
Recent Theoretical Insights into the Oxidative Degradation of Biopolymers and Plastics by Metalloenzymes †

 ,
,  , ,
, ,
Abstract
:1. Introduction
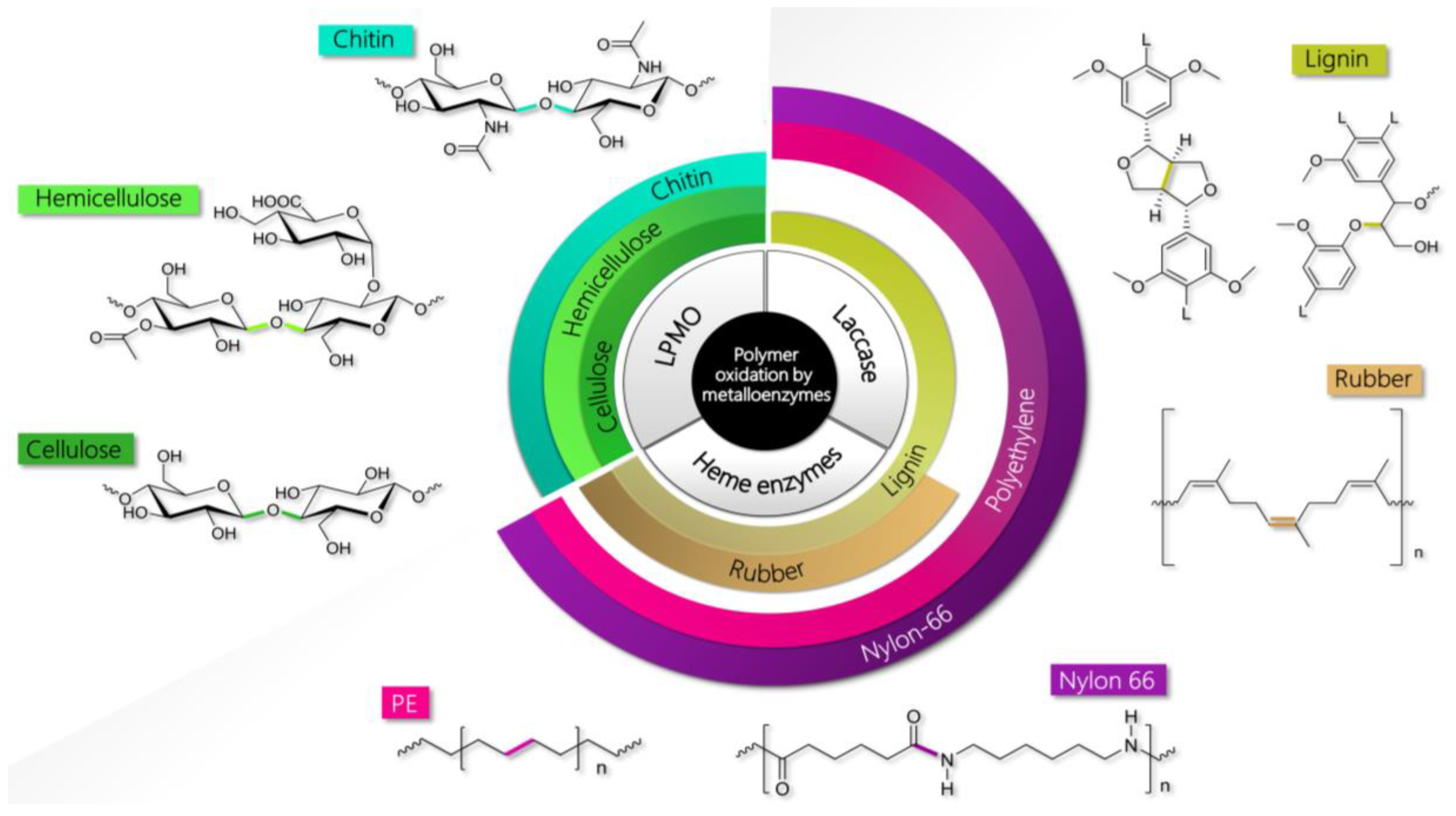
2. Natural and Synthetic Polymers Processed by Oxidative Metalloenzymes
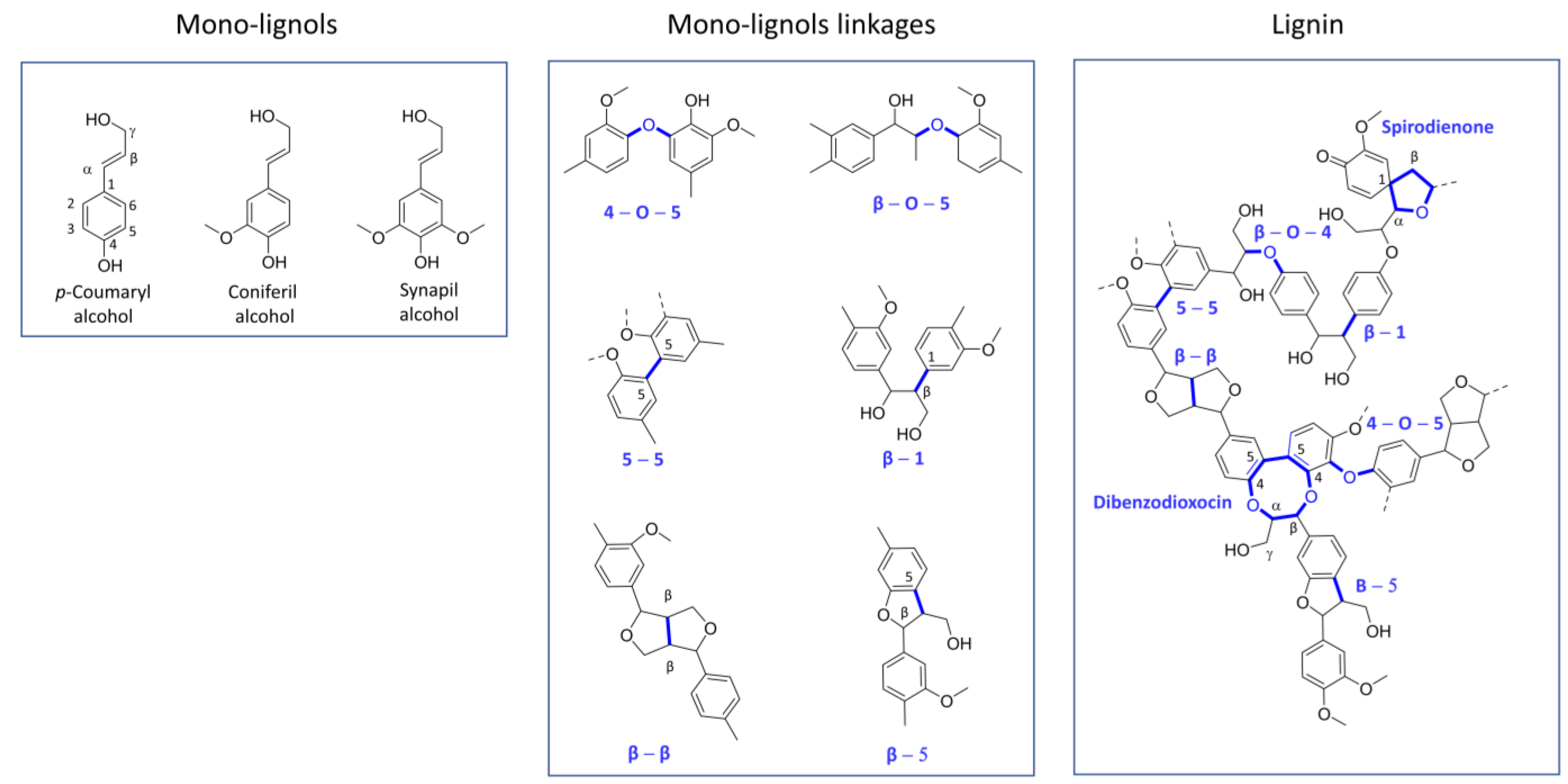
2.1. Lignin
2.2. Cellulose and Hemicellulose
2.3. Chitin
2.4. Natural Rubber
2.5. Plastics
3. Degradation of Polysaccharides by LPMO
3.1. LPMO Classification and Architecture
3.2. Brief Overview of the Catalytic Mechanism
3.3. LPMO-Substrate Interaction Studies
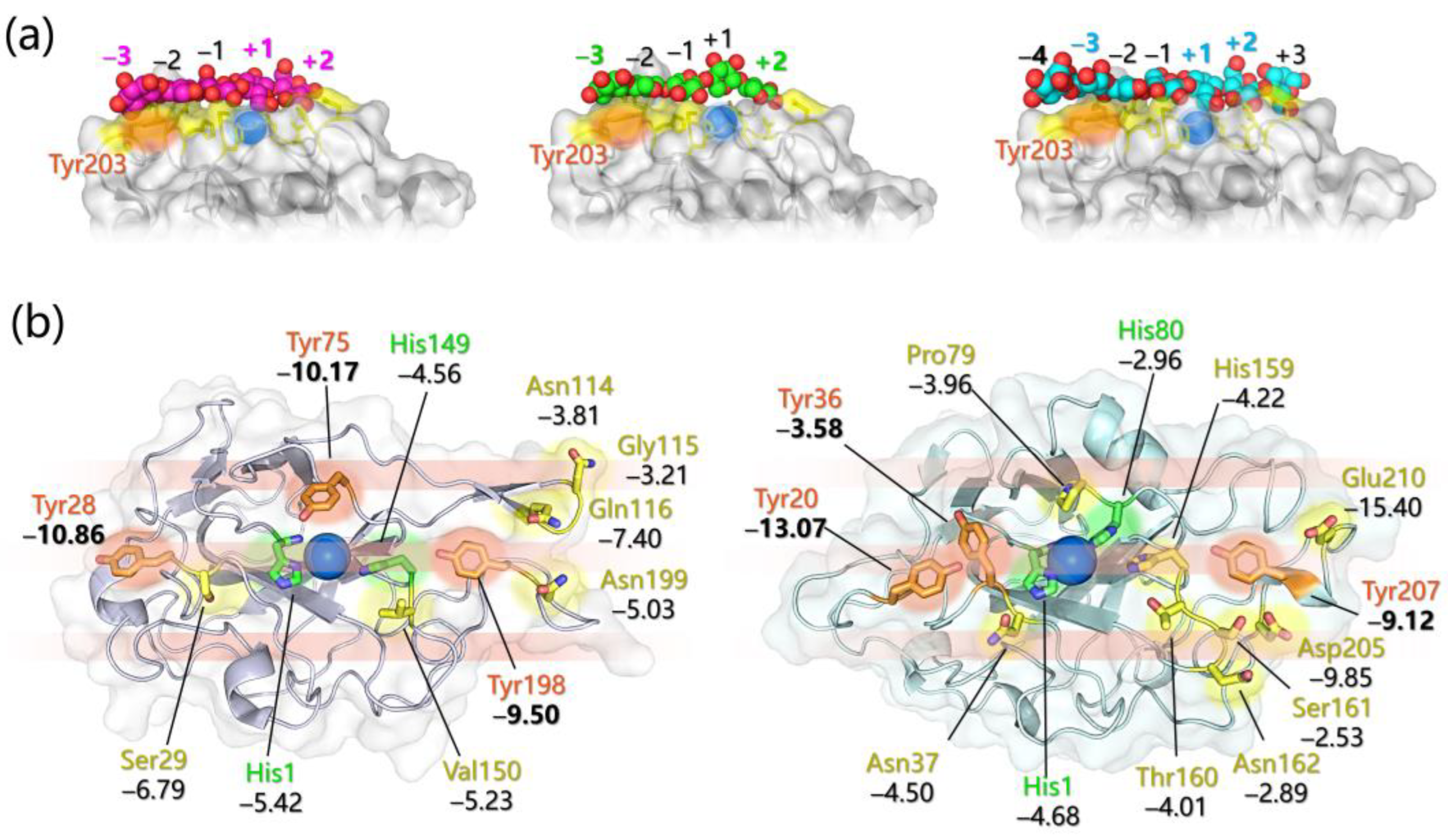
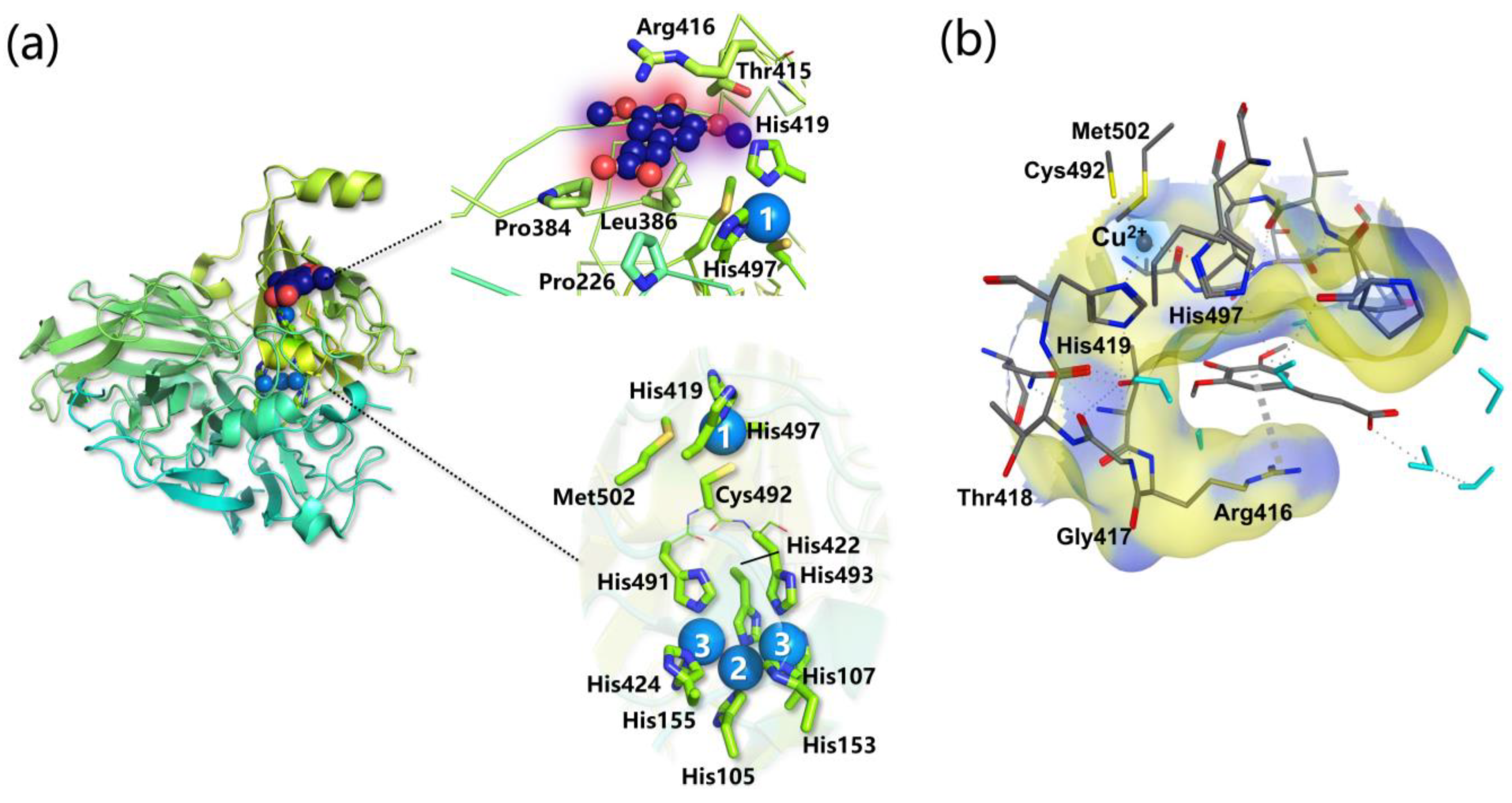
- Specific favorable orientation(s) of cellulose/chitin onto different LPMOs (both AA9 and AA10) have been detected along simulations; two examples are reported in Figure 4b. In particular: (i) in AA9 from Phanerochaete chrysosporium (PcAA9_D) Tyr28 (on L2) and Tyr198 (on LC) are aligned over the same cellulose chain, while the adjacent chain interacts with Tyr75 (on L3, Figure 4b, left) [102]; (ii) for AA9 from the white-rot fungus Heterobasidion irregulare (HiAA9_B), two different cellulose orientations have been found, a first one in which the three surface tyrosines (Tyr20, Tyr36, and Tyr207) all interact with the same cellulose chain (Figure 4b, right) and a second one in which HiAA9_B is rotated by 30° and so the Tyr residues interact with different cellulose chains [103]; (iii) in AA9 LPMO from Myceliophtora thermophila (MtAA9_L), the most involved residues in cellulose binding interact with two adjacent substrate chains [108]; and (iv) in chitin-active AA10 LPMO from Serratia marcescens (SmAA10_A), around 60–70% of contacts involves the to-be-cleaved chitin chain (considering either α- or β-chitin) [104].
- Potential energy surface (PES) for PcAA9_D-cellulose interaction revealed ~10 Å separated energy minima in correspondence to the three surface-exposed Tyr residues. This distance is compatible with the cellobiose unit, so the spacing among the aromatic residues is strategic for the polysaccharide binding, as also mentioned in other investigations [65,99,102].
- The frequency with which surface-exposed residues interact with substrates during simulations allowed determination of the important residues for binding, and interaction energy analysis allowed the quantification of the average energy contribution to the binding of each residue of interest (Figure 4b).
- During MD simulations, the cellulose/chitin slightly shifts so that the Cu-H1 distances are always shorter than the Cu-H4 ones in perfect match with the C1 regioselectivity of the LPMOs considered. This means that MD can effectively reproduce the C1 over C4 (or vice versa) oxidative preference, and thus may be used to design mutants with a desired regiochemistry.
- The SmAA10_A-chitin interaction analysis revealed the formation of a 12 Å channel between the protein and the substrate surfaces [104]. This tunnel reaches a maximum radius of ~1.6 Å suggesting that only small molecules, such as O2, H2O, or H2O2, may have access but not ascorbate or other reducing agents. It was also speculated that Glu60 may act as a gate that regulates the diffusion of substrates along the channel, which is in line with the experimental observation that Glu60 replacement causes a drop in catalytic efficiency [95].
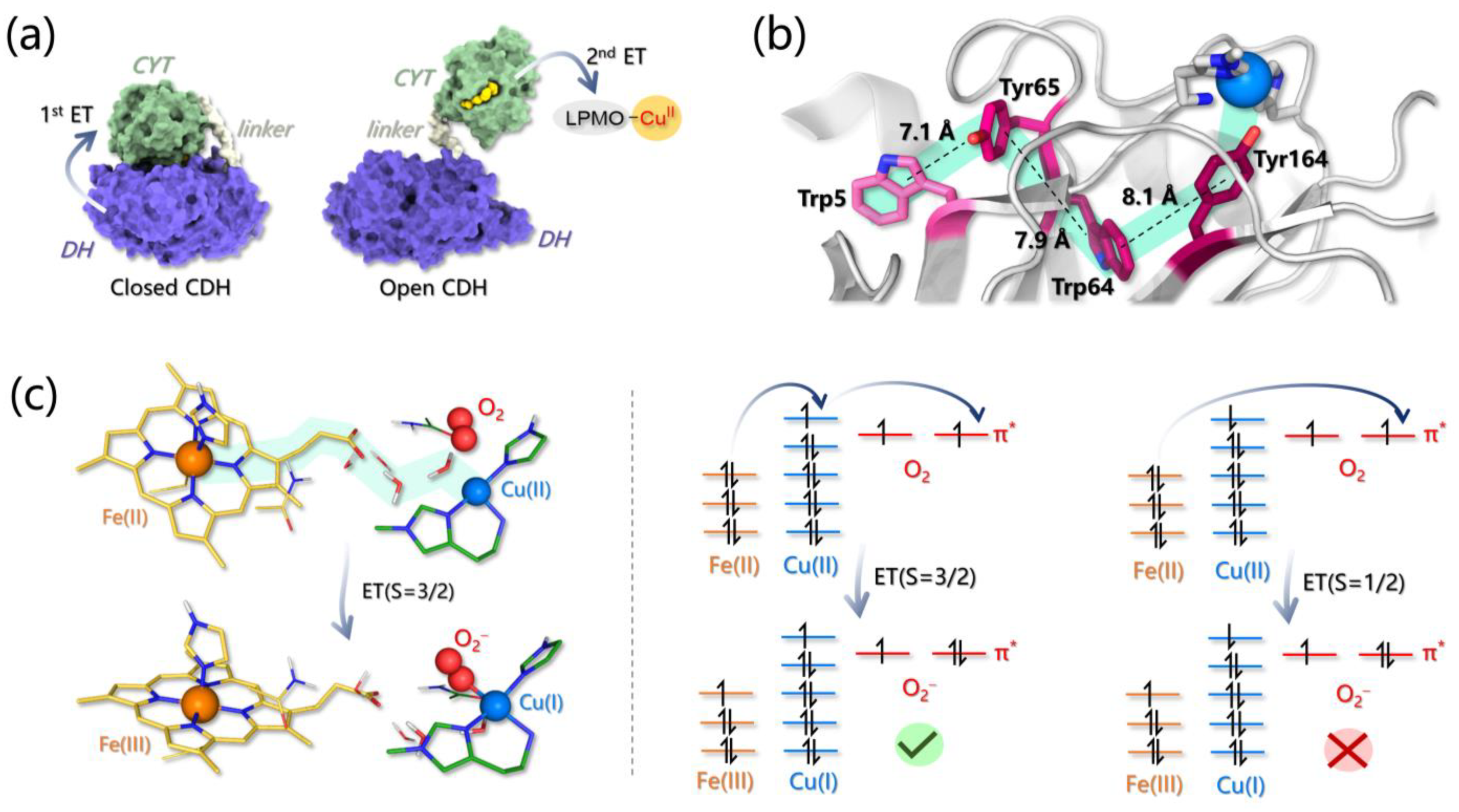
3.4. Electron Transfer Investigations
4. Laccases and Lignin Oxidation
4.1. Classification, Architecture, and Substrates
4.2. Mechanistic Details of Catalysis and Redox Potential Issues Related to Substrates

4.3. Computational Modeling Techniques as Valuable Tools in the Laccase-Lignin Degradation Insights
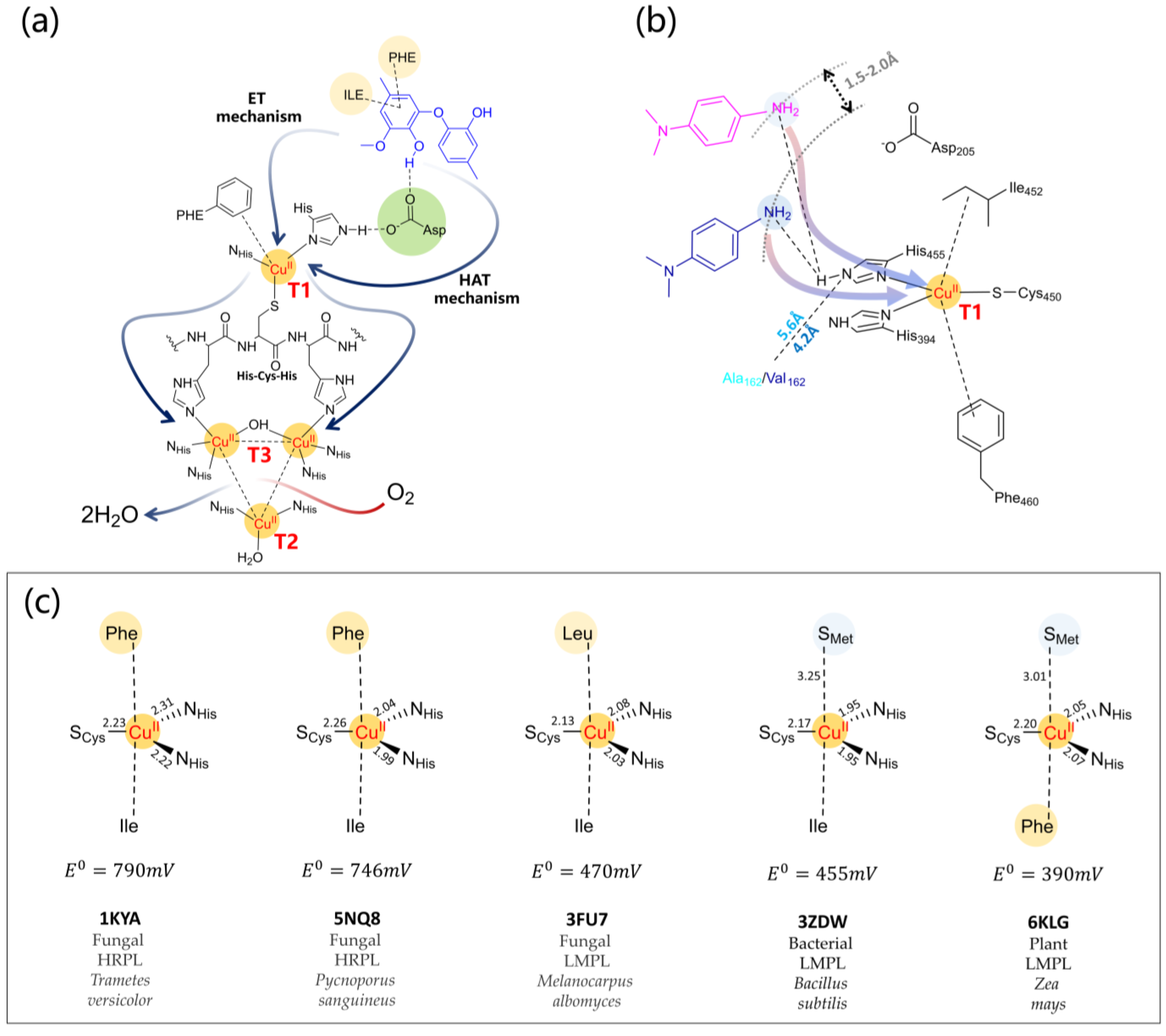
- The reliability of the molecular docking procedure was tested to be able to properly reproduce experimentally available laccase substrate binding with ABTS, 2,6-Dimethoxyphenol, or 2,5-xylidine [192].
- It has been ascertained that observed KM values relate directly to the lifetime of the active substrate of the enzyme and estimated binding free energies [194,195] and kinetic data correlate with the DFT spin population of the substrate, in particular, the kcat value [32,169,215]. Reorganization energies for multi-copper oxidases can be computed at the DFT level [216] and those of laccases for the first mono-electronic ET from the substrate to T1 Cu relates inversely to the enzymatic activity [132] showing that this step could determine the laccase turnover.
- Finally, the literature shows that the full-QM cluster model and QM/MM level were able to reproduce redox potential variations of two mutants compared to the wild-type enzyme. Specifically, the redox potential (a) increases for Escherichia coli CueO mutant L502K [146]; and (b) decreases for the F463M mutant from Trametes Versicolor laccase [217].
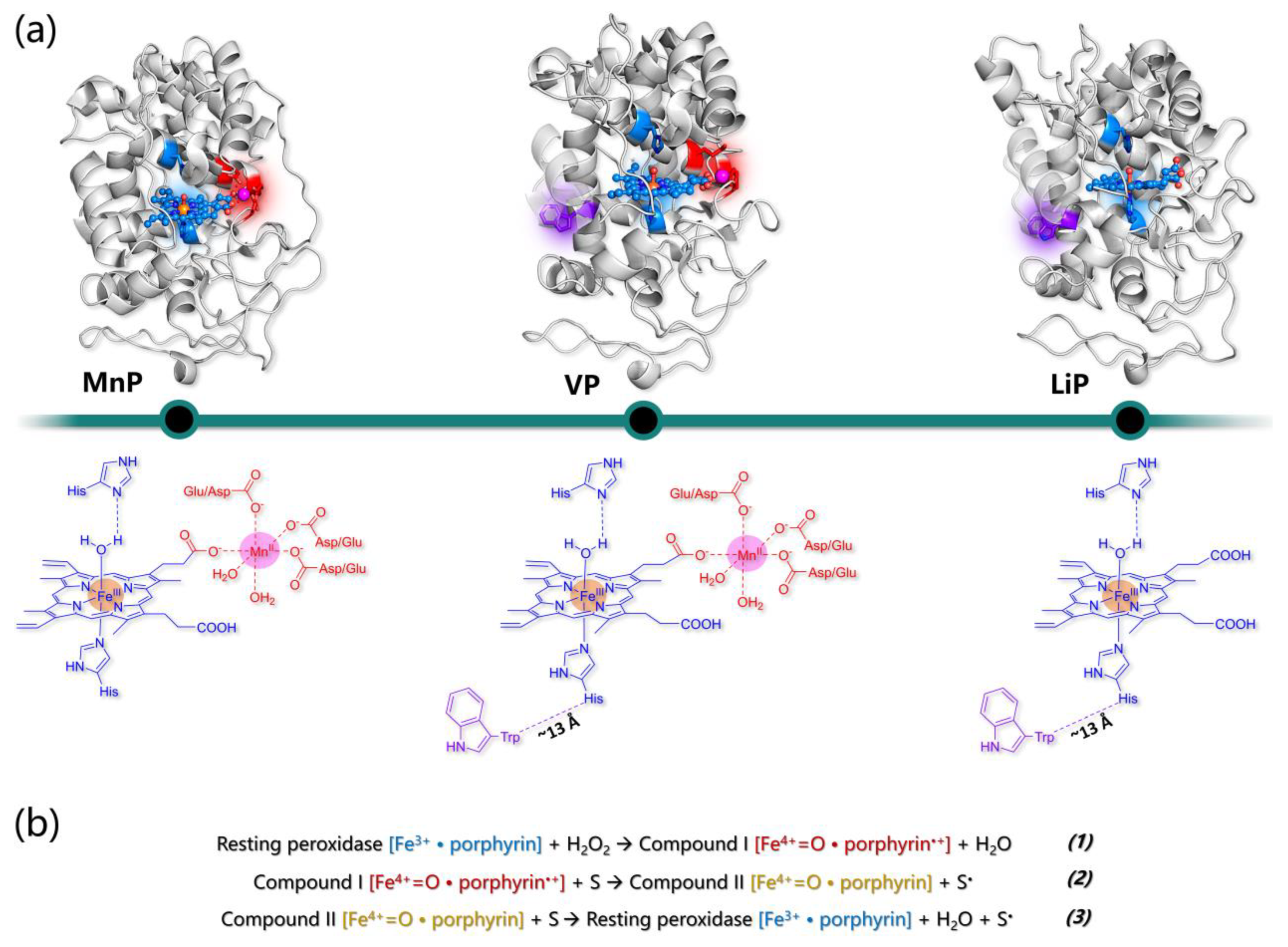
5. Heme Peroxidases for Lignin Oxidation
5.1. The General Structure of Ligninolytic Peroxidases
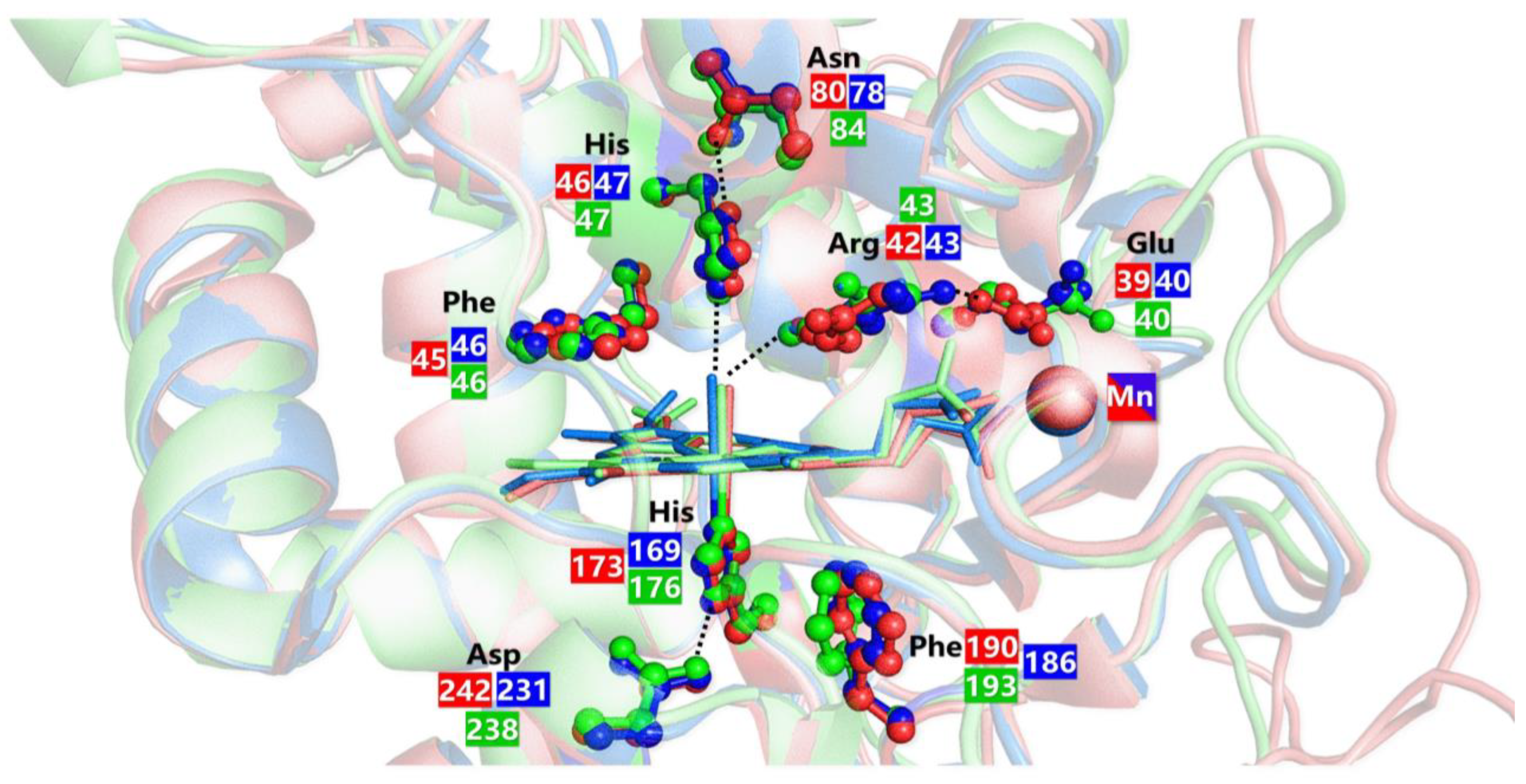
5.2. Manganese Peroxidases
5.3. Lignin Peroxidases
5.4. Versatile Peroxidases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MnP | PDB | Docking | MD, QM, Hybrid | Ref. |
| Phanerodontia chrysosporium | 3M5Q | ABTS | MD | [185] |
| Gelatoporia subvermispora | 4CZN (extralong) | ABTS | MD, QM/MM | [234] |
| Phanerodontia chrysosporium | 4BM1, 4CZN | ABTS | QM/MM | [236] |
| Phanerochaete chrysosporium | 1MNP | ABTS | [209] | |
| LiP | PDB | Docking | MD, QM, Hybrid | Ref. |
| Phanerochaete chrysosporium | 1QPA (HTR171) wt and W171A | VA | MD | [254] |
| Phanerochaete chrysosporium | 1LLP | ABTS | MD | [185] |
| Phanerochaete chrysosporium | 1LLP | VA | MD, QM/MM | [257] |
| Phanerochaete chrysosporium | 1B82 | Atrazine | MD | [256] |
| Phanerochaete chrysosporium | 1LLP wt and E250Q | VA | MD, QM/MM | [260] |
| Bacterial LiP | Homology model | 12 lignin models * | [267] | |
| Phanerochaete chrysosporium | 1LGA | ABTS | MD | [209] |
6. Degradation of Rubber and Plastics by Oxidative Metalloenzymes
6.1. Nature Strategy for Rubber Degradation: Heme Oxygenases

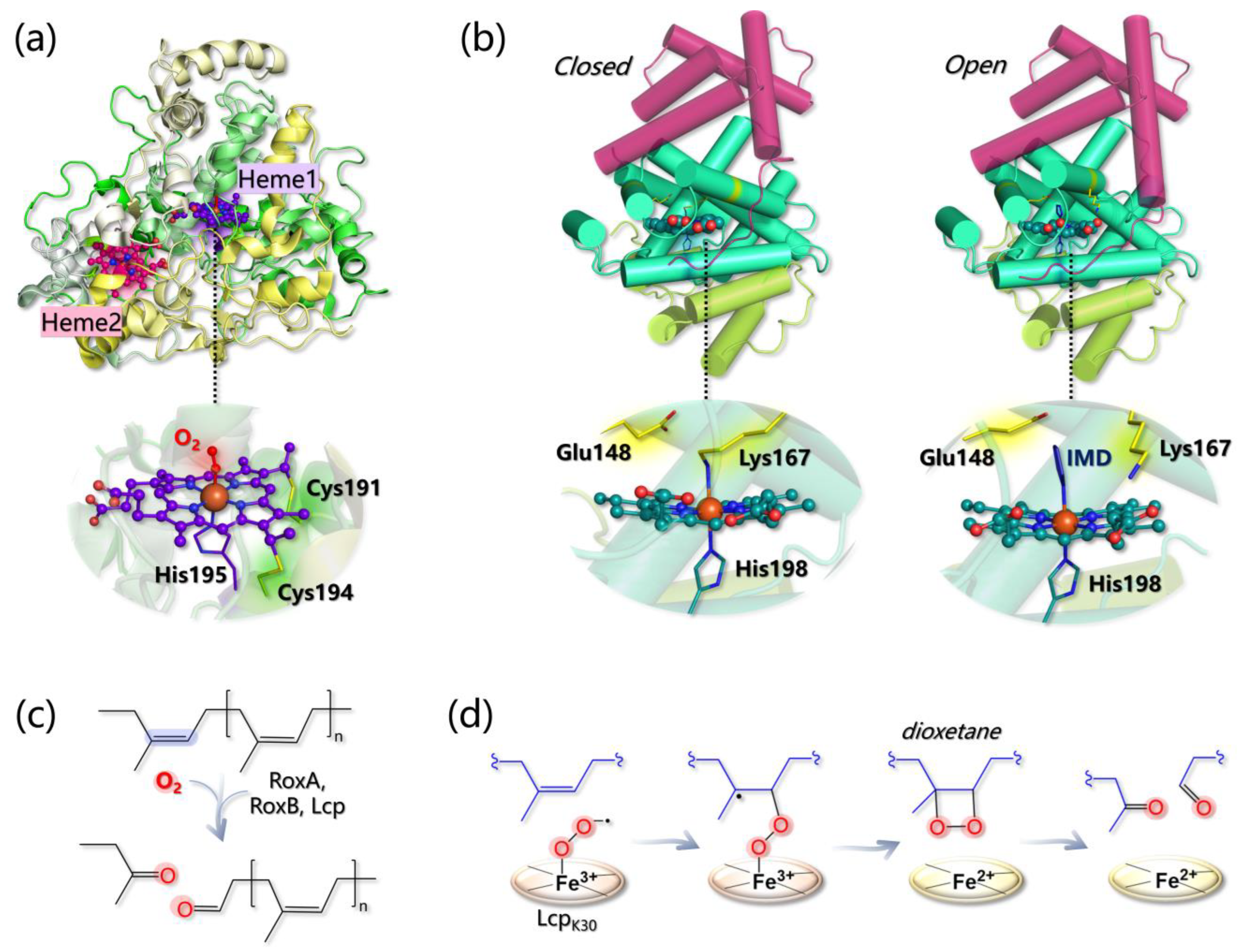
- The Fe(II)-O2 system can be better described as an open shell singlet Fe(III)-O2− species, and the distal O atom of O2− is predicted to be more reactive than the proximal one, as indicated by its larger spin density;
- Glu148 cannot act as a base since the proton abstraction from the polyisoprene allylic position is energetically prohibitive. Geometry optimizations of the Lcp-substrate system considering three models for the mutants E148A, E148Q, and E148H revealed that the distance between the O2 ligand and the double bond of the substrate (undergoing oxidation) increases, suggesting that although Glu148 is not directly involved in the reaction, it can indirectly control substrate positioning within the pocket for productive catalysis.
- The energy profiles for assumed reaction pathways have been calculated, accounting for the different spin states that may originate along catalysis. O2− can react with either one carbon or the other forming the isoprene C=C bond and, in each case, catalysis can proceed by forming a dioxetane or an epoxide intermediate. Therefore, four pathways can be envisioned overall. The most likely one (i) involves the C carbon that is closest to the O2− ligand and (ii) entails the formation of a dioxetane intermediate, with an overall activation barrier of 15.5 kcal/mol (Figure 12d).
6.2. Laccases and Heme Peroxidases for Plastic Degradation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Glossary
References
- Osman, A.I.; Mehta, N.; Elgarahy, A.M.; Al-Hinai, A.; Al-Muhtaseb, A.H.; Rooney, D.W. Conversion of Biomass to Biofuels and Life Cycle Assessment: A Review. Environ. Chem. Lett. 2021, 19, 4075–4118. [Google Scholar] [CrossRef]
- Fülöp, L.; Ecker, J. An Overview of Biomass Conversion: Exploring New Opportunities. PeerJ 2020, 8, e9586. [Google Scholar] [CrossRef] [PubMed]
- Fairley, P. Introduction: Next Generation Biofuels. Nature 2011, 474, S2–S5. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, Q.A.; Al Abdallah, Q.; Tracy Nixon, B.; Fortwendel, J.R. The Enzymatic Conversion of Major Algal and Cyanobacterial Carbohydrates to Bioethanol. Front. Energy Res. 2016, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Xu, F. Biomass Converting Enzymes as Industrial Biocatalysts for Fuels and Chemicals: Recent Developments. Catalysts 2012, 2, 244–263. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wang, Y. Recent Progress in the Conversion of Biomass Wastes into Functional Materials for Value-Added Applications. Sci. Technol. Adv. Mater. 2020, 21, 787–804. [Google Scholar] [CrossRef]
- Wang, Y.; Van Le, Q.; Yang, H.; Lam, S.S.; Yang, Y.; Gu, H.; Sonne, C.; Peng, W. Progress in Microbial Biomass Conversion into Green Energy. Chemosphere 2021, 281, 130835. [Google Scholar] [CrossRef]
- Lu, H.; Yadav, V.; Zhong, M.; Bilal, M.; Taherzadeh, M.J.; Iqbal, H.M.N. Bioengineered Microbial Platforms for Biomass-Derived Biofuel Production—A Review. Chemosphere 2022, 288, 132528. [Google Scholar] [CrossRef]
- Dutta, S.; Wu, K.C.-W. Enzymatic Breakdown of Biomass: Enzyme Active Sites, Immobilization, and Biofuel Production. Green Chem. 2014, 16, 4615–4626. [Google Scholar] [CrossRef]
- Ashokkumar, V.; Venkatkarthick, R.; Jayashree, S.; Chuetor, S.; Dharmaraj, S.; Kumar, G.; Chen, W.-H.; Ngamcharussrivichai, C. Recent Advances in Lignocellulosic Biomass for Biofuels and Value-Added Bioproducts—A Critical Review. Bioresour. Technol. 2022, 344, 126195. [Google Scholar] [CrossRef]
- Yan, N.; Chen, X. Sustainability: Don’t Waste Seafood Waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Chatterjee, K.; Madras, G. Enzymatic Degradation of Polymers: A Brief Review. Mater. Sci. Technol. 2014, 30, 567–573. [Google Scholar] [CrossRef]
- Chen, C.-C.; Dai, L.; Ma, L.; Guo, R.-T. Enzymatic Degradation of Plant Biomass and Synthetic Polymers. Nature Rev. Chem. 2020, 4, 114–126. [Google Scholar]
- Orlando, M.; Molla, G.; Castellani, P.; Pirillo, V.; Torretta, V.; Ferronato, N. Microbial Enzyme Biotechnology to Reach Plastic Waste Circularity: Current Status, Problems and Perspectives. Int. J. Mol. Sci. 2023, 24, 3877. [Google Scholar] [CrossRef]
- Martín, A.J.; Mondelli, C.; Jaydev, S.D.; Pérez-Ramírez, J. Catalytic Processing of Plastic Waste on the Rise. Chem 2021, 7, 1487–1533. [Google Scholar] [CrossRef]
- Mohanan, N.; Montazer, Z.; Sharma, P.K.; Levin, D.B. Microbial and Enzymatic Degradation of Synthetic Plastics. Front. Microbiol. 2020, 11, 580709. [Google Scholar] [CrossRef]
- Iiyoshi, Y.; Tsutsumi, Y.; Nishida, T. Polyethylene Degradation by Lignin-Degrading Fungi and Manganese Peroxidase. J. Wood Sci. 1998, 44, 222–229. [Google Scholar] [CrossRef]
- Yao, C.; Xia, W.; Dou, M.; Du, Y.; Wu, J. Oxidative Degradation of UV-Irradiated Polyethylene by Laccase-Mediator System. J. Hazard. Mater. 2022, 440, 129709. [Google Scholar] [CrossRef]
- Fujisawa, M.; Hirai, H.; Nishida, T. Degradation of Polyethylene and Nylon-66 by the Laccase-Mediator System. J. Polym. Environ. 2001, 9, 103–108. [Google Scholar] [CrossRef]
- Nayanashree, G.; Thippeswamy, B. Biodegradation of Natural Rubber by Laccase and Manganese Peroxidase Enzyme of Bacillus subtilis. Environ. Process. 2015, 2, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Birke, J.; Jendrossek, D. Rubber Oxygenase and Latex Clearing Protein Cleave Rubber to Different Products and Use Different Cleavage Mechanisms. Appl. Environ. Microbiol. 2014, 80, 5012–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somu, P.; Narayanasamy, S.; Gomez, L.A.; Rajendran, S.; Lee, Y.R.; Balakrishnan, D. Immobilization of Enzymes for Bioremediation: A Future Remedial and Mitigating Strategy. Environ. Res. 2022, 212, 113411. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on Their Way to Industrial Biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valetti, F.; Gilardi, G. Improvement of Biocatalysts for Industrial and Environmental Purposes by Saturation Mutagenesis. Biomolecules 2013, 3, 778–811. [Google Scholar] [CrossRef] [Green Version]
- Marchi-Delapierre, C.; Rondot, L.; Cavazza, C.; Ménage, S. Oxidation Catalysis by Rationally Designed Artificial Metalloenzymes. Isr. J. Chem. 2015, 55, 61–75. [Google Scholar] [CrossRef]
- Zhao, Y.; Man, Y.; Wen, J.; Guo, Y.; Lin, J. Advances in Imaging Plant Cell Walls. Trends Plant Sci. 2019, 24, 867–878. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Novaes, E.; Kirst, M.; Chiang, V.; Winter-Sederoff, H.; Sederoff, R. Lignin and Biomass: A Negative Correlation for Wood Formation and Lignin Content in Trees. Plant Physiol. 2010, 154, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Gomes, F.J.B.; de Souza, R.E.; Brito, E.O.; Lelis, R.C.C. A Review on Lignin Sources and Uses. J. Appl. Biotechnol. Bioeng. 2020, 7, 100–105. [Google Scholar]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin Valorization: Improving Lignin Processing in the Biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.T.; Speranza, M.; Ruiz-Dueñas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; del Río, J.C. Biodegradation of Lignocellulosics: Microbial, Chemical, and Enzymatic Aspects of the Fungal Attack of Lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar] [PubMed]
- Xie, S.; Ragauskas, A.J.; Yuan, J.S. Lignin Conversion: Opportunities and Challenges for the Integrated Biorefinery. Ind. Biotechnol. 2016, 12, 161–167. [Google Scholar] [CrossRef]
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; et al. Lignins: Natural Polymers from Oxidative Coupling of 4-Hydroxyphenyl- Propanoids. Phytochem. Rev. 2004, 3, 29–60. [Google Scholar] [CrossRef]
- Vanholme, R.; De Meester, B.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Its Integration into Metabolism. Curr. Opin. Biotechnol. 2019, 56, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Rongpipi, S.; Ye, D.; Gomez, E.D.; Gomez, E.W. Progress and Opportunities in the Characterization of Cellulose—An Important Regulator of Cell Wall Growth and Mechanics. Front. Plant Sci. 2018, 9, 1894. [Google Scholar] [CrossRef] [Green Version]
- Zoghlami, A.; Paës, G. Lignocellulosic Biomass: Understanding Recalcitrance and Predicting Hydrolysis. Front. Chem 2019, 7, 874. [Google Scholar] [CrossRef] [Green Version]
- Michelin, M.; Ruiz, H.A.; Silva, D.P.; Ruzene, D.S.; Teixeira, J.A.; Polizeli, M.L.T. Cellulose from Lignocellulosic Waste. In Polysaccharides; Springer: Cham, Switzerland, 2014; pp. 1–33. [Google Scholar] [CrossRef]
- Khaire, K.C.; Moholkar, V.S.; Goyal, A. Bioconversion of sugarcane tops to bioethanol and other value added products: An overview. Mat. Sci. Energy. Technol. 2021, 4, 54–68. [Google Scholar] [CrossRef]
- Ning, P.; Yang, G.; Hu, L.; Sun, J.; Shi, L.; Zhou, Y.; Wang, Z.; Yang, J. Recent Advances in the Valorization of Plant Biomass. Biotechnol. Biofuels 2021, 14, 102. [Google Scholar] [CrossRef]
- Sanjanwala, D.; Londhe, V.; Trivedi, R.; Bonde, S.; Sawarkar, S.; Kale, V.; Patravale, V. Polysaccharide-Based Hydrogels for Drug Delivery and Wound Management: A Review. Expert Opin. Drug Deliv. 2022, 19, 1664–1695. [Google Scholar] [CrossRef]
- Basik, A.A.; Sanglier, J.-J.; Yeo, C.T.; Sudesh, K. Microbial Degradation of Rubber: Actinobacteria. Polymers 2021, 13, 1989. [Google Scholar] [CrossRef]
- Yao, Z.; Seong, H.J.; Jang, Y.-S. Environmental Toxicity and Decomposition of Polyethylene. Ecotoxicol. Environ. Saf. 2022, 242, 113933. [Google Scholar] [CrossRef]
- Tiseo, I. Polyethylene Market Volume Worldwide 2015–2029; Statista: Hamburg, Germany, 2021. [Google Scholar]
- Friedrich, J.; Zalar, P.; Mohorcic, M.; Klun, U.; Krzan, A. Ability of Fungi to Degrade Synthetic Polymer Nylon-6. Chemosphere 2007, 67, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lin, K.-Y.A.; Kwon, E.E.; Lee, J. Renewable Routes to Monomeric Precursors of Nylon 66 and Nylon 6 from Food Waste. J. Clean. Prod. 2019, 227, 624–633. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Walton, P.H.; Davies, G.J. On the Catalytic Mechanisms of Lytic Polysaccharide Monooxygenases. Curr. Opin. Chem. Biol. 2016, 31, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, Z.; Sørlie, M.; Petrović, D.; Courtade, G.; Aachmann, F.L.; Vaaje-Kolstad, G.; Bissaro, B.; Røhr, Å.K.; Eijsink, V.G. Polysaccharide Degradation by Lytic Polysaccharide Monooxygenases. Curr. Opin. Struct. Biol. 2019, 59, 54–64. [Google Scholar] [CrossRef]
- Hemsworth, G.R.; Johnston, E.M.; Davies, G.J.; Walton, P.H. Lytic Polysaccharide Monooxygenases in Biomass Conversion. Trends Biotechnol. 2015, 33, 747–761. [Google Scholar] [CrossRef]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.H.; Horn, S.J. A C4-Oxidizing Lytic Polysaccharide Monooxygenase Cleaving Both Cellulose and Cello-Oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef] [Green Version]
- Agger, J.W.; Isaksen, T.; Várnai, A.; Vidal-Melgosa, S.; Willats, W.G.T.; Ludwig, R.; Horn, S.J.; Eijsink, V.G.H.; Westereng, B. Discovery of LPMO Activity on Hemicelluloses Shows the Importance of Oxidative Processes in Plant Cell Wall Degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 6287–6292. [Google Scholar] [CrossRef] [Green Version]
- Bennati-Granier, C.; Garajova, S.; Champion, C.; Grisel, S.; Haon, M.; Zhou, S.; Fanuel, M.; Ropartz, D.; Rogniaux, H.; Gimbert, I.; et al. Substrate Specificity and Regioselectivity of Fungal AA9 Lytic Polysaccharide Monooxygenases Secreted by Podospora Anserina. Biotechnol. Biofuels 2015, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Frandsen, K.E.H.; Lo Leggio, L. Lytic Polysaccharide Monooxygenases: A Crystallographer’s View on a New Class of Biomass-Degrading Enzymes. IUCrJ 2016, 3, 448–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Span, E.A.; Suess, D.L.M.; Deller, M.C.; David Britt, R.; Marletta, M.A. The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem. Biol. 2017, 12, 1095–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburrini, K.C.; Terrapon, N.; Lombard, V.; Bissaro, B.; Longhi, S.; Berrin, J.-G. Bioinformatic Analysis of Lytic Polysaccharide Monooxygenases Reveals the Pan-Families Occurrence of Intrinsically Disordered C-Terminal Extensions. Biomolecules 2021, 11, 1632. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Forsberg, Z.; Loose, J.S.; Bissaro, B.; Eijsink, V.G. Structural Diversity of Lytic Polysaccharide Monooxygenases. Curr. Opin. Struct. Biol. 2017, 44, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Aachmann, F.L.; Sørlie, M.; Skjåk-Bræk, G.; Eijsink, V.G.H.; Vaaje-Kolstad, G. NMR Structure of a Lytic Polysaccharide Monooxygenase Provides Insight into Copper Binding, Protein Dynamics, and Substrate Interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 18779–18784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtade, G.; Wimmer, R.; Røhr, Å.K.; Preims, M.; Felice, A.K.G.; Dimarogona, M.; Vaaje-Kolstad, G.; Sørlie, M.; Sandgren, M.; Ludwig, R.; et al. Interactions of a Fungal Lytic Polysaccharide Monooxygenase with β-Glucan Substrates and Cellobiose Dehydrogenase. Proc. Natl. Acad. Sci. USA 2016, 113, 5922–5927. [Google Scholar] [CrossRef] [Green Version]
- Frandsen, K.E.H.; Simmons, T.J.; Dupree, P.; Poulsen, J.-C.N.; Hemsworth, G.R.; Ciano, L.; Johnston, E.M.; Tovborg, M.; Johansen, K.S.; von Freiesleben, P.; et al. The Molecular Basis of Polysaccharide Cleavage by Lytic Polysaccharide Monooxygenases. Nat. Chem. Biol. 2016, 12, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Span, E.A.; Marletta, M.A. The Framework of Polysaccharide Monooxygenase Structure and Chemistry. Curr. Opin. Struct. Biol. 2015, 35, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Ciano, L.; Davies, G.J.; Tolman, W.B.; Walton, P.H. Bracing Copper for the Catalytic Oxidation of C–H Bonds. Nat. Catal. 2018, 1, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Ståhlberg, J.; Sandgren, M.; Paton, R.S.; Beckham, G.T. Quantum Mechanical Calculations Suggest That Lytic Polysaccharide Monooxygenases Use a Copper-Oxyl, Oxygen-Rebound Mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Beeson, W.T.; Vu, V.V.; Span, E.A.; Phillips, C.M.; Marletta, M.A. Cellulose Degradation by Polysaccharide Monooxygenases. Annu. Rev. Biochem. 2015, 84, 923–946. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Beeson, W.T., 4th; Phillips, C.M.; Marletta, M.A.; Cate, J.H.D. Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure 2012, 20, 1051–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracher, D.; Andlar, M.; Furtmüller, P.G.; Ludwig, R. Active-Site Copper Reduction Promotes Substrate Binding of Fungal Lytic Polysaccharide Monooxygenase and Reduces Stability. J. Biol. Chem. 2018, 293, 1676–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Walton, P.H.; Rovira, C. Molecular Mechanisms of Oxygen Activation and Hydrogen Peroxide Formation in Lytic Polysaccharide Monooxygenases. ACS Catal. 2019, 9, 4958–4969. [Google Scholar] [CrossRef] [Green Version]
- Hemsworth, G.R.; Davies, G.J.; Walton, P.H. Recent Insights into Copper-Containing Lytic Polysaccharide Mono-Oxygenases. Curr. Opin. Struct. Biol. 2013, 23, 660–668. [Google Scholar] [CrossRef]
- Forsberg, Z.; Vaaje-Kolstad, G.; Westereng, B.; Bunæs, A.C.; Stenstrøm, Y.; MacKenzie, A.; Sørlie, M.; Horn, S.J.; Eijsink, V.G.H. Cleavage of Cellulose by a CBM33 Protein. Protein Sci. 2011, 20, 1479–1483. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, R.J.; Sweeney, M.D.; Lo Leggio, L.; Otten, H.; Poulsen, J.-C.N.; Johansen, K.S.; Krogh, K.B.R.M.; Jørgensen, C.I.; Tovborg, M.; Anthonsen, A.; et al. Insights into the Oxidative Degradation of Cellulose by a Copper Metalloenzyme That Exploits Biomass Components. Proc. Natl. Acad. Sci. USA 2011, 108, 15079–15084. [Google Scholar] [CrossRef] [Green Version]
- Langston, J.A.; Shaghasi, T.; Abbate, E.; Xu, F.; Vlasenko, E.; Sweeney, M.D. Oxidoreductive Cellulose Depolymerization by the Enzymes Cellobiose Dehydrogenase and Glycoside Hydrolase 61. Appl. Environ. Microbiol. 2011, 77, 7007–7015. [Google Scholar] [CrossRef] [Green Version]
- Várnai, A.; Umezawa, K.; Yoshida, M.; Eijsink, V.G.H. The Pyrroloquinoline-Quinone-Dependent Pyranose Dehydrogenase from Coprinopsis Cinerea Drives Lytic Polysaccharide Monooxygenase Action. Appl. Environ. Microbiol. 2018, 84, e00156-18. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2. Nat. Chem. Biol. 2017, 13, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Kuusk, S.; Väljamäe, P. Kinetics of H2O2-Driven Catalysis by a Lytic Polysaccharide Monooxygenase from the Fungus Trichoderma Reesei. J. Biol. Chem. 2021, 297, 101256. [Google Scholar] [CrossRef]
- Filandr, F.; Man, P.; Halada, P.; Chang, H.; Ludwig, R.; Kracher, D. The HO-Dependent Activity of a Fungal Lytic Polysaccharide Monooxygenase Investigated with a Turbidimetric Assay. Biotechnol. Biofuels 2020, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.M.; Transue, W.J.; Meier, K.K.; Kelemen, B.; Solomon, E.I. Kinetic Analysis of Amino Acid Radicals Formed in HO-Driven Cu LPMO Reoxidation Implicates Dominant Homolytic Reactivity. Proc. Natl. Acad. Sci. USA 2020, 117, 11916–11922. [Google Scholar] [CrossRef] [PubMed]
- Hangasky, J.A.; Iavarone, A.T.; Marletta, M.A. Reactivity of O versus HO with Polysaccharide Monooxygenases. Proc. Natl. Acad. Sci. USA 2018, 115, 4915–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, L.; Breglia, R.; Lambrughi, M.; Fantucci, P.; De Gioia, L.; Borsari, M.; Sola, M.; Bortolotti, C.A.; Bruschi, M. Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations. Inorg. Chem. 2018, 57, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, C.H.; Qayyum, M.F.; Wong, S.D.; Xu, F.; Hemsworth, G.R.; Walton, D.J.; Young, N.A.; Davies, G.J.; Walton, P.H.; Johansen, K.S.; et al. Spectroscopic and Computational Insight into the Activation of O2 by the Mononuclear Cu Center in Polysaccharide Monooxygenases. Proc. Natl. Acad. Sci. USA 2014, 111, 8797–8802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudmundsson, M.; Kim, S.; Wu, M.; Ishida, T.; Momeni, M.H.; Vaaje-Kolstad, G.; Lundberg, D.; Royant, A.; Ståhlberg, J.; Eijsink, V.G.H.; et al. Structural and Electronic Snapshots during the Transition from a Cu(II) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-Ray Photoreduction. J. Biol. Chem. 2014, 289, 18782–18792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedegård, E.D.; Ryde, U. Targeting the Reactive Intermediate in Polysaccharide Monooxygenases. J. Biol. Inorg. Chem. 2017, 22, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Paradisi, A.; Johnston, E.M.; Tovborg, M.; Nicoll, C.R.; Ciano, L.; Dowle, A.; McMaster, J.; Hancock, Y.; Davies, G.J.; Walton, P.H. Formation of a Copper(II)-Tyrosyl Complex at the Active Site of Lytic Polysaccharide Monooxygenases Following Oxidation by HO. J. Am. Chem. Soc. 2019, 141, 18585–18599. [Google Scholar] [CrossRef] [Green Version]
- Courtade, G.; Ciano, L.; Paradisi, A.; Lindley, P.J.; Forsberg, Z.; Sørlie, M.; Wimmer, R.; Davies, G.J.; Eijsink, V.G.H.; Walton, P.H.; et al. Mechanistic Basis of substrate–O2coupling within a Chitin-Active Lytic Polysaccharide Monooxygenase: An Integrated NMR/EPR Study. Proc. Natl. Acad. Sci. USA 2020, 117, 19178–19189. [Google Scholar] [CrossRef] [PubMed]
- Theibich, Y.A.; Sauer, S.P.A.; Leggio, L.L.; Hedegård, E.D. Estimating the Accuracy of Calculated Electron Paramagnetic Resonance Hyperfine Couplings for a Lytic Polysaccharide Monooxygenase. Comput. Struct. Biotechnol. J. 2021, 19, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Lindley, P.J.; Parkin, A.; Davies, G.J.; Walton, P.H. Mapping the Protonation States of the Histidine Brace in an AA10 Lytic Polysaccharide Monooxygenase Using CW-EPR Spectroscopy and DFT Calculations. Faraday Discuss. 2022, 234, 336–348. [Google Scholar] [CrossRef]
- Wang, B.; Johnston, E.M.; Li, P.; Shaik, S.; Davies, G.J.; Walton, P.H.; Rovira, C. QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate. ACS Catal. 2018, 8, 1346–1351. [Google Scholar] [CrossRef] [Green Version]
- Hedegård, E.D.; Ryde, U. Multiscale Modelling of Lytic Polysaccharide Monooxygenases. ACS Omega 2017, 2, 536–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedegård, E.D.; Ryde, U. Molecular Mechanism of Lytic Polysaccharide Monooxygenases. Chem. Sci. 2018, 9, 3866–3880. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Streit, B.; Isaksen, I.; Eijsink, V.G.H.; Beckham, G.T.; DuBois, J.L.; Røhr, Å.K. Molecular Mechanism of the Chitinolytic Peroxygenase Reaction. Proc. Natl. Acad. Sci. USA 2020, 117, 1504–1513. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Z.; Davies, G.J.; Walton, P.H.; Rovira, C. Activation of O2 and H2O2 by Lytic Polysaccharide Monooxygenases. ACS Catal. 2020, 10, 12760–12769. [Google Scholar] [CrossRef]
- Bissaro, B.; Eijsink, V.G.H. Lytic Polysaccharide Monooxygenases: Enzymes for Controlled and Site-Specific Fenton-like Chemistry. Essays Biochem. 2023, EBC20220250. [Google Scholar] [CrossRef]
- Larsson, E.D.; Dong, G.; Veryazov, V.; Ryde, U.; Hedegård, E.D. Is Density Functional Theory Accurate for Lytic Polysaccharide Monooxygenase Enzymes? Dalton Trans. 2020, 49, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, M.M.; Hedegård, E.D. Molecular Mechanism of Substrate Oxidation in Lytic Polysaccharide Monooxygenases: Insight from Theoretical Investigations. Chem.-Eur. J. 2023, 29, e202202379. [Google Scholar]
- Eijsink, V.G.H.; Petrovic, D.; Forsberg, Z.; Mekasha, S.; Røhr, Å.K.; Várnai, A.; Bissaro, B.; Vaaje-Kolstad, G. On the Functional Characterization of Lytic Polysaccharide Monooxygenases (LPMOs). Biotechnol. Biofuels 2019, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaaje-Kolstad, G.; Houston, D.R.; Riemen, A.H.K.; Eijsink, V.G.H.; van Aalten, D.M.F. Crystal Structure and Binding Properties of the Serratia Marcescens Chitin-Binding Protein CBP21. J. Biol. Chem. 2005, 280, 11313–11319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.V.; Welner, D.; McFarland, K.C.; Re, E.; Navarro Poulsen, J.-C.; Brown, K.; Salbo, R.; Ding, H.; Vlasenko, E.; Merino, S.; et al. Stimulation of Lignocellulosic Biomass Hydrolysis by Proteins of Glycoside Hydrolase Family 61: Structure and Function of a Large, Enigmatic Family. Biochemistry 2010, 49, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Vu, V.V.; Beeson, W.T.; Phillips, C.M.; Cate, J.H.D.; Marletta, M.A. Determinants of Regioselective Hydroxylation in the Fungal Polysaccharide Monooxygenases. J. Am. Chem. Soc. 2014, 136, 562–565. [Google Scholar] [CrossRef]
- Book, A.J.; Yennamalli, R.M.; Takasuka, T.E.; Currie, C.R.; Phillips, G.N., Jr.; Fox, B.G. Evolution of Substrate Specificity in Bacterial AA10 Lytic Polysaccharide Monooxygenases. Biotechnol. Biofuels 2014, 7, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, T.J.; Frandsen, K.E.H.; Ciano, L.; Tryfona, T.; Lenfant, N.; Poulsen, J.C.; Wilson, L.F.L.; Tandrup, T.; Tovborg, M.; Schnorr, K.; et al. Structural and Electronic Determinants of Lytic Polysaccharide Monooxygenase Reactivity on Polysaccharide Substrates. Nat. Commun. 2017, 8, 1064. [Google Scholar] [CrossRef] [Green Version]
- Tandrup, T.; Tryfona, T.; Frandsen, K.E.H.; Johansen, K.S.; Dupree, P.; Lo Leggio, L. Oligosaccharide Binding and Thermostability of Two Related AA9 Lytic Polysaccharide Monooxygenases. Biochemistry 2020, 59, 3347–3358. [Google Scholar] [CrossRef]
- Borisova, A.S.; Isaksen, T.; Dimarogona, M.; Kognole, A.A.; Mathiesen, G.; Várnai, A.; Røhr, Å.K.; Payne, C.M.; Sørlie, M.; Sandgren, M.; et al. Structural and Functional Characterization of a Lytic Polysaccharide Monooxygenase with Broad Substrate Specificity. J. Biol. Chem. 2015, 290, 22955–22969. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Beckham, G.T.; Larsson, A.M.; Ishida, T.; Kim, S.; Payne, C.M.; Himmel, M.E.; Crowley, M.F.; Horn, S.J.; Westereng, B.; et al. Crystal Structure and Computational Characterization of the Lytic Polysaccharide Monooxygenase GH61D from the Basidiomycota Fungus Phanerochaete Chrysosporium. J. Biol. Chem. 2013, 288, 12828–12839. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Kognole, A.A.; Wu, M.; Westereng, B.; Crowley, M.F.; Kim, S.; Dimarogona, M.; Payne, C.M.; Sandgren, M. Structural and Molecular Dynamics Studies of a C1-Oxidizing Lytic Polysaccharide Monooxygenase from Heterobasidion Irregulare Reveal Amino Acids Important for Substrate Recognition. FEBS J. 2018, 285, 2225–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissaro, B.; Isaksen, I.; Vaaje-Kolstad, G.; Eijsink, V.G.H.; Røhr, Å.K. How a Lytic Polysaccharide Monooxygenase Binds Crystalline Chitin. Biochemistry 2018, 57, 1893–1906. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.-C.; Kracher, D.; Gandini, R.; Sygmund, C.; Kittl, R.; Haltrich, D.; Hällberg, B.M.; Ludwig, R.; Divne, C. Structural Basis for Cellobiose Dehydrogenase Action during Oxidative Cellulose Degradation. Nat. Commun. 2015, 6, 7542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, C.V.F.P.; Breslmayr, E.; Tunega, D.; Ludwig, R.; Oostenbrink, C. Interaction between Cellobiose Dehydrogenase and Lytic Polysaccharide Monooxygenase. Biochemistry 2019, 58, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Feng, S.; Rovira, C.; Wang, B. How Oxygen Binding Enhances Long-Range Electron Transfer: Lessons From Reduction of Lytic Polysaccharide Monooxygenases by Cellobiose Dehydrogenase. Angew. Chem. Int. Ed. Engl. 2021, 60, 2385–2392. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Li, T.; Tan, H.; Li, G.; Yin, H. Distinct Interaction of Lytic Polysaccharide Monooxygenase with Cellulose Revealed by Computational and Biochemical Studies. J. Phys. Chem. Lett. 2020, 11, 3987–3992. [Google Scholar] [CrossRef]
- McEvoy, A.; Creutzberg, J.; Singh, R.K.; Bjerrum, M.J.; Hedegård, E.D. The Role of the Active Site Tyrosine in the Mechanism of Lytic Polysaccharide Monooxygenase. Chem. Sci. 2020, 12, 352–362. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Functional and Protective Hole Hopping in Metalloenzymes. Chem. Sci. 2021, 12, 13988–14003. [Google Scholar] [CrossRef]
- Kracher, D.; Scheiblbrandner, S.; Felice, A.K.G.; Breslmayr, E.; Preims, M.; Ludwicka, K.; Haltrich, D.; Eijsink, V.G.H.; Ludwig, R. Extracellular Electron Transfer Systems Fuel Cellulose Oxidative Degradation. Science 2016, 352, 1098–1101. [Google Scholar] [CrossRef]
- Arrigoni, F.; Di Carlo, C.; Rovetta, A.; De Gioia, L.; Zampella, G.; Bertini, L. Superoxide Reduction by Cu-Amyloid Beta Peptide Complexes: A Density Functional Theory Study. Eur. J. Inorg. Chem. 2022, 2022, e202200245. [Google Scholar] [CrossRef]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.M.; Solomon, E.I. Electron Transfer and Reaction Mechanism of Laccases. Cell. Mol. Life Sci. 2015, 72, 869–883. [Google Scholar] [CrossRef] [Green Version]
- Hakulinen, N.; Rouvinen, J. Three-Dimensional Structures of Laccases. Cell. Mol. Life Sci. 2015, 72, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.S.; Goradia, B.; Saxena, A. Bacterial Laccase: Recent Update on Production, Properties and Industrial Applications. 3 Biotech 2017, 7, 323. [Google Scholar] [CrossRef]
- Loi, M.; Glazunova, O.; Fedorova, T.; Logrieco, A.F.; Mulè, G. Fungal Laccases: The Forefront of Enzymes for Sustainability. J. Fungi 2021, 7, 1048. [Google Scholar] [CrossRef]
- Singh, D.; Gupta, N. Microbial Laccase: A Robust Enzyme and Its Industrial Applications. Biologia 2020, 75, 1183–1193. [Google Scholar] [CrossRef]
- Arregui, L.; Ayala, M.; Gómez-Gil, X.; Gutiérrez-Soto, G.; Hernández-Luna, C.E.; Herrera de Los Santos, M.; Levin, L.; Rojo-Domínguez, A.; Romero-Martínez, D.; Saparrat, M.C.N.; et al. Laccases: Structure, Function, and Potential Application in Water Bioremediation. Microb. Cell Fact. 2019, 18, 200. [Google Scholar] [CrossRef] [Green Version]
- Cañas, A.I.; Camarero, S. Laccases and Their Natural Mediators: Biotechnological Tools for Sustainable Eco-Friendly Processes. Biotechnol. Adv. 2010, 28, 694–705. [Google Scholar] [CrossRef]
- Mate, D.M.; Alcalde, M. Laccase: A Multi-Purpose Biocatalyst at the Forefront of Biotechnology. Microb. Biotechnol. 2017, 10, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.-B.; Luo, Q.; Wang, H.-R.; Chen, Y.; Liao, X.-R. Bacterial Laccases: Promising Biological Green Tools for Industrial Applications. Cell. Mol. Life Sci. 2018, 75, 3569–3592. [Google Scholar] [CrossRef]
- Wang, J.; Feng, J.; Jia, W.; Chang, S.; Li, S.; Li, Y. Lignin Engineering through Laccase Modification: A Promising Field for Energy Plant Improvement. Biotechnol. Biofuels 2015, 8, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassanini, I.; Ferrandi, E.E.; Riva, S.; Monti, D. Biocatalysis with Laccases: An Updated Overview. Catalysts 2020, 11, 26. [Google Scholar] [CrossRef]
- Cardullo, N.; Muccilli, V.; Tringali, C. Laccase-Mediated Synthesis of Bioactive Natural Products and Their Analogues. RSC Chem. Biol. 2022, 3, 614–647. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, U.N.; Singh, P.; Pandey, V.P.; Kumar, A. Structure–function Relationship among Bacterial, Fungal and Plant Laccases. J. Mol. Catal. B Enzym. 2011, 68, 117–128. [Google Scholar] [CrossRef]
- Janusz, G.; Pawlik, A.; Świderska-Burek, U.; Polak, J.; Sulej, J.; Jarosz-Wilkołazka, A.; Paszczyński, A. Laccase Properties, Physiological Functions, and Evolution. Int. J. Mol. Sci. 2020, 21, 966. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Go, N. Function and Molecular Evolution of Multicopper Blue Proteins. Cell. Mol. Life Sci. 2005, 62, 2050–2066. [Google Scholar] [CrossRef]
- Gräff, M.; Buchholz, P.C.F.; Le Roes-Hill, M.; Pleiss, J. Multicopper Oxidases: Modular Structure, Sequence Space, and Evolutionary Relationships. Proteins 2020, 88, 1329–1339. [Google Scholar] [CrossRef]
- Olbrich, A.C.; Schild, J.N.; Urlacher, V.B. Correlation between the T1 Copper Reduction Potential and Catalytic Activity of a Small Laccase. J. Inorg. Biochem. 2019, 201, 110843. [Google Scholar] [CrossRef]
- Mehra, R.; Kepp, K.P. Contribution of Substrate Reorganization Energies of Electron Transfer to Laccase Activity. Phys. Chem. Chem. Phys. 2019, 21, 15805–15814. [Google Scholar] [CrossRef]
- Kallio, J.P.; Rouvinen, J.; Kruus, K.; Hakulinen, N. Probing the Dioxygen Route in Melanocarpus Albomyces Laccase with Pressurized Xenon Gas. Biochemistry 2011, 50, 4396–4398. [Google Scholar] [CrossRef] [PubMed]
- Fulop, V.; Wilkinson, R. Structural and Functional Characterisation of a Bacterial Laccase-like Multi-Copper Oxidase CueO from Lignin-Degrading Bacterium Ochrobactrum Sp. with Oxidase Activity towards Lignin Model Compounds and Lignosulfonate. FEBS Lett. 2018, 285, 1684–1700. [Google Scholar]
- Damas, J.M.; Baptista, A.M.; Soares, C.M. The Pathway for O2 Diffusion inside CotA Laccase and Possible Implications on the Multicopper Oxidases Family. J. Chem. Theory Comput. 2014, 10, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Valles, M.; Kamaruddin, A.F.; Wong, L.S.; Blanford, C.F. Inhibition in Multicopper Oxidases: A Critical Review. Catal. Sci. Technol. 2020, 10, 5386–5410. [Google Scholar] [CrossRef]
- Xu, F. Oxidation of Phenols, Anilines, and Benzenethiols by Fungal Laccases: Correlation between Activity and Redox Potentials as Well as Halide Inhibition. Biochemistry 1996, 35, 7608–7614. [Google Scholar] [CrossRef] [PubMed]
- Mate, D.M.; Alcalde, M. Laccase Engineering: From Rational Design to Directed Evolution. Biotechnol. Adv. 2015, 33, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, Y.; Zhan, J.; Lin, Y.; Yang, X. Axial Bonds at the T1 Cu Site of Thermus Thermophilus SG0.5JP17-16 Laccase Influence Enzymatic Properties. FEBS Open Bio 2019, 9, 986–995. [Google Scholar] [CrossRef] [Green Version]
- Durão, P.; Bento, I.; Fernandes, A.T.; Melo, E.P.; Lindley, P.F.; Martins, L.O. Perturbations of the T1 Copper Site in the CotA Laccase from Bacillus subtilis: Structural, Biochemical, Enzymatic and Stability Studies. J. Biol. Inorg. Chem. 2006, 11, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Aza, P.; de Salas, F.; Molpeceres, G.; Rodríguez-Escribano, D.; de la Fuente, I.; Camarero, S. Protein Engineering Approaches to Enhance Fungal Laccase Production in S. cerevisiae. Int. J. Mol. Sci. 2021, 22, 1157. [Google Scholar] [CrossRef]
- Pardo, I.; Camarero, S. Exploring the Oxidation of Lignin-Derived Phenols by a Library of Laccase Mutants. Molecules 2015, 20, 15929–15943. [Google Scholar] [CrossRef] [Green Version]
- Maté, D.; García-Burgos, C.; García-Ruiz, E.; Ballesteros, A.O.; Camarero, S.; Alcalde, M. Laboratory Evolution of High-Redox Potential Laccases. Chem. Biol. 2010, 17, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Christopher, L.P.; Yao, B.; Ji, Y. Lignin Biodegradation with Laccase-Mediator Systems. Front. Energy Res. 2014, 2. [Google Scholar] [CrossRef]
- Frasconi, M.; Favero, G.; Boer, H.; Koivula, A.; Mazzei, F. Kinetic and Biochemical Properties of High and Low Redox Potential Laccases from Fungal and Plant Origin. Biochim. Biophys. Acta 2010, 1804, 899–908. [Google Scholar] [CrossRef]
- Mateljak, I.; Monza, E.; Lucas, M.F.; Guallar, V.; Aleksejeva, O.; Ludwig, R.; Leech, D.; Shleev, S.; Alcalde, M. Increasing Redox Potential, Redox Mediator Activity, and Stability in a Fungal Laccase by Computer-Guided Mutagenesis and Directed Evolution. ACS Catal. 2019, 9, 4561–4572. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, T.; Jolivalt, C.; Briozzo, P.; Caminade, E.; Joly, N.; Madzak, C.; Mougin, C. Crystal Structure of a Four-Copper Laccase Complexed with an Arylamine: Insights into Substrate Recognition and Correlation with Kinetics. Biochemistry 2002, 41, 7325–7333. [Google Scholar] [CrossRef]
- Enguita, F.J.; Marçal, D.; Martins, L.O.; Grenha, R.; Henriques, A.O.; Lindley, P.F.; Carrondo, M.A. Substrate and Dioxygen Binding to the Endospore Coat Laccase from Bacillus subtilis. J. Biol. Chem. 2004, 279, 23472–23476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matera, I.; Gullotto, A.; Tilli, S.; Ferraroni, M.; Scozzafava, A.; Briganti, F. Crystal Structure of the Blue Multicopper Oxidase from the White-Rot Fungus Trametes Trogii Complexed with P-Toluate. Inorg. Chim. Acta 2008, 361, 4129–4137. [Google Scholar] [CrossRef]
- Kallio, J.P.; Auer, S.; Jänis, J.; Andberg, M.; Kruus, K.; Rouvinen, J.; Koivula, A.; Hakulinen, N. Structure–Function Studies of a Melanocarpus Albomyces Laccase Suggest a Pathway for Oxidation of Phenolic Compounds. J. Mol. Biol. 2009, 392, 895–909. [Google Scholar] [CrossRef]
- Orlikowska, M.; de J. Rostro-Alanis, M.; Bujacz, A.; Hernández-Luna, C.; Rubio, R.; Parra, R.; Bujacz, G. Structural Studies of Two Thermostable Laccases from the White-Rot Fungus Pycnoporus Sanguineus. Int. J. Biol. Macromol. 2018, 107, 1629–1640. [Google Scholar] [CrossRef]
- Xie, T.; Liu, Z.; Wang, G. Structural Basis for Monolignol Oxidation by a Maize Laccase. Nat. Plants 2020, 6, 231–237. [Google Scholar] [CrossRef]
- Liu, Z.C.; Xie, T.; Wang, G.G. Crystal Structure of CotA Laccase Complexed with ABTS at a Novel Binding Site. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72 Pt 4, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Piontek, K.; Antorini, M.; Choinowski, T. Crystal Structure of a Laccase from the Fungus Trametes Versicolor at 1.90-A Resolution Containing a Full Complement of Coppers. J. Biol. Chem. 2002, 277, 37663–37669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambria, M.T.; Gullotto, D.; Garavaglia, S.; Cambria, A. In Silico Study of Structural Determinants Modulating the Redox Potential ofRigidoporus Lignosusand Other Fungal Laccases. J. Biomol. Struct. Dyn. 2012, 30, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Quintanar, L.; Stoj, C.; Taylor, A.B.; Hart, P.J.; Kosman, D.J.; Solomon, E.I. Shall We Dance? How a Multicopper Oxidase Chooses Its Electron Transfer Partner. Acc. Chem. Res. 2007, 40, 445–452. [Google Scholar] [CrossRef]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 Reduction to H2O by the Multicopper Oxidases. Dalton Trans. 2008, 30, 3921–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, C.; Madzak, C.; Vadalà, R.; Jolivalt, C.; Gentili, P. Concerted Electron/proton Transfer Mechanism in the Oxidation of Phenols by Laccase. Chembiochem 2013, 14, 2500–2505. [Google Scholar] [CrossRef]
- Li, J.; Farrokhnia, M.; Rulíšek, L.; Ryde, U. Catalytic Cycle of Multicopper Oxidases Studied by Combined Quantum- and Molecular-Mechanical Free-Energy Perturbation Methods. J. Phys. Chem. B 2015, 119, 8268–8284. [Google Scholar] [CrossRef] [Green Version]
- Heppner, D.E.; Kjaergaard, C.H.; Solomon, E.I. Mechanism of the Reduction of the Native Intermediate in the Multicopper Oxidases: Insights into Rapid Intramolecular Electron Transfer in Turnover. J. Am. Chem. Soc. 2014, 136, 17788–17801. [Google Scholar] [CrossRef]
- Rulíšek, L.; Ryde, U. Theoretical Studies of the Active-Site Structure, Spectroscopic and Thermodynamic Properties, and Reaction Mechanism of Multicopper Oxidases. Coord. Chem. Rev. 2013, 257, 445–458. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, M.A.; D’Annibale, A.; Galli, C.; Gentili, P.; Sergi, F. An Assessment of the Relative Contributions of Redox and Steric Issues to Laccase Specificity towards Putative Substrates. Org. Biomol. Chem. 2008, 6, 868–878. [Google Scholar] [CrossRef]
- Astolfi, P.; Brandi, P.; Galli, C.; Gentili, P.; Gerini, M.F.; Greci, L.; Lanzalunga, O. New Mediators for the Enzyme Laccase: Mechanistic Features and Selectivity in the Oxidation of Non-Phenolic Substrates. New J. Chem. 2005, 29, 1308. [Google Scholar] [CrossRef]
- Majeau, J.-A.; Brar, S.K.; Tyagi, R.D. Laccases for Removal of Recalcitrant and Emerging Pollutants. Bioresour. Technol. 2010, 101, 2331–2350. [Google Scholar] [CrossRef] [PubMed]
- Bourbonnais, R.; Paice, M.G. Oxidation of Non-Phenolic Substrates. FEBS Lett. 1990, 267, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Arora, D.S.; Sharma, R.K. Ligninolytic Fungal Laccases and Their Biotechnological Applications. Appl. Biochem. Biotechnol. 2010, 160, 1760–1788. [Google Scholar] [CrossRef] [PubMed]
- Plácido, J.; Capareda, S. Ligninolytic Enzymes: A Biotechnological Alternative for Bioethanol Production. Bioresour. Bioprocess. 2015, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, M.; Jaiswal, N.; Singh, S.; Pandey, V.P.; Dwivedi, U.N. Molecular Docking and Dynamics Simulation Analyses Unraveling the Differential Enzymatic Catalysis by Plant and Fungal Laccases with Respect to Lignin Biosynthesis and Degradation. J. Biomol. Struct. Dyn. 2015, 33, 1835–1849. [Google Scholar] [CrossRef]
- De Salas, F.; Cañadas, R.; Santiago, G.; Virseda-Jerez, A.; Vind, J.; Gentili, P.; Martínez, A.T.; Guallar, V.; Muñoz, I.G.; Camarero, S. Structural and Biochemical Insights into an Engineered High-Redox Potential Laccase Overproduced in Aspergillus. Int. J. Biol. Macromol. 2019, 141, 855–867. [Google Scholar] [CrossRef] [PubMed]
- de Salas, F.; Pardo, I.; Salavagione, H.J.; Aza, P.; Amougi, E.; Vind, J.; Martínez, A.T.; Camarero, S. Advanced Synthesis of Conductive Polyaniline Using Laccase as Biocatalyst. PLoS ONE 2016, 11, e0164958. [Google Scholar] [CrossRef] [Green Version]
- Longe, L.F.; Couvreur, J.; Grandchamp, M.L.; Garnier, G.; Allais, F.; Saito, K. Importance of Mediators for Lignin Degradation by Fungal Laccase. ACS Sustain. Chem. Eng. 2018, 6, 10097–10107. [Google Scholar] [CrossRef]
- Bourbonnais, R.; Leech, D.; Paice, M.G. Electrochemical Analysis of the Interactions of Laccase Mediators with Lignin Model Compounds. Biochim. Biophys. Acta 1998, 1379, 381–390. [Google Scholar] [CrossRef]
- Galli, C.; Gentili, P. Chemical Messengers: Mediated Oxidations with the Enzyme Laccase. J. Phys. Org. Chem. 2004, 17, 973–977. [Google Scholar] [CrossRef]
- Branchi, B.; Galli, C.; Gentili, P. Kinetics of Oxidation of Benzyl Alcohols by the Dication and Radical Cation of ABTS. Comparison with Laccase-ABTS Oxidations: An Apparent Paradox. Org. Biomol. Chem. 2005, 3, 2604–2614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, M.; Zheng, Y.; Zhao, X.; Li, B.; Huan, W. Atomic-Scale Investigation of the Interaction between Coniferyl Alcohol and Laccase for Lignin Degradation Using Molecular Dynamics Simulations and Spectroscopy. J. Dispers. Sci. Technol. 2019, 40, 686–694. [Google Scholar] [CrossRef]
- Morozova, O.V.; Shumakovich, G.P.; Gorbacheva, M.A.; Shleev, S.V.; Yaropolov, A.I. “Blue” Laccases. Biochemistry 2007, 72, 1136–1150. [Google Scholar] [CrossRef]
- Munk, L.; Sitarz, A.K.; Kalyani, D.C.; Mikkelsen, J.D.; Meyer, A.S. Can Laccases Catalyze Bond Cleavage in Lignin? Biotechnol. Adv. 2015, 33, 13–24. [Google Scholar] [CrossRef]
- Zhu, D.; Liang, N.; Zhang, R.; Ahmad, F.; Zhang, W.; Yang, B.; Wu, J.; Geng, A.; Gabriel, M.; Sun, J. Insight into Depolymerization Mechanism of Bacterial Laccase for Lignin. ACS Sustain. Chem. Eng. 2020, 8, 12920–12933. [Google Scholar] [CrossRef]
- Hilgers, R.; Vincken, J.-P.; Gruppen, H.; Kabel, M.A. Laccase/Mediator Systems: Their Reactivity toward Phenolic Lignin Structures. ACS Sustain Chem Eng 2018, 6, 2037–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, M.; Galli, C.; Gentili, P. Comparing the Catalytic Efficiency of Some Mediators of Laccase. J. Mol. Catal. B Enzym. 2002, 16, 231–240. [Google Scholar] [CrossRef]
- Mot, A.C.; Silaghi-Dumitrescu, R. Laccases: Complex Architectures for One-Electron Oxidations. Biochemistry 2012, 77, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Lahtinen, M.; Kruus, K.; Boer, H.; Kemell, M.; Andberg, M.; Viikari, L.; Sipilä, J. The Effect of Lignin Model Compound Structure on the Rate of Oxidation Catalyzed by Two Different Fungal Laccases. J. Mol. Catal. B Enzym. 2009, 57, 204–210. [Google Scholar] [CrossRef]
- Cambria, M.T.; Di Marino, D.; Falconi, M.; Garavaglia, S.; Cambria, A. Docking Simulation and Competitive Experiments Validate the Interaction between the 2,5-Xylidine Inhibitor and Rigidoporus Lignosus Laccase. J. Biomol. Struct. Dyn. 2010, 27, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Galli, C.; Gentili, P.; Jolivalt, C.; Madzak, C.; Vadalà, R. How Is the Reactivity of Laccase Affected by Single-Point Mutations? Engineering Laccase for Improved Activity towards Sterically Demanding Substrates. App. Microbiol. Biotechnol. 2011, 91, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zeng, G.; Tan, Z.; Jiang, M.; Li, H.; Liu, L.; Zhu, Y.; Yu, Z.; Wei, Z.; Liu, Y.; et al. Understanding Lignin-Degrading Reactions of Ligninolytic Enzymes: Binding Affinity and Interactional Profile. PLoS ONE 2011, 6, e25647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, N.J.; Kepp, K.P. Setting the Stage for Electron Transfer: Molecular Basis of ABTS-Binding to Four Laccases from Trametes Versicolor at Variable pH and Protein Oxidation State. J. Mol. Catal. B Enzym. 2014, 100, 68–77. [Google Scholar] [CrossRef]
- Martínez-Sotres, C.; Rutiaga-Quiñones, J.G.; Herrera-Bucio, R.; Gallo, M.; López-Albarrán, P. Molecular Docking Insights into the Inhibition of Laccase Activity by Medicarpin. Wood Sci. Technol. 2015, 49, 857–868. [Google Scholar] [CrossRef]
- Monza, E.; Fatima Lucas, M.; Camarero, S.; Alejaldre, L.C.; Martínez, A.T.; Guallar, V. Insights into Laccase Engineering from Molecular Simulations: Toward a Binding-Focused Strategy. J. Phys. Chem. Lett. 2015, 6, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zeng, G.; Lai, C.; Li, J.; Xu, P.; Wu, H. Molecular Basis of Laccase Bound to Lignin: Insight from Comparative Studies on the Interaction of Trametes Versicolor Laccase with Various Lignin Model Compounds. RSC Adv. 2015, 5, 52307–52313. [Google Scholar] [CrossRef]
- Takur, K.R.; Kohli, M.; Pande, K.; Malik, A.; Deshmukh, A.; Kayal, A.; Kommoju, P.R.; Kulkarni, N. In Silico Studies Disclose the Underlying Link between Binding Affinity and Redox Potential in Laccase Isoforms. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef]
- Pardo, I.; Santiago, G.; Gentili, P.; Lucas, F.; Monza, E.; Medrano, F.J.; Galli, C.; Martínez, A.T.; Guallar, V.; Camarero, S. Re-Designing the Substrate Binding Pocket of Laccase for Enhanced Oxidation of Sinapic Acid. Catal. Sci. Technol. 2016, 6, 3900–3910. [Google Scholar] [CrossRef] [Green Version]
- Dellafiora, L.; Galaverna, G.; Reverberi, M.; Dall’Asta, C. Degradation of Aflatoxins by Means of Laccases from Trametes Versicolor: An In Silico Insight. Toxins 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Lucas, M.F.; Monza, E.; Jørgensen, L.J.; Ernst, H.A.; Piontek, K.; Bjerrum, M.J.; Martinez, Á.T.; Camarero, S.; Guallar, V. Simulating Substrate Recognition and Oxidation in Laccases: From Description to Design. J. Chem. Theory Comput. 2017, 13, 1462–1467. [Google Scholar] [CrossRef]
- Mehra, R.; Meyer, A.S.; Kepp, K.P. Molecular Dynamics Derived Life Times of Active Substrate Binding Poses Explain of Laccase Mutants. RSC Adv. 2018, 8, 36915–36926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehra, R.; Muschiol, J.; Meyer, A.S.; Kepp, K.P. A Structural-Chemical Explanation of Fungal Laccase Activity. Sci. Rep. 2018, 8, 17285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulistyaningdyah, W.T.; Ogawa, J.; Tanaka, H.; Maeda, C.; Shimizu, S. Characterization of Alkaliphilic Laccase Activity in the Culture Supernatant of Myrothecium Verrucaria 24G-4 in Comparison with Bilirubin Oxidase. FEMS Microbiol. Lett. 2004, 230, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Mo, D.; Zeng, G.; Yuan, X.; Chen, M.; Hu, L.; Li, H.; Wang, H.; Xu, P.; Lai, C.; Wan, J.; et al. Molecular Docking Simulation on the Interactions of Laccase from Trametes Versicolor with Nonylphenol and Octylphenol Isomers. Bioprocess Biosyst. Eng. 2018, 41, 331–343. [Google Scholar] [CrossRef]
- Kameshwar, A.K.S.; Barber, R.; Qin, W. Comparative Modeling and Molecular Docking Analysis of White, Brown and Soft Rot Fungal Laccases Using Lignin Model Compounds for Understanding the Structural and Functional Properties of Laccases. J. Mol. Graph. Model. 2018, 79, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Zeng, G.; Chen, M.; Jiang, Y.; Shao, B.; Li, Z.; Liu, Y. Effect of Surfactants on the Interaction of Phenol with Laccase: Molecular Docking and Molecular Dynamics Simulation Studies. J. Hazard. Mater. 2018, 357, 10–18. [Google Scholar] [CrossRef]
- Avelar, M.; Pastor, N.; Ramirez-Ramirez, J.; Ayala, M. Replacement of Oxidizable Residues Predicted by QM-MM Simulation of a Fungal Laccase Generates Variants with Higher Operational Stability. J. Inorg. Biochem. 2018, 178, 125–133. [Google Scholar] [CrossRef]
- Sharma, K.K.; Singh, D.; Rawat, S. Molecular Dynamics Simulation Studies Suggests Unconventional Roles of Non-Secretary Laccases from Enteropathogenic Gut Bacteria and Cryptococcus Neoformans Serotype D. Comput. Biol. Chem. 2018, 73, 41–48. [Google Scholar] [CrossRef]
- Hongyan, L.; Zexiong, Z.; Shiwei, X.; He, X.; Yinian, Z.; Haiyun, L.; Zhongsheng, Y. Study on Transformation and Degradation of Bisphenol A by Trametes Versicolor Laccase and Simulation of Molecular Docking. Chemosphere 2019, 224, 743–750. [Google Scholar] [CrossRef]
- Yosberto, C.-M. Theoretical Study on Binding Interactions of Laccase-Enzyme from Ganoderma Weberianum with Multiples Ligand Substrates with Environmental Impact. Ann. Proteom. Bioinform. 2019, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, K.; Shankar, J.; Verma, P. Multicopper Oxidase (MCO) Laccase from Stropharia Sp. ITCC-8422: An Apparent Authentication Using Integrated Experimental and in Silico Analysis. 3 Biotech 2020, 10, 413. [Google Scholar]
- Sun, R. Lignin Source and Structural Characterization. ChemSusChem 2020, 13, 4174. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Li, J.; Wang, M.; Ren, L.; Luo, Y. Genes Identification, Molecular Docking and Dynamics Simulation Analysis of Laccases from Amylostereum Areolatum Provides Molecular Basis of Laccase Bound to Lignin. Int. J. Mol. Sci. 2020, 21, 8845. [Google Scholar] [CrossRef]
- Liu, Y.; Mao, H.; Hu, C.; Tron, T.; Lin, J.; Wang, J.; Sun, B. Molecular Docking Studies and in Vitro Degradation of Four Aflatoxins (AFB, AFB, AFG, and AFG) by a Recombinant Laccase from Saccharomyces Cerevisiae. J. Food Sci. 2020, 85, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Santacruz-Juárez, E.; Buendia-Corona, R.E.; Ramírez, R.E.; Sánchez, C. Fungal Enzymes for the Degradation of Polyethylene: Molecular Docking Simulation and Biodegradation Pathway Proposal. J. Hazard. Mater. 2021, 411, 125118. [Google Scholar] [CrossRef]
- Bhatt, P.; Tiwari, M.; Parmarick, P.; Bhatt, K.; Gangola, S.; Adnan, M.; Singh, Y.; Bilal, M.; Ahmed, S.; Chen, S. Insights into the Catalytic Mechanism of Ligninolytic Peroxidase and Laccase in Lignin Degradation. Bioremediat. J. 2022, 26, 281–291. [Google Scholar] [CrossRef]
- Mahuri, M.; Paul, M.; Sahu, S.K.; Thatoi, H. In Silico Homology Modeling, Docking and Sequence Analysis of Some Bacterial Laccases to Unravel Enzymatic Specificity towards Lignin Biodegradation. J. Biomol. Struct. Dyn. 2022, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Khan, M.A.; Khan, A.; Ahmad, S.; Zofair, S.F.F.; Younus, H. In Silico and in Vitro Studies on the Inhibition of Laccase Activity by Ellagic Acid: Implications in Drug Designing for the Treatment of Cryptococcal Infections. Int. J. Biol. Macromol. 2022, 209, 642–654. [Google Scholar] [CrossRef]
- Wang, L.; Xue, C.; Owens, G.; Chen, Z. Artificial Intelligence Modeling and Molecular Docking to Analyze the Laccase Delignification Process of Rice Straw by Comamonas Testosteroni FJ17. Bioresour. Technol. 2022, 345, 126565. [Google Scholar] [CrossRef]
- Bhatt, P.; Bhatt, K.; Chen, W.-J.; Huang, Y.; Xiao, Y.; Wu, S.; Lei, Q.; Zhong, J.; Zhu, X.; Chen, S. Bioremediation Potential of Laccase for Catalysis of Glyphosate, Isoproturon, Lignin, and Parathion: Molecular Docking, Dynamics, and Simulation. J. Hazard. Mater. 2023, 443, 130319. [Google Scholar] [CrossRef]
- Zaccaria, M.; Dawson, W.; Kish, D.R.; Reverberi, M.; di Patti, M.C.B.; Domin, M.; Cristiglio, V.; Chan, B.; Dellafiora, L.; Gabel, F.; et al. Experimental–theoretical Study of Laccase as a Detoxifier of Aflatoxins. Sci. Rep. 2023, 13, 860. [Google Scholar] [CrossRef] [PubMed]
- Santiago, G.; de Salas, F.; Fátima Lucas, M.; Monza, E.; Acebes, S.; Martinez, Á.T.; Camarero, S.; Guallar, V. Computer-Aided Laccase Engineering: Toward Biological Oxidation of Arylamines. ACS Catal. 2016, 6, 5415–5423. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.; Ryde, U.; Roos, B.O. Quantum Chemical Calculations of the Reorganization Energy of Blue-Copper Proteins. Protein Sci. 1998, 7, 2659–2668. [Google Scholar] [CrossRef] [Green Version]
- Götze, J.P.; Bühl, M. Laccase Redox Potentials: pH Dependence and Mutants, a QM/MM Study. J. Phys. Chem. B 2016, 120, 9265–9276. [Google Scholar] [CrossRef] [Green Version]
- Olmeda, I.; Casino, P.; Collins, R.E.; Sendra, R.; Callejón, S.; Huesa, J.; Soares, A.S.; Ferrer, S.; Pardo, I. Structural Analysis and Biochemical Properties of Laccase Enzymes from Two Pediococcus Species. Microb. Biotechnol. 2021, 14, 1026–1043. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Weichsel, A.; Grass, G.; Thakali, K.; Hazzard, J.T.; Tollin, G.; Rensing, C.; Montfort, W.R. Crystal Structure and Electron Transfer Kinetics of CueO, a Multicopper Oxidase Required for Copper Homeostasis in Escherichia Coli. Proc. Natl. Acad. Sci. USA 2002, 99, 2766–2771. [Google Scholar] [CrossRef] [Green Version]
- Glazunova, O.; Trushkin, N.; Moiseenko, K.; Filimonov, I.; Fedorova, T. Catalytic Efficiency of Basidiomycete Laccases: Redox Potential versus Substrate-Binding Pocket Structure. Catalysts 2018, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tan, Y.; Sun, S.; Zhou, L.; Wu, G.; Shao, Y.; Wang, M.; Xin, Z. Improving Degradation of Polycyclic Aromatic Hydrocarbons by Bacillus Atrophaeus Laccase Fused with Vitreoscilla Hemoglobin and a Novel Strong Promoter Replacement. Biology 2022, 11, 1129. [Google Scholar] [CrossRef]
- Singh, D.; Sharma, K.K.; Jacob, S.; Gakhar, S.K. Molecular Docking of Laccase Protein from Bacillus Safensis DSKK5 Isolated from Earthworm Gut: A Novel Method to Study Dye Decolorization Potential. Water Air Soil Pollut. 2014, 225, 2175. [Google Scholar] [CrossRef]
- Giacobelli, V.G.; Monza, E.; Fatima Lucas, M.; Pezzella, C.; Piscitelli, A.; Guallar, V.; Sannia, G. Repurposing Designed Mutants: A Valuable Strategy for Computer-Aided Laccase Engineering—The Case of POXA1b. Catal. Sci. Technol. 2017, 7, 515–523. [Google Scholar] [CrossRef]
- Chiadò, A.; Bosco, F.; Bardelli, M.; Simonelli, L.; Pedotti, M.; Marmo, L.; Varani, L. Rational Engineering of the Lccβ T. versicolor Laccase for the Mediator-Less Oxidation of Large Polycyclic Aromatic Hydrocarbons. Comput. Struct.l Biotechnol. J. 2021, 19, 2213–2222. [Google Scholar] [CrossRef]
- Camarero, S.; Pardo, I.; Cañas, A.I.; Molina, P.; Record, E.; Martínez, A.T.; Martínez, M.J.; Alcalde, M. Engineering Platforms for Directed Evolution of Laccase from Pycnoporus Cinnabarinus. Appl. Environ. Microbiol. 2012, 78, 1370–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofrichter, M.; Ullrich, R.; Pecyna, M.J.; Liers, C.; Lundell, T. New and Classic Families of Secreted Fungal Heme Peroxidases. Appl. Microbiol. Biotechnol. 2010, 87, 871–897. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.T. Molecular Biology and Structure-Function of Lignin-Degrading Heme Peroxidases. Enzym. Microb. Technol. 2002, 30, 425–444. [Google Scholar] [CrossRef]
- Hammel, K.E.; Cullen, D. Role of fungal peroxidases in biological ligninolysis. Curr. Opin. Plant. Biol. 2008, 11, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.S. Structure and Action Mechanism of Ligninolytic Enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef]
- Hofrichter, M. Review: Lignin Conversion by Manganese Peroxidase (MnP). Enzy. Microb. Technol. 2002, 30, 454–466. [Google Scholar] [CrossRef]
- Kumar, A.; Arora, P.K. Biotechnological Applications of Manganese Peroxidases for Sustainable Management. Front. Environ. Sci. 2022, 10, 875157. [Google Scholar] [CrossRef]
- Chowdhary, P.; Shukla, G.; Raj, G.; Ferreira, L.F.R.; Bharagava, R.N. Microbial Manganese Peroxidase: A Ligninolytic Enzyme and Its Ample Opportunities in Research. SN Appl. Sci. 2019, 1, 45. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Cui, K.; Guo, Z.; Cui, M.; Chen, Y. Manganese Peroxidase Mediated Oxidation of Sulfamethoxazole: Integrating the Computational Analysis to Reveal the Reaction Kinetics, Mechanistic Insights, and Oxidation Pathway. J. Hazard. Mater. 2021, 415, 125719. [Google Scholar] [CrossRef]
- Fernández-Fueyo, E.; Acebes, S.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Romero, A.; Medrano, F.J.; Guallar, V.; Martínez, A.T. Structural Implications of the C-Terminal Tail in the Catalytic and Stability Properties of Manganese Peroxidases from Ligninolytic Fungi. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Romero, A.; Hammel, K.E.; Medrano, F.J.; Martínez, A.T. Ligninolytic Peroxidase Genes in the Oyster Mushroom Genome: Heterologous Expression, Molecular Structure, Catalytic and Stability Properties, and Lignin-Degrading Ability. Biotechnol. Biofuels 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acebes, S.; Fernandez-Fueyo, E.; Monza, E.; Fatima Lucas, M.; Almendral, D.; Ruiz-Dueñas, F.J.; Lund, H.; Martinez, A.T.; Guallar, V. Rational Enzyme Engineering Through Biophysical and Biochemical Modeling. ACS Catal. 2016, 6, 1624–1629. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Dueñas, F.J.; Lundell, T.; Floudas, D.; Nagy, L.G.; Barrasa, J.M.; Hibbett, D.S.; Martínez, A.T. Lignin-Degrading Peroxidases in Polyporales: An Evolutionary Survey Based on 10 Sequenced Genomes. Mycologia 2013, 105, 1428–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso-Fernández, I.; Martínez, A.T.; Ruiz-Dueñas, F.J. Experimental Recreation of the Evolution of Lignin-Degrading Enzymes from the Jurassic to Date. Biotechnol. Biofuels 2017, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Ayuso-Fernández, I.; De Lacey, A.L.; Cañada, F.J.; Ruiz-Dueñas, F.J.; Martínez, A.T. Increase of Redox Potential during the Evolution of Enzymes Degrading Recalcitrant Lignin. Chemistry 2019, 25, 2708–2712. [Google Scholar] [CrossRef] [Green Version]
- Tuomela, M.; Hatakka, A. Oxidative Fungal Enzymes for Bioremediation. Compr. Biotechnol. 2011, 6, 183–196. [Google Scholar]
- Wen, X.; Jia, Y.; Li, J. Degradation of Tetracycline and Oxytetracycline by Crude Lignin Peroxidase Prepared from Phanerochaete Chrysosporium—A White Rot Fungus. Chemosphere 2009, 75, 1003–1007. [Google Scholar] [CrossRef]
- Zhang, Y.; Geißen, S.-U. In Vitro Degradation of Carbamazepine and Diclofenac by Crude Lignin Peroxidase. J. Hazard. Mater. 2010, 176, 1089–1092. [Google Scholar] [CrossRef]
- Guo, J.; Liu, X.; Zhang, X.; Wu, J.; Chai, C.; Ma, D.; Chen, Q.; Xiang, D.; Ge, W. Immobilized Lignin Peroxidase on FeO@SiO@polydopamine Nanoparticles for Degradation of Organic Pollutants. Int. J. Biol. Macromol. 2019, 138, 433–440. [Google Scholar] [CrossRef]
- Ding, Y.; Cui, K.; Liu, X.; Xie, Q.; Guo, Z.; Chen, Y. Lignin Peroxidase-Catalyzed Direct Oxidation of Trace Organic Pollutants through a Long-Range Electron Transfer Mechanism: Using Propranolol as an Example. J. Hazard. Mater. 2022, 431, 128544. [Google Scholar] [CrossRef] [PubMed]
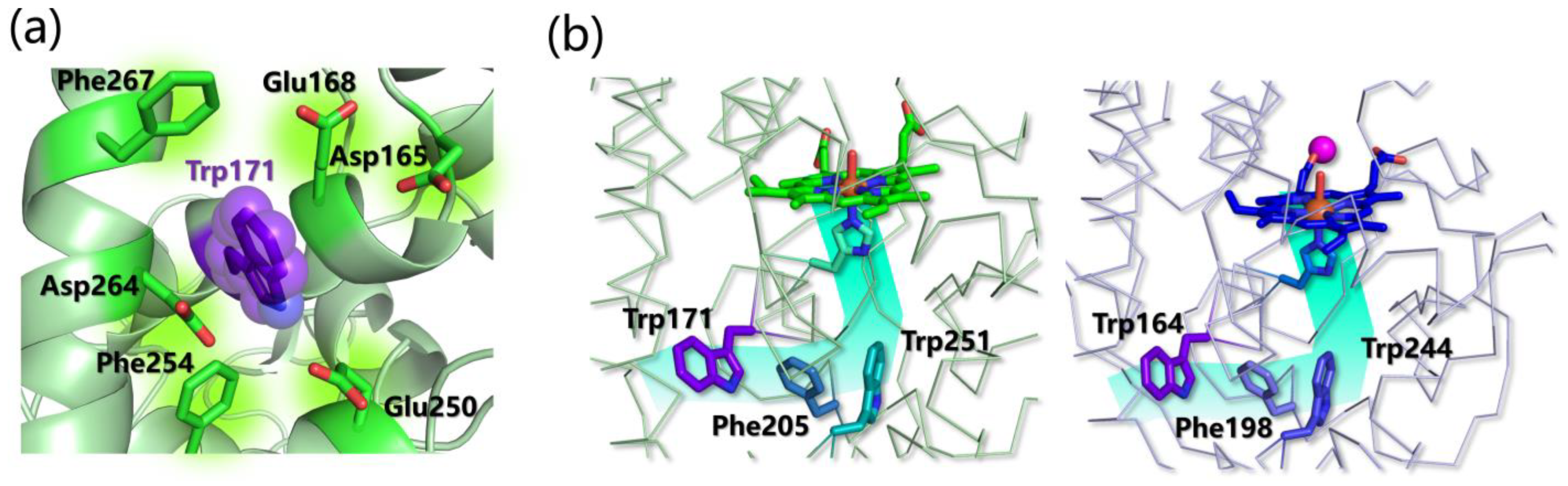
- Choinowski, T.; Blodig, W.; Winterhalter, K.H.; Piontek, K. The Crystal Structure of Lignin Peroxidase at 1.70 Å Resolution Reveals a Hydroxy Group on the C β of Tryptophan 171: A Novel Radical Site Formed during the Redox Cycle. Huber. J. Mol. Biol. 1999, 286, 809–827. [Google Scholar] [CrossRef]
- Sáez-Jiménez, V.; Rencoret, J.; Rodríguez-Carvajal, M.A.; Gutiérrez, A.; Ruiz-Dueñas, F.J.; Martínez, A.T. Role of Surface Tryptophan for Peroxidase Oxidation of Nonphenolic Lignin. Biotechnol. Biofuels 2016, 9, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, L.T.M.; Kim, S.J.; Kim, Y.H. Improvement of Catalytic Performance of Lignin Peroxidase for the Enhanced Degradation of Lignocellulose Biomass Based on the Imbedded Electron-Relay in Long-Range Electron Transfer Route. Biotechnol. Biofuels 2016, 9, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acebes, S.; Ruiz-Dueñas, F.J.; Toubes, M.; Sáez-Jiménez, V.; Pérez-Boada, M.; Lucas, M.F.; Martínez, A.T.; Guallar, V. Mapping the Long-Range Electron Transfer Route in Ligninolytic Peroxidases. J. Phys. Chem. B 2017, 121, 3946–3954. [Google Scholar] [CrossRef] [PubMed]
- Bernini, C.; Pogni, R.; Basosi, R.; Sinicropi, A. The Nature of Tryptophan Radicals Involved in the Long-Range Electron Transfer of Lignin Peroxidase and Lignin Peroxidase-like Systems: Insights from Quantum Mechanical/molecular Mechanics Simulations. Proteins 2012, 80, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Blodig, W.; Smith, A.T.; Doyle, W.A.; Piontek, K. Crystal Structures of Pristine and Oxidatively Processed Lignin Peroxidase Expressed in Escherichia Coli and of the W171F Variant That Eliminates the Redox Active Tryptophan 171. Implications for the Reaction Mechanism. J. Mol. Biol. 2001, 305, 851–861. [Google Scholar] [CrossRef]
- Pham, L.T.M.; Seo, H.; Kim, K.-J.; Kim, Y.H. In Silico-Designed Lignin Peroxidase from Phanerochaete Chrysosporium Shows Enhanced Acid Stability for Depolymerization of Lignin. Biotechnol. Biofuels 2018, 11, 325. [Google Scholar] [CrossRef] [Green Version]
- Pham, L.T.M.; Deng, K.; Northen, T.R.; Singer, S.W.; Adams, P.D.; Simmons, B.A.; Sale, K.L. Experimental and Theoretical Insights into the Effects of pH on Catalysis of Bond-Cleavage by the Lignin Peroxidase Isozyme H8 from Phanerochaete Chrysosporium. Biotechnol. Biofuels 2021, 14, 108. [Google Scholar] [CrossRef]
- Valli, K.; Wariishi, H.; Gold, M.H. Oxidation of Monomethoxylated Aromatic Compounds by Lignin Peroxidase: Role of Veratryl Alcohol in Lignin Biodegradation. Biochemistry 1990, 29, 8535–8539. [Google Scholar] [CrossRef] [PubMed]
- Gerini, M.F.; Francesca Gerini, M.; Roccatano, D.; Baciocchi, E.; Di Nola, A. Molecular Dynamics Simulations of Lignin Peroxidase in Solution. Biophys. J. 2003, 84, 3883–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, W.A.; Blodig, W.; Veitch, N.C.; Piontek, K.; Smith, A.T. Two Substrate Interaction Sites in Lignin Peroxidase Revealed by Site-Directed Mutagenesis. Biochemistry 1998, 37, 15097–15105. [Google Scholar] [CrossRef]
- Ecker, J.; Fülöp, L. Lignin Peroxidase Ligand Access Channel Dysfunction in the Presence of Atrazine. Sci. Rep. 2018, 8, 5989. [Google Scholar] [CrossRef] [Green Version]
- Recabarren, R.; Fuenzalida-Valdivia, I.; Alzate-Morales, J. Studying the Binding Mechanisms of Veratryl Alcohol to P. chrysosporium Lignin Peroxidase: Insights from Theoretical Approaches. Theor. Chem. Acc. 2016, 135, 71. [Google Scholar] [CrossRef]
- Miki, Y.; Calviño, F.R.; Pogni, R.; Giansanti, S.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Basosi, R.; Romero, A.; Martínez, A.T. Crystallographic, Kinetic, and Spectroscopic Study of the First Ligninolytic Peroxidase Presenting a Catalytic Tyrosine. J. Biol. Chem. 2011, 286, 15525–15534. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Pogni, R.; Acebes, S.; Lucas, F.; Fernández-Fueyo, E.; Baratto, M.C.; Fernández, M.I.; de los Ríos, V.; Ruiz-Dueñas, F.J.; Sinicropi, A.; et al. Formation of a Tyrosine Adduct Involved in Lignin Degradation by Trametopsis Cervina Lignin Peroxidase: A Novel Peroxidase Activation Mechanism. Biochem. J. 2013, 452, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Romero, J.O.; Fernández-Fueyo, E.; Avila-Salas, F.; Recabarren, R.; Alzate-Morales, J.; Martínez, A.T. Binding and Catalytic Mechanisms of Veratryl Alcohol Oxidation by Lignin Peroxidase: A Theoretical and Experimental Study. Comput. Struct. Biotechnol. J. 2019, 17, 1066–1074. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Martínez, M.J.; Martínez, A.T. Molecular Characterization of a Novel Peroxidase Isolated from the Ligninolytic Fungus Pleurotus Eryngii. Mol. Microbiol. 1999, 31, 223–235. [Google Scholar] [CrossRef]
- Pérez-Boada, M.; Ruiz-Dueñas, F.J.; Pogni, R.; Basosi, R.; Choinowski, T.; Martínez, M.J.; Piontek, K.; Martínez, A.T. Versatile Peroxidase Oxidation of High Redox Potential Aromatic Compounds: Site-Directed Mutagenesis, Spectroscopic and Crystallographic Investigation of Three Long-Range Electron Transfer Pathways. J. Mol. Biol. 2005, 354, 385–402. [Google Scholar] [CrossRef]
- Bernini, C.; Pogni, R.; Ruiz-Dueñas, F.J.; Martínez, A.T.; Basosi, R.; Sinicropi, A. EPR Parameters of Amino Acid Radicals in P. Eryngii Versatile Peroxidase and Its W164Y Variant Computed at the QM/MM Level. Phys. Chem. Chem. Phys. 2011, 13, 5078. [Google Scholar]
- Kohler, A.C.; Simmons, B.A.; Sale, K.L. Structure-Based Engineering of a Plant-Fungal Hybrid Peroxidase for Enhanced Temperature and pH Tolerance. Cell Chem. Biol. 2018, 25, 974–983.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáez-Jiménez, V.; Fernández-Fueyo, E.; Medrano, F.J.; Romero, A.; Martínez, A.T.; Ruiz-Dueñas, F.J. Improving the pH-Stability of Versatile Peroxidase by Comparative Structural Analysis with a Naturally-Stable Manganese Peroxidase. PLoS ONE 2015, 10, e0140984. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Martínez, A.T. Engineering a Fungal Peroxidase That Degrades Lignin at Very Acidic pH. Biotechnol. Biofuels 2014, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Katari, S.K.; Umamaheswari, A.; Raj, A. In silico exploration of lignin peroxidase for unraveling the degradation mechanism employing lignin model compounds. RSC Adv. 2021, 11, 14632–14653. [Google Scholar] [CrossRef] [PubMed]
- Jendrossek, D.; Birke, J. Rubber Oxygenases. Appl. Microbiol. Biotechnol. 2019, 103, 125–142. [Google Scholar] [CrossRef] [Green Version]
- Rose, K.; Steinbüchel, A. Biodegradation of Natural Rubber and Related Compounds: Recent Insights into a Hardly Understood Catabolic Capability of Microorganisms. Appl. Environ. Microbiol. 2005, 71, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Mutti, F.G. Alkene Cleavage Catalysed by Heme and Nonheme Enzymes: Reaction Mechanisms and Biocatalytic Applications. Bioinorg. Chem. Appl. 2012, 2012, 626909. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Bolados, H.; Bascuñan-Heredia, A.; Alvarez, G. Sustainable Approach of the Natural Rubber. Green-Based Nanocomposite Mater. Appl. 2023, 279–294. [Google Scholar] [CrossRef]
- Andler, R. Bacterial and Enzymatic Degradation of Poly(cis-1,4-Isoprene) Rubber: Novel Biotechnological Applications. Biotechnol. Adv. 2020, 44, 107606. [Google Scholar] [CrossRef]
- Vivod, R.; Andler, R.; Oetermann, S.; Altenhoff, A.-L.; Seipel, N.; Holtkamp, M.; Hogeback, J.; Karst, U.; Steinbüchel, A. Characterization of the Latex Clearing Protein of the Poly(cis-1,4-Isoprene) and Poly(trans-1,4-Isoprene) Degrading Bacterium Nocardia Nova SH22a. J. Gen. Appl. Microbiol. 2020, 65, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braaz, R.; Fischer, P.; Jendrossek, D. Novel Type of Heme-Dependent Oxygenase Catalyzes Oxidative Cleavage of Rubber (poly-Cis-1,4-Isoprene). Appl. Environ. Microbiol. 2004, 70, 7388–7395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braaz, R.; Armbruster, W.; Jendrossek, D. Heme-Dependent Rubber Oxygenase RoxA of Xanthomonas Sp. Cleaves the Carbon Backbone of Poly(cis -1,4-Isoprene) by a Dioxygenase Mechanism. Appl. Environ. Microbiol. 2005, 71, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Kasai, D.; Imai, S.; Asano, S.; Tabata, M.; Iijima, S.; Kamimura, N.; Masai, E.; Fukuda, M. Identification of Natural Rubber Degradation Gene in Rhizobacter Gummiphilus NS21. Biosci. Biotechnol. Biochem. 2017, 81, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Birke, J.; Röther, W.; Jendrossek, D. RoxB Is a Novel Type of Rubber Oxygenase That Combines Properties of Rubber Oxygenase RoxA and Latex Clearing Protein (Lcp). Appl. Environ. Microbiol. 2017, 83, e00721-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birke, J.; Röther, W.; Jendrossek, D. Latex Clearing Protein (Lcp) of Streptomyces Sp. Strain K30 Is a b -Type Cytochrome and Differs from Rubber Oxygenase A (RoxA) in Its Biophysical Properties. Appl. Environ. Microbiol. 2015, 81, 3793–3799. [Google Scholar] [CrossRef] [Green Version]
- Röther, W.; Austen, S.; Birke, J.; Jendrossek, D. Cleavage of Rubber by the Latex Clearing Protein (Lcp) of Streptomyces Sp. Strain K30: Molecular Insights. Appl. Environ. Microbiol. 2016, 82, 6593–6602. [Google Scholar] [CrossRef] [Green Version]
- Chia, K.-H.; Nanthini, J.; Thottathil, G.P.; Najimudin, N.; Haris, M.R.H.; Sudesh, K. Identification of New Rubber-Degrading Bacterial Strains from Aged Latex. Polym Degrad. Stab. 2014, 109, 354–361. [Google Scholar] [CrossRef]
- Seidel, J.; Schmitt, G.; Hoffmann, M.; Jendrossek, D.; Einsle, O. Structure of the Processive Rubber Oxygenase RoxA from Xanthomonas Sp. Proc. Natl. Acad. Sci. USA 2013, 110, 13833–13838. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, G.; Seiffert, G.; Kroneck, P.M.H.; Braaz, R.; Jendrossek, D. Spectroscopic Properties of Rubber Oxygenase RoxA from Xanthomonas Sp., a New Type of Dihaem Dioxygenase. Microbiology 2010, 156, 2537–2548. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Chen, Q.; Wang, B. Unraveling the Mechanism of Latex Clearing Protein LcpK30 Catalysed Oxidative Cleavage of Poly(cis-1,4-Isoprene). Sci. Bull. 2021, 66, 2887–2897. [Google Scholar]
- Ilcu, L.; Röther, W.; Birke, J.; Brausemann, A.; Einsle, O.; Jendrossek, D. Structural and Functional Analysis of Latex Clearing Protein (Lcp) Provides Insight into the Enzymatic Cleavage of Rubber. Sci. Rep. 2017, 7, 6179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, Y. Mechanical Insights into the Enzymatic Cleavage of Double C–C Bond in Poly(cis-1,4-Isoprene) by the Latex Clearing Protein. Inorg. Chem. 2020, 59, 9627–9637. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Honda, Y.; Kuwahara, M.; Watanabe, T. Degradation of Vulcanized and Nonvulcanized Polyisoprene Rubbers by Lipid Peroxidation Catalyzed by Oxidative Enzymes and Transition Metals. Biomacromolecules 2003, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Nayanashree, G.; Thippeswamy, B. Natural Rubber Degradation by Laccase and Manganese Peroxidase Enzymes of Penicillium Chrysogenum. Int. J. Environ. Sci. Technol. 2015, 12, 2665–2672. [Google Scholar] [CrossRef]
- Andler, R.; D’Afonseca, V.; Pino, J.; Valdés, C.; Salazar-Viedma, M. Assessing the Biodegradation of Vulcanised Rubber Particles by Fungi Using Genetic, Molecular and Surface Analysis. Front. Bioeng. Biotechnol. 2021, 9, 761510. [Google Scholar] [CrossRef]
- Kushwaha, A.; Goswami, L.; Singhvi, M.; Kim, B.S. Biodegradation of Poly(ethylene Terephthalate): Mechanistic Insights, Advances, and Future Innovative Strategies. J. Chem. Eng. 2023, 457, 141230. [Google Scholar] [CrossRef]
- Ghatge, S.; Yang, Y.; Ahn, J.-H.; Hur, H.-G. Biodegradation of Polyethylene: A Brief Review. Appl. Biol. Chem. 2020, 63, 27. [Google Scholar] [CrossRef]
- Bardají, D.K.R.; Moretto, J.A.S.; Furlan, J.P.R.; Stehling, E.G. A Mini-Review: Current Advances in Polyethylene Biodegradation. World J. Microbiol. Biotechnol. 2020, 36, 32. [Google Scholar] [CrossRef]
- Santo, M.; Weitsman, R.; Sivan, A. The Role of the Copper-Binding Enzyme—Laccase—in the Biodegradation of Polyethylene by the Actinomycete Rhodococcus Ruber. Int. Biodeterior. Biodegrad. 2013, 84, 204–210. [Google Scholar] [CrossRef]
- Goudriaan, M.; Morales, V.H.; van der Meer, M.T.J.; Mets, A.; Ndhlovu, R.T.; van Heerwaarden, J.; Simon, S.; Heuer, V.B.; Hinrichs, K.-U.; Niemann, H. A Stable Isotope Assay with C-Labeled Polyethylene to Investigate Plastic Mineralization Mediated by Rhodococcus Ruber. Mar. Pollut. Bull. 2023, 186, 114369. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Lv, J.; Wei, W.; Guo, S. Effects of Adding Laccase to Bacterial Consortia Degrading Heavy Oil. Processes 2021, 9, 2025. [Google Scholar] [CrossRef]
- Zampolli, J.; Orro, A.; Manconi, A.; Ami, D.; Natalello, A.; Di Gennaro, P. Transcriptomic Analysis of Rhodococcus Opacus R7 Grown on Polyethylene by RNA-Seq. Sci. Rep. 2021, 11, 21311. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Zimmermann, W. Microbial Enzymes for the Recycling of Recalcitrant Petroleum-Based Plastics: How Far Are We? Microb. Biotechnol. 2017, 10, 1308–1322. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.; Moussavi, G.; Rezaei, M. Enhanced Peroxidase-Mediated Biodegradation of Polyethylene Using the Bacterial Consortia under H2O2-Biostimulation. Polymer 2022, 240, 124508. [Google Scholar] [CrossRef]
- Deguchi, T.; Kakezawa, M.; Nishida, T. Nylon Biodegradation by Lignin-Degrading Fungi. Appl. Environ. Microbiol. 1997, 63, 329–331. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Kitaoka, Y.; Kakezawa, M.; Nishida, T. Purification and Characterization of a Nylon-Degrading Enzyme. Appl. Environ. Microbiol. 1998, 64, 1366–1371. [Google Scholar] [CrossRef] [Green Version]
- Ojha, N.; Pradhan, N.; Singh, S.; Barla, A.; Shrivastava, A.; Khatua, P.; Rai, V.; Bose, S. Evaluation of HDPE and LDPE Degradation by Fungus, Implemented by Statistical Optimization. Sci. Rep. 2017, 7, 39515. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, C. Fungal Potential for the Degradation of Petroleum-Based Polymers: An Overview of Macro- and Microplastics Biodegradation. Biotechnol. Adv. 2020, 40, 107501. [Google Scholar] [CrossRef]
- Ehara, K.; Iiyoshi, Y.; Tsutsumi, Y.; Nishida, T. Polyethylene Degradation by Manganese Peroxidase in the Absence of Hydrogen Peroxide. J. Wood Sci. 2000, 46, 180–183. [Google Scholar] [CrossRef]
- Zhang, X.-B.; Li, Z.-S.; Lu, Z.-Y.; Sun, C.-C. Molecular Dynamics Simulation of the Linear Low-Density Polyethylene Crystallization. J. Chem. Phys. 2001, 115, 3916–3922. [Google Scholar] [CrossRef]
- Perna, V.; Meyer, A.S.; Holck, J.; Eltis, L.D.; Eijsink, V.G.H.; Agger, J.W. Laccase-Catalyzed Oxidation of Lignin Induces Production of H2O2. ACS Sustain. Chem. Eng. 2020, 8, 831–841. [Google Scholar] [CrossRef]
- Stripp, S.T.; Duffus, B.R.; Fourmond, V.; Léger, C.; Leimkuhler, S.; Hirota, S.; Hu, Y.; Jasniewski, A.; Ogata, H.; Ribbe, M.W. Second and Outer Coordination Sphere Effects in Nitrogenase, Hydrogenase, Formate Dehydrogenase, and CO Dehydrogenase. Chem. Rev. 2022, 122, 11900–11973. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Saikia, S.; Bordoloi, M. Molecular Docking: Challenges, Advances and Its Use in Drug Discovery Perspective. Curr. Drug Targets 2019, 20, 501–521. [Google Scholar] [CrossRef]
- Kalos, M.H.; Whitlock, P.A. Monte Carlo Methods Volume I: Basics; Wiley: New York, NY, USA, 1986; pp. 78–88. [Google Scholar]
- Chib, S.; Greenberg, E. Understanding the Metropolis-Hastings Algorithm. Am. Stat. 1995, 49, 327–335. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 9781118712276. [Google Scholar]
- Andersoon, K.; Malmqvist, P.; Roos, B.O. Second Order Perturbation Theory with a Complete Active Space Self Consistent Field Reference Function. J. Chem. Phys. 1992, 96, 1218. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM Methods for Biological Systems. In Atomistic Approaches in Modern Biology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 173–290. [Google Scholar]
- Ahmadi, S.; Herrera, L.B.; Chehelamirani, M.; Hostaš, J.; Jalife, S.; Salahub, D.R. Multiscale Modeling of Enzymes: QM-Cluster, QM/MM, and QM/MM/MD: A Tutorial Review. Int. J. Quantum Chem. 2018, 118, e25558. [Google Scholar] [CrossRef]










| LPMO | PDB | Docking | MD, QM, Hybrid | Ref. |
|---|---|---|---|---|
| Neurospora crassa AA9 | 4D7U | Xyloglucan, cellotetraose, cellopentaose, cellohexaose | [101] | |
| Neurospora crassa AA9 | 4D7U | Cellohexaose | [59] | |
| Phanerochaete chrysosporium AA9 | 4B5Q | MD with crystalline cellulose (1β) | [102] | |
| Heterobasidion irregulare AA9 | 5NNS | MD with crystalline cellulose (1β) | [103] | |
| Serratia marcescens AA10 | 2BEM | MD with crystalline chitin (α[100], α[110], β[100]) | [104] | |
| Neurospora crassa AA9 | 4EIR | CDH | [105] | |
| Four Neurospora crassa AA9 | 4D7U, 4EIR, 4QI8, 4EIS | Five CYT structures * | MD, Umbrella sampling | [106] |
| Lentinus similis AA9 | 5ACF | CDH | MD, QM/MM/metadynamics | [107] |
| Lentinus similis AA9 | 5ACF | Ascorbate | QM/MM/metadynamics | [67] |
| Laccase | PDB | Ligands | Residues | Ref. |
|---|---|---|---|---|
| Bacillus subtilis | 4Q8B | Sinapic acid | H419, H497 P226,R416,G417,G376,T337, T415, L386, 3H2O (see Figure 6a,b) | |
| Bacillus subtilis | 1OF0 | ABTS | H419, H497 G417,T260, T415,L386 | |
| Trametes versicolor | 1KYA | 2,5-xylidine | H458 D206,F162, F265,L164,G392,P391 1H2O | [147] |
| Bacillus subtilis | 3ZDW | ABTS | H497 F226,F384,C229, C322, G323,A227 | [148] |
| Trametes trogii | 2HRG | p-methylbenzoate | H455 A205,G391,V162,F331 1H2O | [149] |
| Melanocarpus albomyces | 3FU9 | 2,6 dimethoxyphenol | H508 F371,F427,L363,L429 | [150] |
| Pycnoporus sanguineus | 5NQ8 | Phenol | H477 F183,F353,H2O | [151] |
| Zea mays (Plant Laccase) | 6KLI | Sinapic alcohol | H519 E449,V348 N273 L446 G447 R518 D162 V276, W522, P362 G361 | [152] |
| 6KLJ | Coniferyl alcohol | H519 E449,Val348 N273 L446,L367,R518 D162 V276, W522, P362 G361,A360,H2O | [152] | |
| Bacillus subtilis | 4YWN | ABTS | Novel binding site far from T1 | [153] |
| Laccase | PDB | Docking | MD, QM, Hybrid | Ref. |
|---|---|---|---|---|
| Trametes hirsuta Q02497 | Homology model | 2,5-xylidine and syringaldehyde | [182] | |
| Rigidoporus lignosus | 1V10 | 2,5-xylidine | [183] | |
| Trametes versicolor | 1KYA wt and F162A, F332A | 2,5-xylidine, 3,5-Di-t-Bu-phenol, and 2,6-di-t-Bu-phenol | [184] | |
| Trametes versicolor | 1GYC | Di-lignol model | MD | [185] |
| Trametes versicolor | Four isoforms | ABTS | MD | [186] |
| Trametes versicolor | 1GYC | Di- and tri-lignols | [187] | |
| Pycnoporus cinnabarinus | 2XYB | ABTS and 2,6-dimethoxyphenol | QM/MM | [188] |
| Trametes versicolor | 1GYC | Monolignols, 2,6-dimethoxyphenol, ferulic acid, guaiacol, sinapic acid, and vanillyl alcohol | [189,190] | |
| Populus trichocarpa (plant laccase) Trametes versicolor (fungal laccase) | 1AOZ 1KYA | Mono-, di-, tri- and tetramer lignin models | [168] | |
| Homologus chimeric 3A4 laccase from basidiomycetes PM1 | 5ANH | Sinapic acid,methyl syringate, dehydrodisinapic acid, and dilactone | [191] | |
| Homologus from Trametes versicolor | 1KYA | Aflatoxin B1 and M1 | [192] | |
| Myceliophthora thermophila Pycnoporus cinnabarinus | 6F5K 2XYB | Syringaldazine and four phenols | QM/MM | [193] |
| Trametes versicolor | 1KYA | 2,6 dimetoxy phenol | MD | [194] |
| Trametes versicolor Cerrena unicolor | 1GYC and homology model | Sinapic acid, syringaldazine, 2,6-dichlorophenol, hyrocaffeic acid, catechol, 2.6-dimethoxyphenol, ABTS, n-(1-Naphtyl) ethylendiamine, 3-amino-4-hydroxy benzenesulfonic acid, and 2-methoxyhydroquinone | MD | [195] |
| Trametes versicolor | 1GYC | 54 substrates from [137,196] | DFT | [132] |
| Trametes versicolor | 1GYC | Nonylphenol and octylophenol isomers | [197] | |
| Six fungal laccase sequences | Homology model | Mono-, di-, tri-, and tetramer lignin models | [198] | |
| Trametes versicolor | 1GYC | Phenol, surfactant triton X-100, and rhamnolipid | MD | [199] |
| Coriolopsis gallica | 4A2E | MD QM/MM | [200] | |
| Cryptococcus neoformans | Prostaglandin and phospholipids | MD | [201] | |
| Trametes versicolor | 1GYC | Bisphenol | [202] | |
| Trametes versicolor | 1GYC | Coniferyl alcohol | [175] | |
| Trametes versicolor | 1GYC | 15 phenolic compounds | QM | [132] |
| Ganoderma weberianum | Homology model | 2,4-dichlorophenol, benzidine, sulϐisoxazole, tetracycline, trimethoprim, ABTS, and 2,6-dimethoxyphenol | [203] | |
| Saccharomyces cerevisiae | 6H5Y | N,N-dimethyl-p-phenylenediamine, and 1-hydroxybenzotriazole | [169] | |
| Stropharia sp. ITCC-8422 | Homology model | ABTS, 2,6-dimethoxyphenol guaiacol, and syringaldazine | [204] | |
| Amylostereum areolatum | Homology model | Mono-, di-, tri-, and tetramer lignin models | [205,206] | |
| Trametes sp. C30 | Homology model | Four aflatoxins | [207] | |
| Trametes versicolor | 1KYA | Dodecane | [208] | |
| Antrodiella faginea | 5EHF | Lignosulfonic acid | [209] | |
| High, medium, and low redox potential laccase | 2HZH (805 mV) 1KYA (780 mV) 1GYC (780 mV) 3FU7 (460 mV) | Lignin tetramer | [190] | |
| Five bacterial laccases | Homology model | Guaiacol, ABTS, and DMP | MD | [189,210] |
| Cryptococcus neoformans | Homology model | Ellagic acid | MD | [211] |
| Comamonas testosteroni | Homology model | Mono-, di-, tri-, and tetramer lignin models | [212] | |
| Trametes versicolor | 1KYA | Glyphosate, isoproturn, dilignol model, and parathion | MD | [213] |
| Trametes versicolor | 1KYA | Two aflatoxins | MD | [214] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovaletti, A.; De Gioia, L.; Fantucci, P.; Greco, C.; Vertemara, J.; Zampella, G.; Arrigoni, F.; Bertini, L. Recent Theoretical Insights into the Oxidative Degradation of Biopolymers and Plastics by Metalloenzymes. Int. J. Mol. Sci. 2023, 24, 6368. https://doi.org/10.3390/ijms24076368
Rovaletti A, De Gioia L, Fantucci P, Greco C, Vertemara J, Zampella G, Arrigoni F, Bertini L. Recent Theoretical Insights into the Oxidative Degradation of Biopolymers and Plastics by Metalloenzymes. International Journal of Molecular Sciences. 2023; 24(7):6368. https://doi.org/10.3390/ijms24076368
Chicago/Turabian StyleRovaletti, Anna, Luca De Gioia, Piercarlo Fantucci, Claudio Greco, Jacopo Vertemara, Giuseppe Zampella, Federica Arrigoni, and Luca Bertini. 2023. "Recent Theoretical Insights into the Oxidative Degradation of Biopolymers and Plastics by Metalloenzymes" International Journal of Molecular Sciences 24, no. 7: 6368. https://doi.org/10.3390/ijms24076368
APA StyleRovaletti, A., De Gioia, L., Fantucci, P., Greco, C., Vertemara, J., Zampella, G., Arrigoni, F., & Bertini, L. (2023). Recent Theoretical Insights into the Oxidative Degradation of Biopolymers and Plastics by Metalloenzymes. International Journal of Molecular Sciences, 24(7), 6368. https://doi.org/10.3390/ijms24076368