V.; Hannun, Y.A.; Costa, V. The Ceramide Activated Protein Phosphatase Sit4 Impairs Sphingolipid Dynamics, Mitochondrial Function and Lifespan in a Yeast Model of Niemann-Pick Type C1. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Wanikawa, M.; Nakamura, H.; Emori, S.; Hashimoto, N.; Murayama, T. Accumulation of Sphingomyelin in Niemann-Pick Disease Type C Cells Disrupts Rab9-Dependent Vesicular Trafficking of Cholesterol. J. Cell. Physiol. 2020, 235, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Feyder, S.; De Craene, J.-O.; Bär, S.; Bertazzi, D.; Friant, S. Membrane Trafficking in the Yeast Saccharomyces cerevisiae Model. Int. J. Mol. Sci. 2015, 16, 1509–1525. [Google Scholar] [CrossRef] [Green Version]
- Kama, R.; Kanneganti, V.; Ungermann, C.; Gerst, J.E. The Yeast Batten Disease Orthologue Btn1 Controls Endosome-Golgi Retrograde Transport via SNARE Assembly. J. Cell Biol. 2011, 195, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Bonangelino, C.J.; Chavez, E.M.; Bonifacino, J.S. Genomic Screen for Vacuolar Protein Sorting Genes in Saccharomyces cerevisiae. Mol. Biol. Cell 2002, 13, 2486–2501. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Zubenko, G.S.; Hasilik, A.; Jones, E.W. Mutant Defective in Processing of an Enzyme Located in the Lysosome-like Vacuole of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1981, 78, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Ren, J.; Li, H.; Zhang, Q.; Armstrong, J.S.; Munn, A.L.; Yang, H. Ncr1p, the Yeast Ortholog of Mammalian Niemann Pick C1 Protein, Is Dispensable for Endocytic Transport. Traffic 2004, 5, 1017–1030. [Google Scholar] [CrossRef]
- Lynch-Day, M.A.; Klionsky, D.J. The Cvt Pathway as a Model for Selective Autophagy. FEBS Lett. 2010, 584, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Torggler, R.; Papinski, D.; Kraft, C. Assays to Monitor Autophagy in Saccharomyces cerevisiae. Cells 2017, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Hall, M.N. Nutrient Sensing and TOR Signaling in Yeast and Mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrick, M.J.; Yu, T.; Chung, C.; Lieberman, A.P. Impaired Proteolysis Underlies Autophagic Dysfunction in Niemann-Pick Type C Disease. Hum. Mol. Genet. 2012, 21, 4876–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.; Shimobayashi, M.; Eisenberg, T.; Merle, D.A.; Pendl, T.; Hall, M.N.; Moustafa, T. TORC1 Promotes Phosphorylation of Ribosomal Protein S6 via the AGC Kinase Ypk3 in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0120250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular Iron Transport and Storage: From Molecular Mechanisms to Health Implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.V.; Ganley, I.G.; Pfeffer, S.R. Clues to Neuro-Degeneration in Niemann-Pick Type C Disease from Global Gene Expression Profiling. PLoS ONE 2006, 1, e19. [Google Scholar] [CrossRef] [Green Version]
- Christomanou, H.; Harzer, K. Ouchterlony Double Immunodiffusion Method Demonstrates Absence of Ferritin Immunoreactivity in Visceral Organs from Nine Patients with Niemann-Pick Disease Type C. Biochem. Mol. Med. 1996, 58, 176–183. [Google Scholar] [CrossRef]
- Christomanou, H.; Kellermann, J.; Link, R.P.; Harzer, K. Deficient Ferritin Immunoreactivity in Visceral Organs from Four Patients with Niemann-Pick Disease Type C. Biochem. Mol. Med. 1995, 55, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Pierzynowska, K.; Rintz, E.; Gaffke, L.; Węgrzyn, G. Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells 2021, 10, 365. [Google Scholar] [CrossRef]
- Martins, T.S.; Costa, V.; Pereira, C. Signaling Pathways Governing Iron Homeostasis in Budding Yeast. Mol. Microbiol. 2018, 109, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Martins, T.S.; Pereira, C.; Canadell, D.; Vilaça, R.; Teixeira, V.; Moradas-Ferreira, P.; de Nadal, E.; Posas, F.; Costa, V. The Hog1p Kinase Regulates Aft1p Transcription Factor to Control Iron Accumulation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, P.K.; Beaupere, C.; Barlit, H.; Romero, A.M.; Tsuchiya, M.; Muir, M.; Martínez-Pastor, M.T.; Puig, S.; Kaeberlein, M.; Labunskyy, V.M. Deficiency of the RNA-Binding Protein Cth2 Extends Yeast Replicative Lifespan by Alleviating Its Repressive Effects on Mitochondrial Function. Cell Rep. 2022, 40, 111113. [Google Scholar] [CrossRef] [PubMed]
- Berthelet, S.; Usher, J.; Shulist, K.; Hamza, A.; Maltez, N.; Johnston, A.; Fong, Y.; Harris, L.J.; Baetz, K. Functional Genomics Analysis of the Saccharomyces cerevisiae Iron Responsive Transcription Factor Aft1 Reveals Iron-Independent Functions. Genetics 2010, 185, 1111–1128. [Google Scholar] [CrossRef] [Green Version]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.-G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione Revisited: A Vital Function in Iron Metabolism and Ancillary Role in Thiol-Redox Control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef] [Green Version]
- Philpott, C.C.; Protchenko, O. Response to Iron Deprivation in Saccharomyces cerevisiae. Eukaryot. Cell 2008, 7, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Jordá, T.; Rozès, N.; Puig, S. Sterol Composition Modulates the Response of Saccharomyces cerevisiae to Iron Deficiency. J. Fungi 2021, 7, 901. [Google Scholar] [CrossRef] [PubMed]
- Montellà-Manuel, S.; Pujol-Carrion, N.; Torre-Ruiz, M.A. Aft1 Nuclear Localization and Transcriptional Response to Iron Starvation Rely upon TORC2/Ypk1 Signaling and Sphingolipid Biosynthesis. Int. J. Mol. Sci. 2023, 24, 2438. [Google Scholar] [CrossRef]
- Brett, C.L.; Kallay, L.; Hua, Z.; Green, R.; Chyou, A.; Zhang, Y.; Graham, T.R.; Donowitz, M.; Rao, R. Genome-Wide Analysis Reveals the Vacuolar PH-Stat of Saccharomyces cerevisiae. PLoS ONE 2011, 6, e17619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montella-Manuel, S.; Pujol-Carrion, N.; Mechoud, M.A.; de la Torre-Ruiz, M.A. Bulk Autophagy Induction and Life Extension Is Achieved When Iron Is the Only Limited Nutrient in Saccharomyces cerevisiae. Biochem. J. 2021, 478, 811–837. [Google Scholar] [CrossRef] [PubMed]
- Gascon, J.M.; Oliveri, V.; McGown, A.; Kaya, E.; Chen, Y.L.; Austin, C.; Walker, M.; Platt, F.M.; Vecchio, G.; Spencer, J. Synthesis and Study of Multifunctional Cyclodextrin–Deferasirox Hybrids. ChemMedChem 2019, 14, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakoury-Elizeh, M.; Tiedeman, J.; Rashford, J.; Ferea, T.; Demeter, J.; Garcia, E.; Rolfes, R.; Brown, P.O.; Botstein, D.; Philpott, C.C. Transcriptional Remodeling in Response to Iron Deprivation in Saccharomyces cerevisiae. Mol. Biol. Cell 2004, 15, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the Signaling Pathways That Control Longevity and Health Span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef] [PubMed]
- Mangan, D. Iron: An Underrated Factor in Aging. Aging 2021, 13, 23407–23415. [Google Scholar] [CrossRef]
- Ndong, M.; Kazami, M.; Suzuki, T.; Uehara, M.; Katsumata, S.I.; Inoue, H.; Kobayashi, K.I.; Tadokoro, T.; Suzuki, K.; Yamamoto, Y. Iron Deficiency Down-Regulates the Akt/TSC1-TSC2/Mammalian Target of Rapamycin Signaling Pathway in Rats and in COS-1 Cells. Nutr. Res. 2009, 29, 640–647. [Google Scholar] [CrossRef]
- Carosi, J.M.; Fourrier, C.; Bensalem, J.; Sargeant, T.J. The mTOR–Lysosome Axis at the Centre of Ageing. FEBS Open Bio 2022, 12, 739–757. [Google Scholar] [CrossRef]
- Sharma, R.; Kumar, R.; Sharma, A.; Goel, A.; Padwad, Y. Long-Term Consumption of Green Tea EGCG Enhances Murine Health Span by Mitigating Multiple Aspects of Cellular Senescence in Mitotic and Post-Mitotic Tissues, Gut Dysbiosis, and Immunosenescence. J. Nutr. Biochem. 2022, 107, 109068. [Google Scholar] [CrossRef]
- Niu, Y.; Na, L.; Feng, R.; Gong, L.; Zhao, Y.; Li, Q.; Li, Y.; Sun, C. The Phytochemical, EGCG, Extends Lifespan by Reducing Liver and Kidney Function Damage and Improving Age-Associated Inflammation and Oxidative Stress in Healthy Rats. Aging Cell 2013, 12, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Huang, X.; Kropat, J.; Henras, A.; Merchant, S.S.; Dickson, R.C.; Chanfreau, G.F. Sphingolipid Signaling Mediates Iron Toxicity. Cell Metab. 2012, 16, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Grilley, M.M.; Stock, S.D.; Dickson, R.C.; Lester, R.L.; Takemoto, J.Y. Syringomycin Action Gene SYR2 Is Essential for Sphingolipid 4- Hydroxylation in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 11062–11068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakoury-Elizeh, M.; Protchenko, O.; Berger, A.; Cox, J.; Gable, K.; Dunn, T.M.; Prinz, W.A.; Bard, M.; Philpott, C.C. Metabolic Response to Iron Deficiency in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 14823–14833. [Google Scholar] [CrossRef] [Green Version]
- Lester, R.L.; Withers, B.R.; Schultz, M.A.; Dickson, R.C. Iron, Glucose and Intrinsic Factors Alter Sphingolipid Composition as Yeast Cells Enter Stationary Phase. Biochim. Biophys. Acta 2013, 1831, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gietz, R.D.; Schiestl, R.H. High-Efficiency Yeast Transformation Using the LiAc/SS Carrier DNA/PEG Method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, E.; Gandhi, T.; Permentier, H.P.; Breitling, R.; Poolman, B.; Slotboom, D.J. The Yeast Vacuolar Membrane Proteome. Mol. Cell. Proteom. 2009, 8, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-Pot, Solid-Phase-Enhanced Sample Preparation for Proteomics Experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Osório, H.; Silva, C.; Ferreira, M.; Gullo, I.; Máximo, V.; Barros, R.; Mendonça, F.; Oliveira, C.; Carneiro, F. Proteomics Analysis of Gastric Cancer Patients with Diabetes Mellitus. J. Clin. Med. 2021, 10, 407. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 Is a Major Target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Medeiros, T.; Vilaca, R.; Moradas-Ferreira, P.; Costa, V. Reduced TORC1 Signaling Abolishes Mitochondrial Dysfunctions and Shortened Chronological Lifespan of Isc1p-Deficient Cells. Microb. Cell 2014, 1, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Puig, S.; Askeland, E.; Thiele, D.J. Coordinated Remodeling of Cellular Metabolism during Iron Deficiency through Targeted mRNA Degradation. Cell 2005, 120, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Fabrizio, P.; Longo, V.D. The Chronological Life Span of Saccharomyces cerevisiae. Aging Cell 2003, 2, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Source |
|---|---|---|
| BY4741 a,b,c,d | Mata, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | EUROSCARF |
| ncr1Δ a,b,c,d | BY4741 ncr1::KanMX4 | [56] |
| yck3∆ a | BY4741 yck3::KanMX4 | This study |
| pep4∆ | BY4741 pep4::KanMX4 | EUROSCARF |
| aft1Δ | BY4741 aft1::HIS3 | [75] |
| ncr1Δaft1Δ | BY4741 ncr1::KanMX4 aft1::HIS3 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, T.S.; Costa, R.S.; Vilaça, R.; Lemos, C.; Teixeira, V.; Pereira, C.; Costa, V. Iron Limitation Restores Autophagy and Increases Lifespan in the Yeast Model of Niemann–Pick Type C1. Int. J. Mol. Sci. 2023, 24, 6221. https://doi.org/10.3390/ijms24076221
Martins TS, Costa RS, Vilaça R, Lemos C, Teixeira V, Pereira C, Costa V. Iron Limitation Restores Autophagy and Increases Lifespan in the Yeast Model of Niemann–Pick Type C1. International Journal of Molecular Sciences. 2023; 24(7):6221. https://doi.org/10.3390/ijms24076221
Chicago/Turabian StyleMartins, Telma S., Rafaela S. Costa, Rita Vilaça, Carolina Lemos, Vitor Teixeira, Clara Pereira, and Vítor Costa. 2023. "Iron Limitation Restores Autophagy and Increases Lifespan in the Yeast Model of Niemann–Pick Type C1" International Journal of Molecular Sciences 24, no. 7: 6221. https://doi.org/10.3390/ijms24076221
APA StyleMartins, T. S., Costa, R. S., Vilaça, R., Lemos, C., Teixeira, V., Pereira, C., & Costa, V. (2023). Iron Limitation Restores Autophagy and Increases Lifespan in the Yeast Model of Niemann–Pick Type C1. International Journal of Molecular Sciences, 24(7), 6221. https://doi.org/10.3390/ijms24076221