
The Succession of the Cellulolytic Microbial Community from the Soil during Oat Straw Decomposition

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
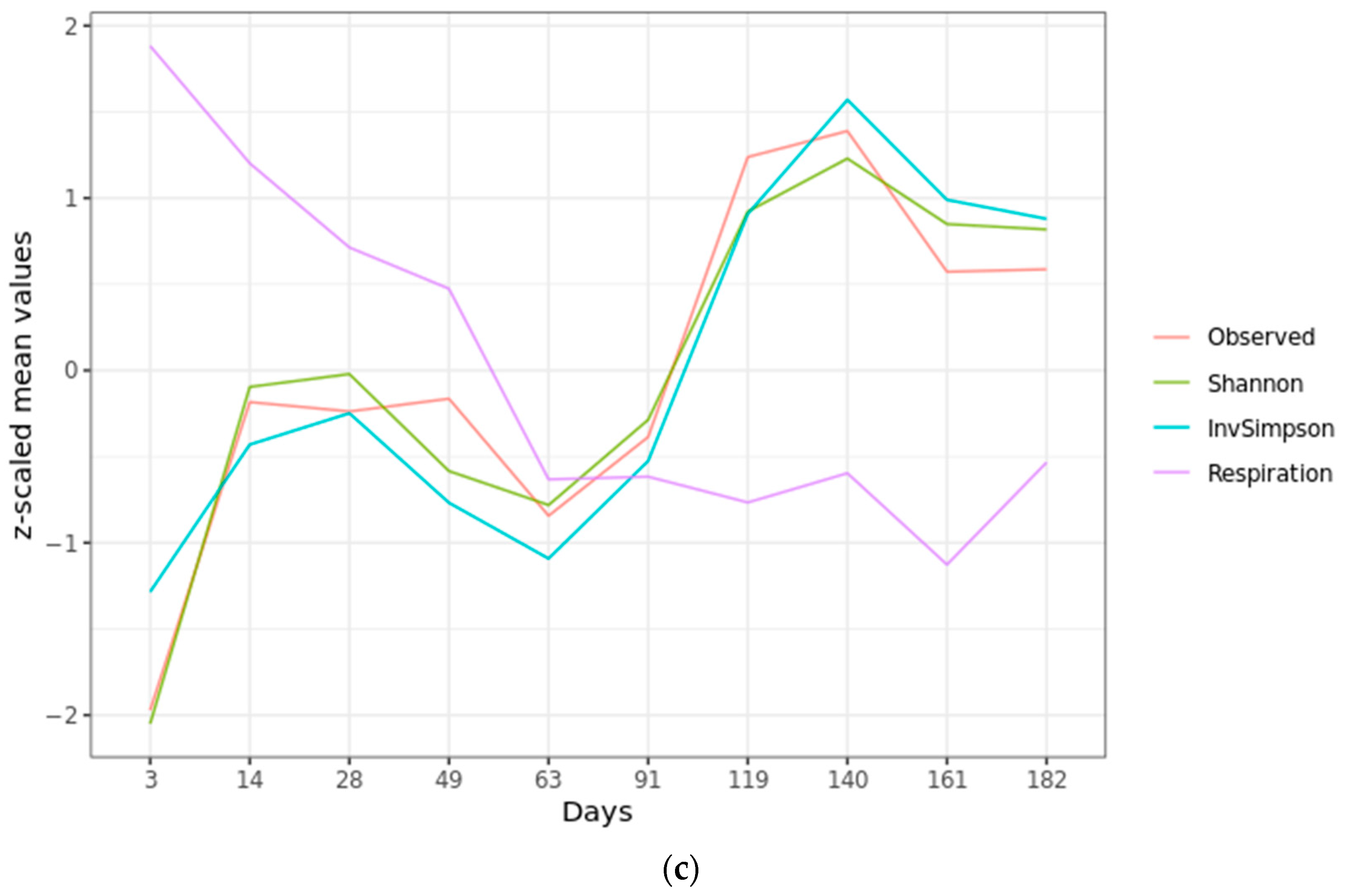
2.1. Microbial Activity
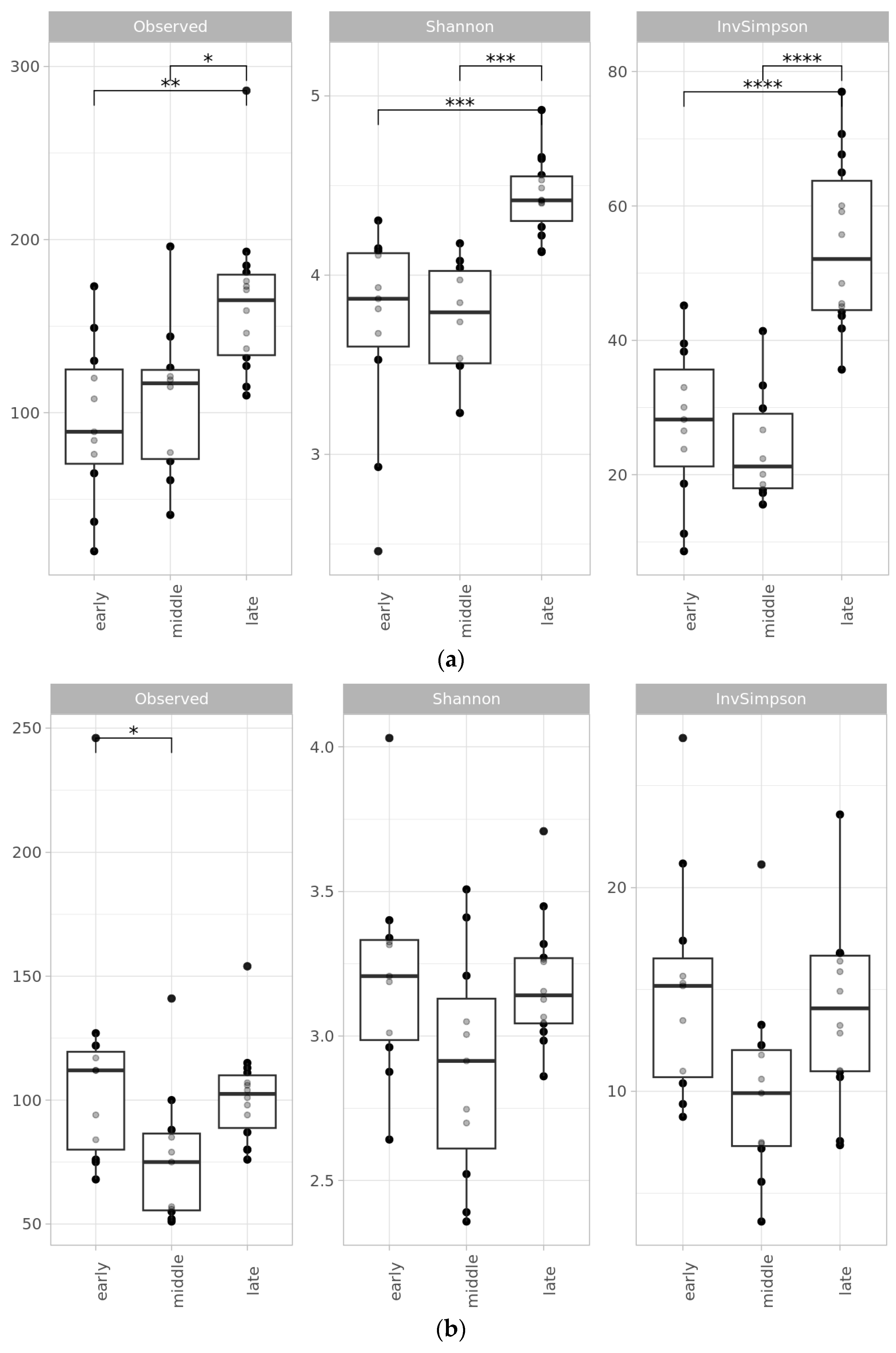
2.2. Microbial Diversity
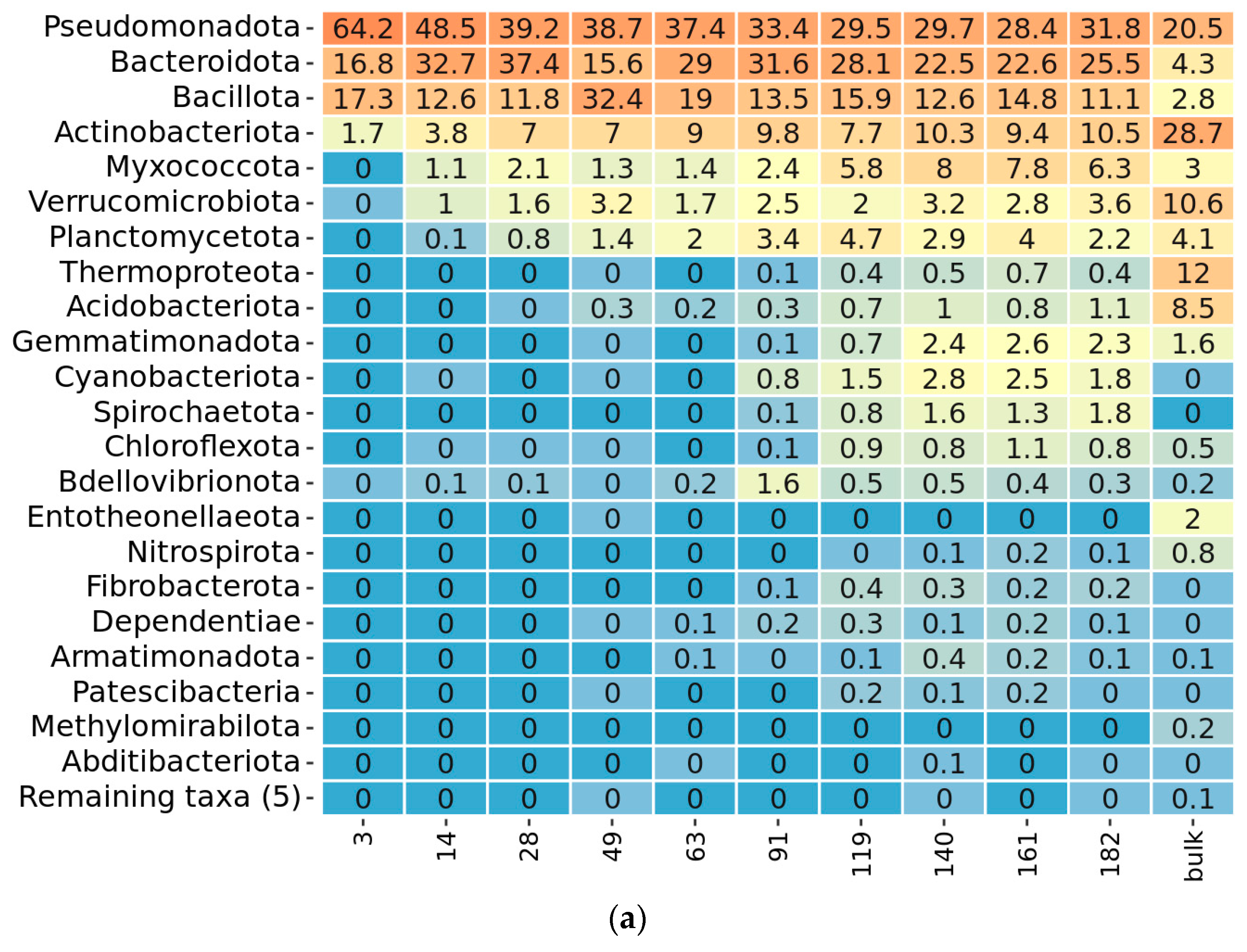
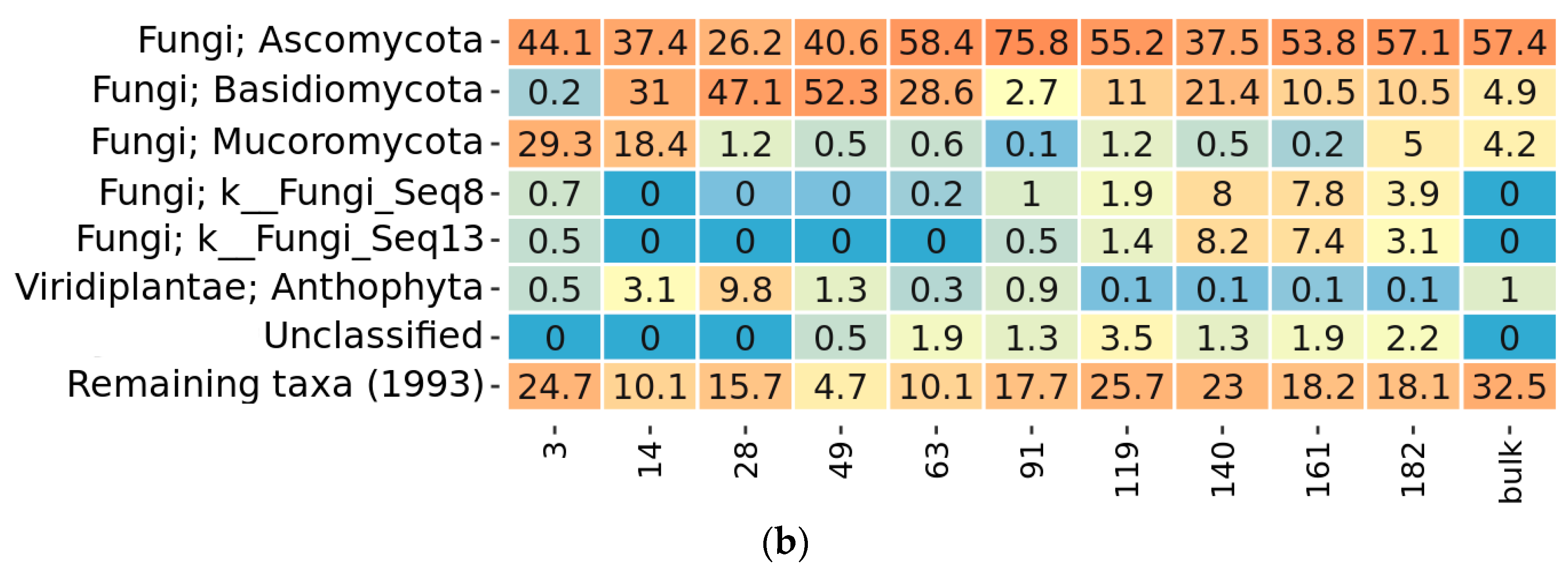
2.3. Taxonomy Overview
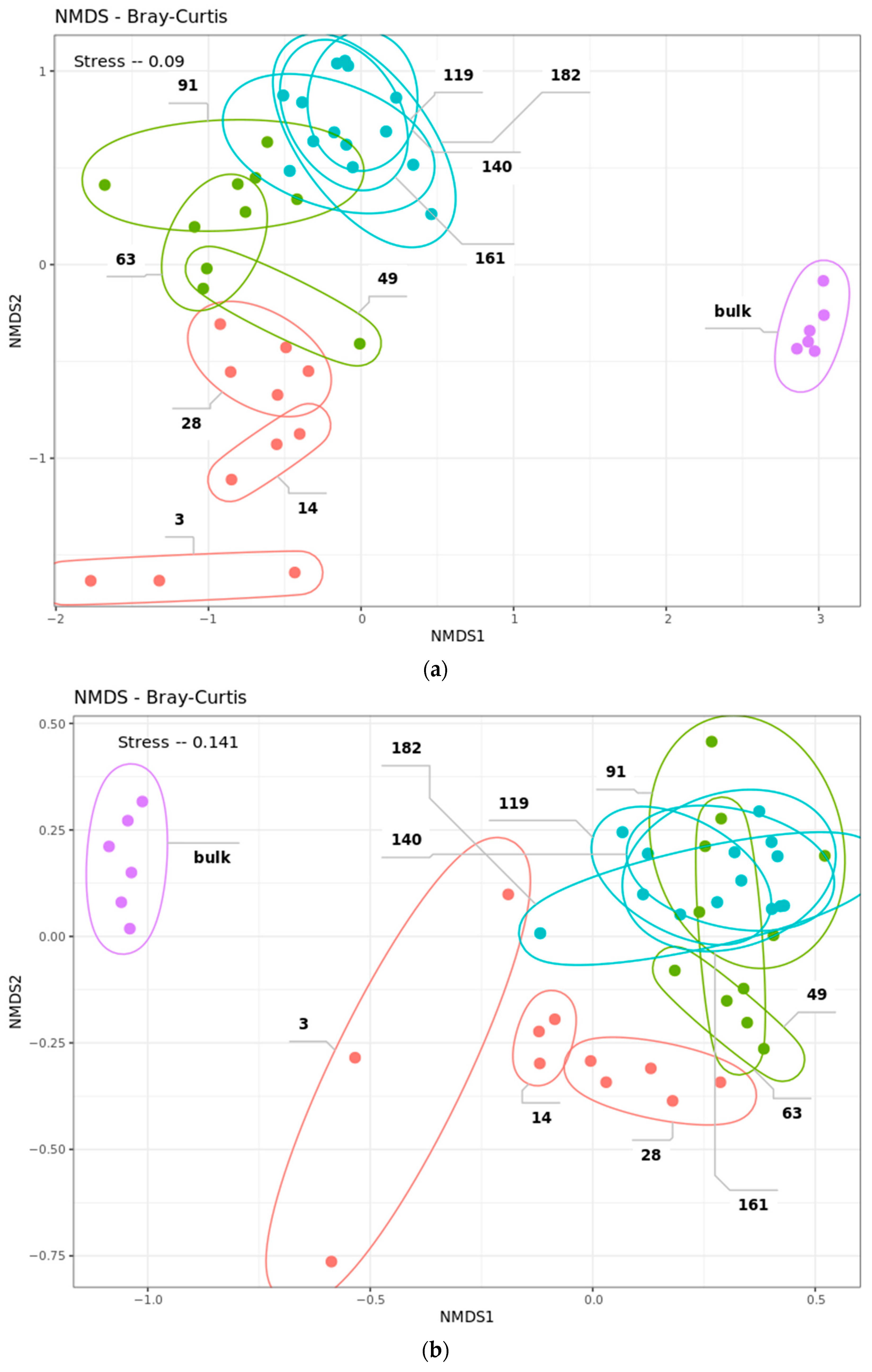
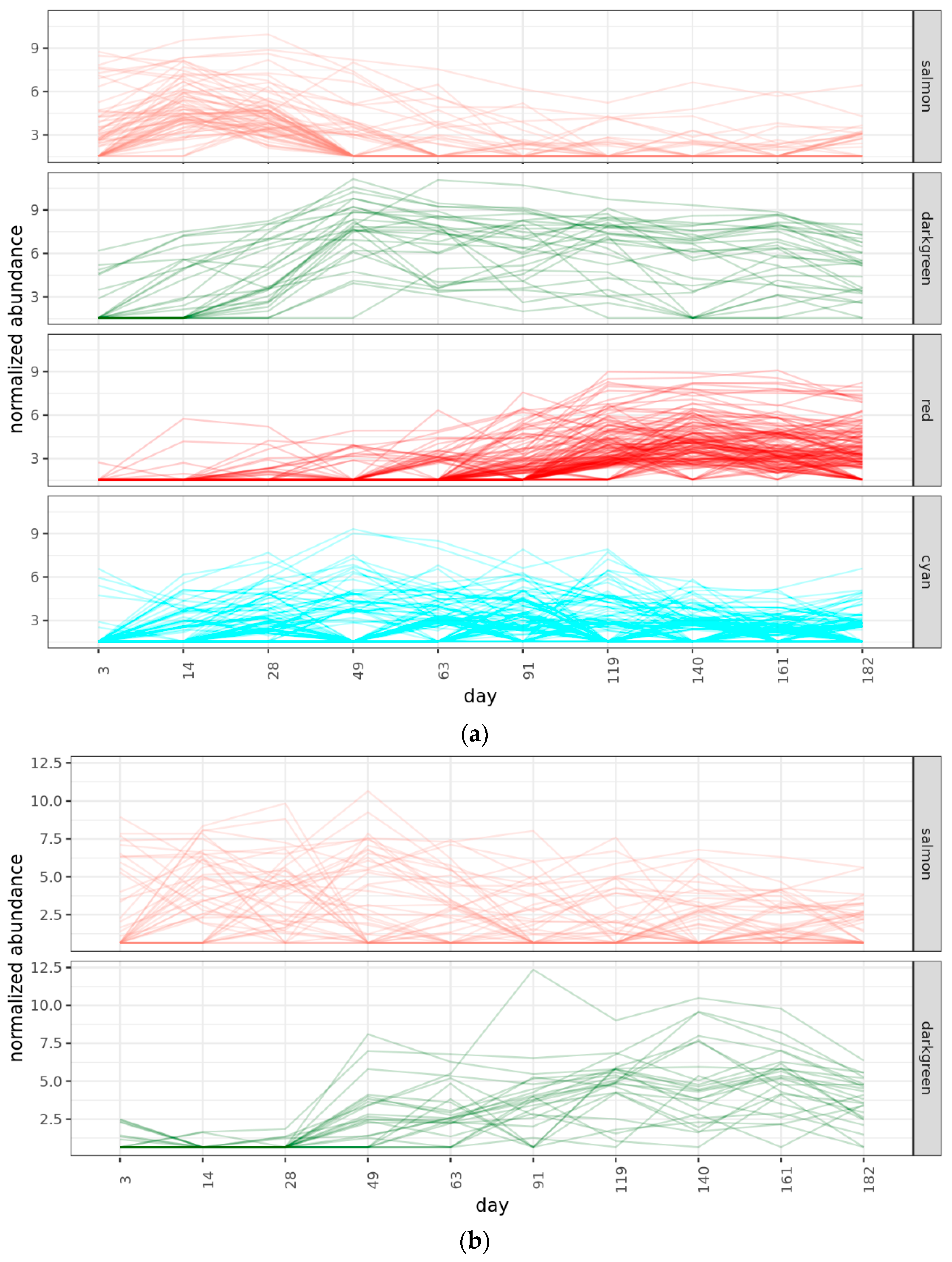
2.4. Community Succession
2.4.1. Data Filtering
2.4.2. Bacterial Phases
2.4.3. Fungal Phases
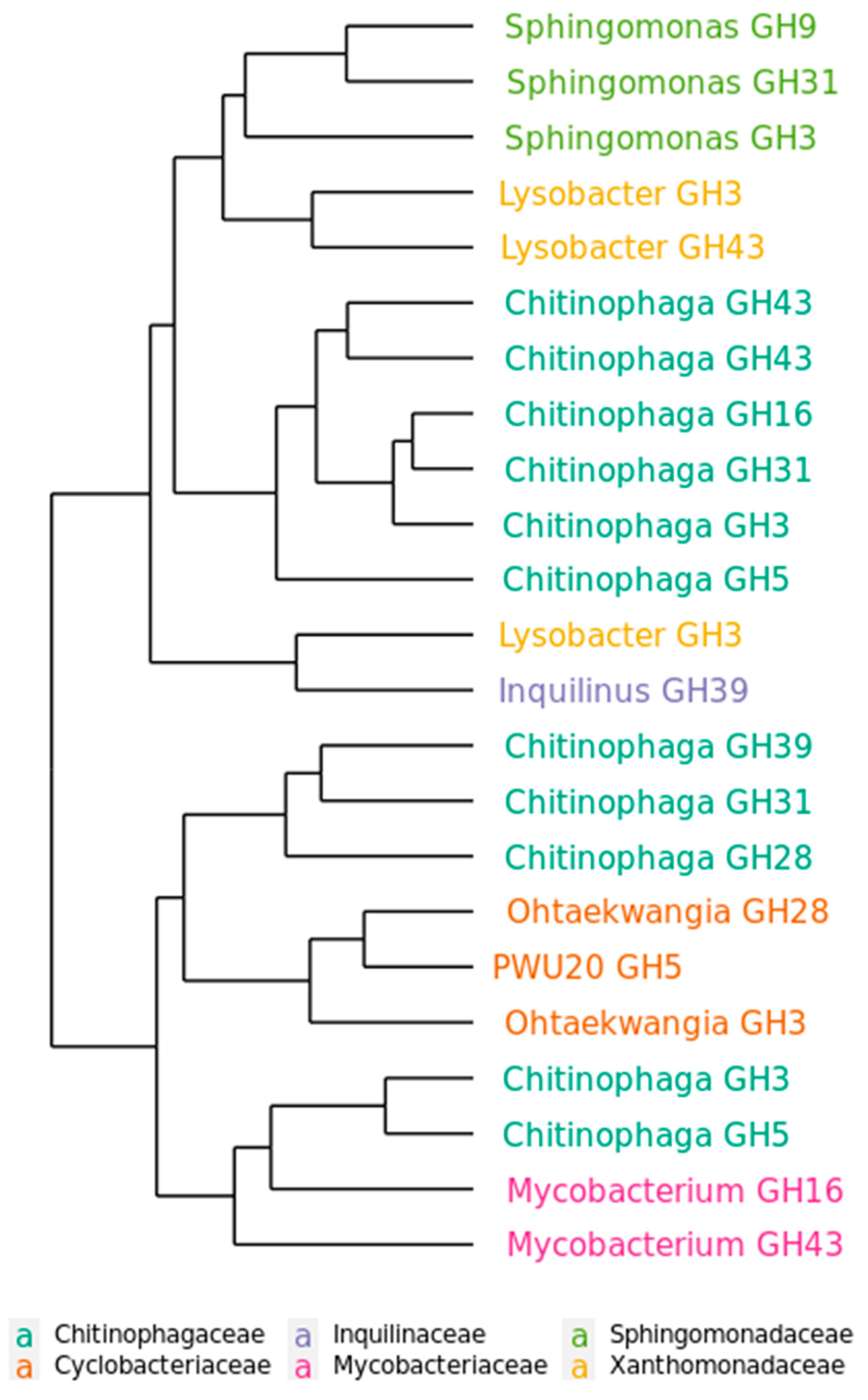
2.5. Functional Distribution of Glycoside hydrolases in the Mature Decomposing Community
2.6. Succession of GH Genes during Phases of Decomposition
3. Discussion
4. Materials and Methods
4.1. Experiment Design
4.2. Microbial Activity Test by the SR Measurement
4.3. Sample Collecting and Amplicon Sequencing
4.4. Amplicon Data Analysis
4.5. Full Metagenome Sequencing and GH Gene Analysis
4.6. Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sokhansanj, S.; Mani, S.; Stumborg, M.; Samson, R.; Fenton, J. Production and Distribution of Cereal Straw on the Canadian Prairies. Can. Biosyst. Eng. 2006, 48, 39–44. [Google Scholar]
- Seglah, P.A.; Wang, Y.; Wang, H.; Bi, Y.; Zhou, K.; Wang, Y.; Wang, H.; Feng, X. Crop Straw Utilization and Field Burning in Northern Region of Ghana. J. Clean. Prod. 2020, 261, 121191. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, F.; Nan, Z. Comparative Grain Yield, Straw Yield, Chemical Composition, Carbohydrate and Protein Fractions, In Vitro Digestibility and Rumen Degradability of Four Common Vetch Varieties Grown on the Qinghai-Tibetan Plateau. Animals 2019, 9, 505. [Google Scholar] [CrossRef] [Green Version]
- Romasanta, R.R.; Sander, B.O.; Gaihre, Y.K.; Alberto, M.C.; Gummert, M.; Quilty, J.; Nguyen, V.H.; Castalone, A.G.; Balingbing, C.; Sandro, J.; et al. How Does Burning of Rice Straw Affect CH4 and N2O Emissions? A Comparative Experiment of Different on-Field Straw Management Practices. Agric. Ecosyst. Environ. 2017, 239, 143–153. [Google Scholar] [CrossRef]
- Ren, J.; Yu, P.; Xu, X. Straw Utilization in China—Status and Recommendations. Sustainability 2019, 11, 1762. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, S.; Gao, J.; Chen, L.; Ning, H.; Hu, Z.; Lu, J.; Tan, X.; Zeng, Y.; Pan, X.; et al. Long-Term Straw Return with Reducing Chemical Fertilizers Application Improves Soil Nitrogen Mineralization in a Double Rice-Cropping System. Agronomy 2022, 12, 1767. [Google Scholar] [CrossRef]
- Han, Y.; Ma, W.; Zhou, B.; Salah, A.; Geng, M.; Cao, C.; Zhan, M.; Zhao, M. Straw Return Increases Crop Grain Yields and K-Use Efficiency under a Maize-Rice Cropping System. Crop J. 2021, 9, 168–180. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The Carbohydrate-Active Enzyme Database: Functions and Literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Henrissat, B. A Classification of Glycosyl Hydrolases Based on Amino Acid Sequence Similarities. Biochem. J. 1991, 280 Pt 2, 309–316. [Google Scholar] [CrossRef]
- Bourne, Y.; Henrissat, B. Glycoside Hydrolases and Glycosyltransferases: Families and Functional Modules. Curr. Opin. Struct. Biol. 2001, 11, 593–600. [Google Scholar] [CrossRef]
- Yeoman, C.J.; Han, Y.; Dodd, D.; Schroeder, C.M.; Mackie, R.I.; Cann, I.K.O. Chapter 1—Thermostable Enzymes as Biocatalysts in the Biofuel Industry. In Advances in Applied Microbiology; Academic Press: Cambridge, MA, USA, 2010; Volume 70, pp. 1–55. [Google Scholar]
- Nguyen, S.T.C.; Freund, H.L.; Kasanjian, J.; Berlemont, R. Function, Distribution, and Annotation of Characterized Cellulases, Xylanases, and Chitinases from CAZy. Appl. Microbiol. Biotechnol. 2018, 102, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Tolalpa, L.; Jiménez, D.J.; de Lima Brossi, M.J.; Salles, J.F.; van Elsas, J.D. Different Inocula Produce Distinctive Microbial Consortia with Similar Lignocellulose Degradation Capacity. Appl. Microbiol. Biotechnol. 2016, 100, 7713–7725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzyński, J.; Wróbel, B.; Ewa, G. Cellulolytic Properties of a Potentially Lignocellulose-Degrading Bacillus Sp. 8E1A Strain Isolated from Bulk Soil. Agronomy 2022, 12, 665. [Google Scholar] [CrossRef]
- Ghazanfar, M.; Irfan, M.; Nadeem, M.; Shakir, H.; Khan, D.-M.; Ali, S.; Saeed, S.; Mehmood, T. Isolation of cellulolytic bacteria from soil and valorization of different lignocellulosic wastes for cellulase production by submerged fermentation. Cellul. Chem. Technol. 2021, 55, 821–828. [Google Scholar] [CrossRef]
- Harindintwali, J.D.; Zhou, J.; Habimana, I.; Dong, X.; Sun, C.; Nwamba, M.C.; Yang, W.; Yu, X. Biotechnological Potential of Cellulolytic Nitrogen-Fixing Klebsiella Sp. C-3 Isolated from Paddy Soil. Bioresour. Technol. Rep. 2021, 13, 100624. [Google Scholar] [CrossRef]
- Shrestha, B.G.; Ghimire, S.; Bhattarai, S.; Phuyal, S.; Thapa, B. Isolation and Screening of Potential Cellulolytic and Xylanolytic Bacteria From Soil Sample for Degradation of Lignocellulosic Biomass. JTLS 2016, 6, 93192. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, R.C.; Cardenas, E.; Leung, H.; Szeitz, A.; Jensen, L.D.; Mohn, W.W. Long-Term Enrichment of Stress-Tolerant Cellulolytic Soil Populations Following Timber Harvesting Evidenced by Multi-Omic Stable Isotope Probing. Front. Microbiol. 2017, 8, 537. [Google Scholar]
- Berlemont, R.; Allison, S.D.; Weihe, C.; Lu, Y.; Brodie, E.L.; Martiny, J.B.H.; Martiny, A.C. Cellulolytic Potential under Environmental Changes in Microbial Communities from Grassland Litter. Front. Microbiol. 2014, 5, 639. [Google Scholar] [CrossRef]
- Avellaneda-Torres, L.M.; Pulido, C.P.G.; Rojas, E.T. Assessment of Cellulolytic Microorganisms in Soils of Nevados Park, Colombia. Braz. J. Microbiol. 2014, 45, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, L.; Hassan, M.; Xie, B. Succession of the Functional Microbial Communities and the Metabolic Functions in Maize Straw Composting Process. Bioresour. Technol. 2018, 256, 333–341. [Google Scholar] [CrossRef]
- Zhang, X.; Borjigin, Q.; Gao, J.; Yu, X.; Zhang, B.; Hu, S.; Han, S.; Liu, R.; Zhang, S. Community Succession and Straw Degradation Characteristics Using a Microbial Decomposer at Low Temperature. PLoS ONE 2022, 17, e0270162. [Google Scholar] [CrossRef]
- Chen, L.; Sun, S.; Yao, B.; Peng, Y.; Gao, C.; Qin, T.; Zhou, Y.; Sun, C.; Quan, W. Effects of Straw Return and Straw Biochar on Soil Properties and Crop Growth: A Review. Front. Plant Sci. 2022, 13, 986763. [Google Scholar] [CrossRef] [PubMed]
- Shuliko, N.N.; Khamova, O.F.; Timokhin, A.Y.; Boiko, V.S.; Tukmacheva, E.V.; Krempa, A. Author Correction: Influence of Long-Term Intensive Use of Irrigated Meadow-Chernozem Soil on the Biological Activity and Productivity of the Arable Layer. Sci. Rep. 2022, 12, 19755. [Google Scholar] [CrossRef]
- Melnichuk, T.N.; Egovtseva, A.Y.; Abdurashytov, S.F.; Abdurashytova, E.R.; Turin, E.N.; Gongalo, A.A.; Zubochenko, A.A.; Pashtetskiy, V.S. State of Microbiocenosis of Southern Chernozem under the No-till System. E3S Web Conf. 2020, 224, 04009. [Google Scholar] [CrossRef]
- Zverev, A.O.; Pershina, E.V.; Provorov, N.A.; Andronov, E.E.; Serikova, E.N. Metagenomic characteristic of rhizosphere effect on cereals in black and sod-podzolic soils. Agric. Biol. 2016, 51, 654–663. [Google Scholar] [CrossRef]
- Evdokimova, E.V.; Gladkov, G.V.; Kuzina, N.I.; Ivanova, E.A.; Kimeklis, A.K.; Zverev, A.O.; Kichko, A.A.; Aksenova, T.S.; Pinaev, A.G.; Andronov, E.E. The Difference between Cellulolytic ‘Culturomes’ and Microbiomes Inhabiting Two Contrasting Soil Types. PLoS ONE 2020, 15, e0242060. [Google Scholar] [CrossRef]
- Brabcová, V.; Nováková, M.; Davidová, A.; Baldrian, P. Dead Fungal Mycelium in Forest Soil Represents a Decomposition Hotspot and a Habitat for a Specific Microbial Community. New Phytol. 2016, 210, 1369–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, C.; Hartmann, M.; Frey, B.; Stierli, B.; Rumpel, C.; Buchmann, N.; Brunner, I. Microbial Succession on Decomposing Root Litter in a Drought-Prone Scots Pine Forest. ISME J. 2019, 13, 2346–2362. [Google Scholar] [CrossRef] [Green Version]
- Khitrov, N.B.; Khaydapova, D.D. Viscoelastic Behavior of Vertic Solonetz in the Kamennaya Steppe. Eurasian Soil Sci. 2019, 52, 808–821. [Google Scholar] [CrossRef]
- Wolińska, A. Chapter 2—Metagenomic Achievements in Microbial Diversity Determination in Croplands: A Review. In Microbial Diversity in the Genomic Era; Das, S., Dash, H.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 15–35. [Google Scholar]
- Elmajdoub, B.; Marschner, P. Response of Microbial Activity and Biomass to Soil Salinity When Supplied with Glucose and Cellulose. Soil Sci. Plant Nutr. 2015, 15, 816–832. [Google Scholar] [CrossRef]
- Cuddeford, D. Oats for Animal Feed. In The Oat Crop; Springer: Berlin/Heidelberg, Germany, 1995; pp. 321–368. [Google Scholar]
- Ma, B.-L.; Zheng, Z.; Ren, C. Chapter 6—Oat. In Crop Physiology Case Histories for Major Crops; Sadras, V.O., Calderini, D.F., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 222–248. ISBN 978-0-12-819194-1. [Google Scholar]
- Wahdan, S.F.M.; Ji, L.; Schädler, M.; Wu, Y.-T.; Sansupa, C.; Tanunchai, B.; Buscot, F.; Purahong, W. Future Climate Conditions Accelerate Wheat Straw Decomposition alongside Altered Microbial Community Composition, Assembly Patterns, and Interaction Networks. ISME J. 2023, 17, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xing, K.; Jiang, J.-H.; Xu, L.-H.; Li, W.-J. Biodiversity, Bioactive Natural Products and Biotechnological Potential of Plant-Associated Endophytic Actinobacteria. Appl. Microbiol. Biotechnol. 2011, 89, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, P.H.; Karthik, C.; Kadirvelu, K.; Divya, K.L.; Venkatasalam, E.P.; Srinivasan, S.; Ramkumar, G.; Saranya, C.; Shanmuganathan, R. Plant Growth Promoting Rhizobacteria (PGPR): A Potential Alternative Tool for Nematodes Bio-Control. Biocatal. Agric. Biotechnol. 2019, 17, 119–128. [Google Scholar] [CrossRef]
- Huang, S.; Sheng, P.; Zhang, H. Isolation and Identification of Cellulolytic Bacteria from the Gut of Holotrichia Parallela Larvae (Coleoptera: Scarabaeidae). Int. J. Mol. Sci. 2012, 13, 2563–2577. [Google Scholar] [CrossRef]
- Oh, H.-W.; Heo, S.-Y.; Kim, D.Y.; Park, D.-S.; Bae, K.S.; Park, H.-Y. Biochemical Characterization and Sequence Analysis of a Xylanase Produced by an Exo-Symbiotic Bacterium of Gryllotalpa Orientalis, Cellulosimicrobium Sp. HY-12. Antonie Van Leeuwenhoek 2008, 93, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Opazo, R.; Ortúzar, F.; Navarrete, P.; Espejo, R.; Romero, J. Reduction of Soybean Meal Non-Starch Polysaccharides and α-Galactosides by Solid-State Fermentation Using Cellulolytic Bacteria Obtained from Different Environments. PLoS ONE 2012, 7, e44783. [Google Scholar] [CrossRef]
- Villas-Boas, S.; Esposito, E.; Mitchell, D. Microbial Conversion of Lignocellulosic Residues for Production of Animal Feeds. Anim. Feed Sci. Technol. 2002, 98, 1–12. [Google Scholar] [CrossRef]
- Sadhu, S.; Saha, P.; Mayilraj, S.; Maiti, T.K. Lactose-Enhanced Cellulase Production by Microbacterium Sp. Isolated from Fecal Matter of Zebra (Equus Zebra). Curr. Microbiol. 2011, 62, 1050–1055. [Google Scholar] [CrossRef]
- Ventorino, V.; Ionata, E.; Birolo, L.; Montella, S.; Marcolongo, L.; de Chiaro, A.; Espresso, F.; Faraco, V.; Pepe, O. Lignocellulose-Adapted Endo-Cellulase Producing Streptomyces Strains for Bioconversion of Cellulose-Based Materials. Front. Microbiol. 2016, 7, 2061. [Google Scholar]
- Tan, H.; Miao, R.; Liu, T.; Yang, L.; Yang, Y.; Chen, C.; Lei, J.; Li, Y.; He, J.; Sun, Q.; et al. A Bifunctional Cellulase-Xylanase of a New Chryseobacterium Strain Isolated from the Dung of a Straw-Fed Cattle. Microb. Biotechnol. 2018, 11, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Photphisutthiphong, Y.; Vatanyoopaisarn, S. Dyadobacter and Sphingobacterium Isolated from Herbivore Manure in Thailand and Their Cellulolytic Activity in Various Organic Waste Substrates. Agric. Nat. Resour. 2019, 53, 89–98. [Google Scholar] [CrossRef]
- Gladkov, G.V.; Kimeklis, A.K.; Afonin, A.M.; Lisina, T.O.; Orlova, O.V.; Aksenova, T.S.; Kichko, A.A.; Pinaev, A.G.; Andronov, E.E. The Structure of Stable Cellulolytic Consortia Isolated from Natural Lignocellulosic Substrates. Int. J. Mol. Sci. 2022, 23, 10779. [Google Scholar] [CrossRef]
- Meng, Q.; Yang, W.; Men, M.; Bello, A.; Xu, X.; Xu, B.; Deng, L.; Jiang, X.; Sheng, S.; Wu, X.; et al. Microbial Community Succession and Response to Environmental Variables During Cow Manure and Corn Straw Composting. Front. Microbiol. 2019, 10, 529. [Google Scholar] [PubMed] [Green Version]
- Kim, Y.-K.; Lee, S.-C.; Cho, Y.-Y.; Oh, H.-J.; Ko, Y.H. Isolation of Cellulolytic Bacillus Subtilis Strains from Agricultural Environments. ISRN Microbiol. 2012, 2012, 650563. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wang, X.; Zhou, J.; Lü, X. Degradation of Switchgrass by Bacillus Subtilis 1AJ3 and Expression of a Beta-Glycoside Hydrolase. Front. Microbiol. 2022, 13, 922371. [Google Scholar] [PubMed]
- Yadav, S.; Dubey, S.K. Cellulose Degradation Potential of Paenibacillus Lautus Strain BHU3 and Its Whole Genome Sequence. Bioresour. Technol. 2018, 262, 124–131. [Google Scholar] [CrossRef]
- Duan, H.; Fu, C.; Du, G.; Xie, S.; Liu, M.; Zhang, B.; Shi, J.; Sun, J. Dynamic Microstructure Assembly Driven by Lysinibacillus Sp. LF-N1 and Penicillium Oxalicum DH-1 Inoculants Corresponds to Composting Performance. Microorganisms 2022, 10, 709. [Google Scholar] [CrossRef]
- McKee, L.S.; La Rosa, S.L.; Westereng, B.; Eijsink, V.G.; Pope, P.B.; Larsbrink, J. Polysaccharide Degradation by the Bacteroidetes: Mechanisms and Nomenclature. Environ. Microbiol. Rep. 2021, 13, 559–581. [Google Scholar] [CrossRef]
- Terrapon, N.; Lombard, V.; Drula, É.; Lapébie, P.; Al-Masaudi, S.; Gilbert, H.J.; Henrissat, B. PULDB: The Expanded Database of Polysaccharide Utilization Loci. Nucleic Acids Res. 2018, 46, D677–D683. [Google Scholar] [CrossRef]
- Pareek, S.; Azuma, J.I.; Matsui, S.; Shimizu, Y. Degradation of Lignin and Lignin Model Compound under Sulfate Reducing Condition. Water Sci. Technol. 2001, 44, 351–358. [Google Scholar]
- Halsall, D.M.; Gibson, A.H. Cellulose Decomposition and Associated Nitrogen Fixation by Mixed Cultures of Cellulomonas Gelida and Azospirillum Species or Bacillus Macerans. Appl. Environ. Microbiol. 1985, 50, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Kappler, U.; Davenport, K.; Beatson, S.; Lucas, S.; Lapidus, A.; Copeland, A.; Berry, K.W.; Glavina Del Rio, T.; Hammon, N.; Dalin, E.; et al. Complete Genome Sequence of the Facultatively Chemolithoautotrophic and Methylotrophic Alpha Proteobacterium Starkeya Novella Type Strain (ATCC 8093T). Stand. Genomic Sci. 2012, 7, 44–58. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Matsumoto, A.; Shimada, R.; Inahashi, Y.; Ōmura, S.; Takahashi, Y. Conexibacter Arvalis Sp. Nov., Isolated from a Cultivated Field Soil Sample. Int. J. Syst. Evol. Microbiol. 2012, 62, 2400–2404. [Google Scholar] [CrossRef] [Green Version]
- Hungate, B.A.; Marks, J.C.; Power, M.E.; Schwartz, E.; van Groenigen, K.J.; Blazewicz, S.J.; Chuckran, P.; Dijkstra, P.; Finley, B.K.; Firestone, M.K.; et al. The Functional Significance of Bacterial Predators. mBio 2021, 12, e00466-21. [Google Scholar] [CrossRef] [PubMed]
- Johnke, J.; Cohen, Y.; de Leeuw, M.; Kushmaro, A.; Jurkevitch, E.; Chatzinotas, A. Multiple Micro-Predators Controlling Bacterial Communities in the Environment. Curr. Opin. Biotechnol. 2014, 27, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Pepe-Ranney, C.; Weisenhorn, P.; Lipton, M.; Buckley, D.H. Competitive Exclusion and Metabolic Dependency among Microorganisms Structure the Cellulose Economy of an Agricultural Soil. mBio 2021, 12, e03099-20. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Song, K.; Li, S.; Zhang, H.; Bai, N.; Zhang, J.; Zhang, H.; Cai, S.; Lv, W.; Cao, L. Effects of Long-Term Integrated Agri-Aquaculture on the Soil Fungal Community Structure and Function in Vegetable Fields. Sci. Rep. 2021, 11, 10813. [Google Scholar] [CrossRef]
- Tymon, L.S.; Morgan, P.; Gundersen, B.; Inglis, D.A. Potential of Endophytic Fungi Collected from Cucurbita Pepo Roots Grown under Three Different Agricultural Mulches as Antagonistic Endophytes to Verticillium Dahliae in Western Washington. Microbiol. Res. 2020, 240, 126535. [Google Scholar] [CrossRef]
- Wood, M. California Fungi: Coprinellus Flocculosus. Available online: http://www.mykoweb.com/CAF/species/Coprinellus_flocculosus.html (accessed on 24 November 2022).
- Wei, M.-J.; Zhang, H.; Dong, W.; Boonmee, S.; Zhang, D. Introducing Dictyochaeta Aquatica Sp. Nov. and Two New Species of Chloridium (Chaetosphaeriaceae, Sordariomycetes) from Aquatic Habitats. Phytotaxa 2018, 362, 187. [Google Scholar] [CrossRef]
- El-Said, A.H.M.; Saleem, A. Ecological and Physiological Studies on Soil Fungi at Western Region, Libya. Mycobiology 2008, 36, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sivapalan, A.; Metussin, R.; Harndan, F.; Zain, R.M. Fungi Associated with Postharvest Fruit Rots of Durio Graveolens and D. Kutejensis in Brunei Darussalam. Australas. Plant Pathol. 1998, 27, 274–277. [Google Scholar] [CrossRef]
- Matić, S.; Gilardi, G.; Gullino, M.L.; Garibaldi, A. Emergence of Leaf Spot Disease on Leafy Vegetable and Ornamental Crops Caused by Paramyrothecium and Albifimbria Species. Phytopathology 2019, 109, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, R.A.; Held, B.W.; Abdel-Azeem, A.M. New Record of Chaetomium Iranianum MF787598 (Chaetomiaceae) for the Egyptian and African Mycobiota. Microb. Biosyst. 2017, 2, 6–9. [Google Scholar] [CrossRef]
- Darwish, A.; Abdel-Azeem, A. Chaetomium Enzymes and Their Applications. In Recent Developments on Genus Chaetomium; Springer: Berlin/Heidelberg, Germany, 2020; pp. 241–249. [Google Scholar]
- Khunnamwong, P.; Ribeiro, J.R.A.; Garcia, K.M.; Hagler, A.N.; Takashima, M.; Ohkuma, M.; Endoh, R.; Sugita, T.; Jindamorakot, S.; Limtong, S. Occultifur Plantarum f.a., Sp. Nov., a Novel Cystobasidiomycetous Yeast Species. Int. J. Syst. Evol. Microbiol. 2017, 67, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, J.; Bauer, R.; Begerow, D.; Oberwinkler, F. Occultifur Externus Sp. Nov., a New Species of Simple-Pored Auricularioid Heterobasidiomycete from Plant Litter in Portugal. Mycologia 1999, 91, 1094–1101. [Google Scholar] [CrossRef]
- de la Cerda, K.A.; Douhan, G.W.; Wong, F.P. Discovery and Characterization of Waitea Circinata Var. Circinata Affecting Annual Bluegrass from the Western United States. Plant Dis. 2007, 91, 791–797. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Vargas García, M.; Suárez-Estrella, F.; Dien, B.; Moreno, J. Lignocellulose-Degrading Enzymes Produced by the Ascomycete Coniochaeta Ligniaria and Related Species: Application for a Lignocellulosic Substrate Treatment. Enzyme Microb. Technol. 2007, 40, 794–800. [Google Scholar] [CrossRef]
- Mondo, S.J.; Jiménez, D.J.; Hector, R.E.; Lipzen, A.; Yan, M.; LaButti, K.; Barry, K.; van Elsas, J.D.; Grigoriev, I.V.; Nichols, N.N. Genome Expansion by Allopolyploidization in the Fungal Strain Coniochaeta 2T2.1 and Its Exceptional Lignocellulolytic Machinery. Biotechnol. Biofuels 2019, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Han, B.-Z.; Ma, Y.; Rombouts, F.M.; Robert Nout, M.J. Effects of Temperature and Relative Humidity on Growth and Enzyme Production by Actinomucor Elegans and Rhizopus Oligosporus during Sufu Pehtze Preparation. Food Chem. 2003, 81, 27–34. [Google Scholar] [CrossRef]
- O’Toole, D.K. Soybean: Soymilk, Tofu, and Okara. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Moreira, E.A.; Persinoti, G.F.; Menezes, L.R.; Paixão, D.A.A.; Alvarez, T.M.; Cairo, J.P.L.F.; Squina, F.M.; Costa-Leonardo, A.M.; Rodrigues, A.; Sillam-Dussès, D.; et al. Complementary Contribution of Fungi and Bacteria to Lignocellulose Digestion in the Food Stored by a Neotropical Higher Termite. Front. Ecol. Evol. 2021, 9, 632590. [Google Scholar] [CrossRef]
- Wilhelm, R.C.; Singh, R.; Eltis, L.D.; Mohn, W.W. Bacterial Contributions to Delignification and Lignocellulose Degradation in Forest Soils with Metagenomic and Quantitative Stable Isotope Probing. ISME J. 2019, 13, 413–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andlar, M.; Rezić, T.; Marđetko, N.; Kracher, D.; Ludwig, R.; Šantek, B. Lignocellulose Degradation: An Overview of Fungi and Fungal Enzymes Involved in Lignocellulose Degradation. Eng. Life Sci. 2018, 18, 768–778. [Google Scholar] [CrossRef] [PubMed]
- De Souza, W.R. Microbial Degradation of Lignocellulosic Biomass. In Sustainable Degradation of Lignocellulosic Biomass—Techniques, Applications and Commercialization; Chandel, A., Ed.; IntechOpen: London, UK, 2013. [Google Scholar]
- Hobdey, S.E.; Donohoe, B.S.; Brunecky, R.; Himmel, M.E.; Bomble, Y.J. New Insights into Microbial Strategies for Biomass Conversion. In Direct Microbial Conversion of Biomass to Advanced Biofuels; Elsevier: Amsterdam, The Netherlands, 2015; pp. 111–127. [Google Scholar]
- Tláskal, V.; Voříšková, J.; Baldrian, P. Bacterial Succession on Decomposing Leaf Litter Exhibits a Specific Occurrence Pattern of Cellulolytic Taxa and Potential Decomposers of Fungal Mycelia. FEMS Microb. Ecol. 2016, 92, fiw177. [Google Scholar] [CrossRef] [Green Version]
- Orlova, O.V.; Kichko, A.A.; Pershina, E.V.; Pinaev, A.G.; Andronov, E.E. Succession of Bacterial Communities in the Decomposition of Oats Straw in Two Soils with Contrasting Properties. Eurasian Soil Sci. 2020, 53, 1620–1628. [Google Scholar] [CrossRef]
- Van Cleve, K.; Coyne, P.I.; Goodwin, E.; Johnson, C.; Kelley, M. A Comparison of Four Methods for Measuring Respiration in Organic Material. Soil Biol. Biochem. 1979, 11, 237–246. [Google Scholar] [CrossRef]
- Gladkov, G.; Kimeklis, A.; Zverev, A.; Pershina, E.; Ivanova, E.; Kichko, A.; Andronov, E.; Abakumov, E. Soil Microbiome of the Postmining Areas in Polar Ecosystems in Surroundings of Nadym, Western Siberia, Russia. Open Agric. 2019, 4, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Kichko, A.A.; Aksenova, T.S.; Shapkin, V.M.; Zverev, A.O.; Khiutti, A.V.; Andronov, E.E. Analysis of Mycobiome in Damaged Potato (Solanum tuberosum L.) Leaves by Using Metagenomic Approaches. Agric. Biol. 2019, 54, 990–1001. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 9 December 2022).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. Ampvis2: An R Package to Analyse and Visualise 16S RRNA Amplicon Data. bioRxiv 2018, bioRxiv:299537. [Google Scholar]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Champaign, IL, USA, 1949. [Google Scholar]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Picante Package—Rdocumentation. Available online: https://www.rdocumentation.org/packages/picante/versions/1.8.2 (accessed on 1 December 2022).
- Girden, E.R. ANOVA: Repeated Measures; Sage Publications Inc.: Thousand Oaks, CA, USA, 1992. [Google Scholar]
- Kruskal, J.B. Multidimensional Scaling by Optimizing Goodness of Fit to a Nonmetric Hypothesis. Psychometrika 1964, 29, 1–27. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; Wiley: New York, NY, USA, 2017; pp. 1–15. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R package Version 2.4-3. 2017. Available online: https://cran.r-project.org/package=vegan (accessed on 9 December 2022).
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing Bacterial Genome Assemblies with Multiplex MinION Sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. MetaFlye: Scalable Long-Read Metagenome Assembly Using Repeat Graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Medaka: Sequence Correction Provided by ONT Research. Available online: https://github.com/nanoporetech/medaka (accessed on 9 December 2022).
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kingdom | Phylum | “Cellulose” Group | “Carbohydrates” Group | “Chitin” Group |
|---|---|---|---|---|
| Archaea | Euryarchaeota | 0 | 1 | 0 |
| Bacteria | Pseudomonadota | 239 | 113 | 67 |
| Bacteroidota | 126 | 123 | 67 | |
| Actinobacteriota | 60 | 52 | 34 | |
| NA | 46 | 27 | 14 | |
| Bacillota | 19 | 22 | 3 | |
| Acidobacteriota | 14 | 13 | 4 | |
| Planctomycetota | 8 | 11 | 1 | |
| Verrucomicrobiota | 1 | 8 | 0 | |
| Cyanobacterota | 3 | 3 | 0 | |
| Chloroflexota | 3 | 2 | 0 | |
| Fungi | Ascomycota | 55 | 56 | 68 |
| Basidiomycota | 0 | 0 | 1 | |
| NA | 0 | 1 | 0 | |
| Total | 574 | 432 | 259 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimeklis, A.K.; Gladkov, G.V.; Orlova, O.V.; Afonin, A.M.; Gribchenko, E.S.; Aksenova, T.S.; Kichko, A.A.; Pinaev, A.G.; Andronov, E.E. The Succession of the Cellulolytic Microbial Community from the Soil during Oat Straw Decomposition. Int. J. Mol. Sci. 2023, 24, 6342. https://doi.org/10.3390/ijms24076342
Kimeklis AK, Gladkov GV, Orlova OV, Afonin AM, Gribchenko ES, Aksenova TS, Kichko AA, Pinaev AG, Andronov EE. The Succession of the Cellulolytic Microbial Community from the Soil during Oat Straw Decomposition. International Journal of Molecular Sciences. 2023; 24(7):6342. https://doi.org/10.3390/ijms24076342
Chicago/Turabian StyleKimeklis, Anastasiia K., Grigory V. Gladkov, Olga V. Orlova, Alexey M. Afonin, Emma S. Gribchenko, Tatiana S. Aksenova, Arina A. Kichko, Alexander G. Pinaev, and Evgeny E. Andronov. 2023. "The Succession of the Cellulolytic Microbial Community from the Soil during Oat Straw Decomposition" International Journal of Molecular Sciences 24, no. 7: 6342. https://doi.org/10.3390/ijms24076342
APA StyleKimeklis, A. K., Gladkov, G. V., Orlova, O. V., Afonin, A. M., Gribchenko, E. S., Aksenova, T. S., Kichko, A. A., Pinaev, A. G., & Andronov, E. E. (2023). The Succession of the Cellulolytic Microbial Community from the Soil during Oat Straw Decomposition. International Journal of Molecular Sciences, 24(7), 6342. https://doi.org/10.3390/ijms24076342