
E-Cigarette Aerosol Condensate Leads to Impaired Coronary Endothelial Cell Health and Restricted Angiogenesis

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
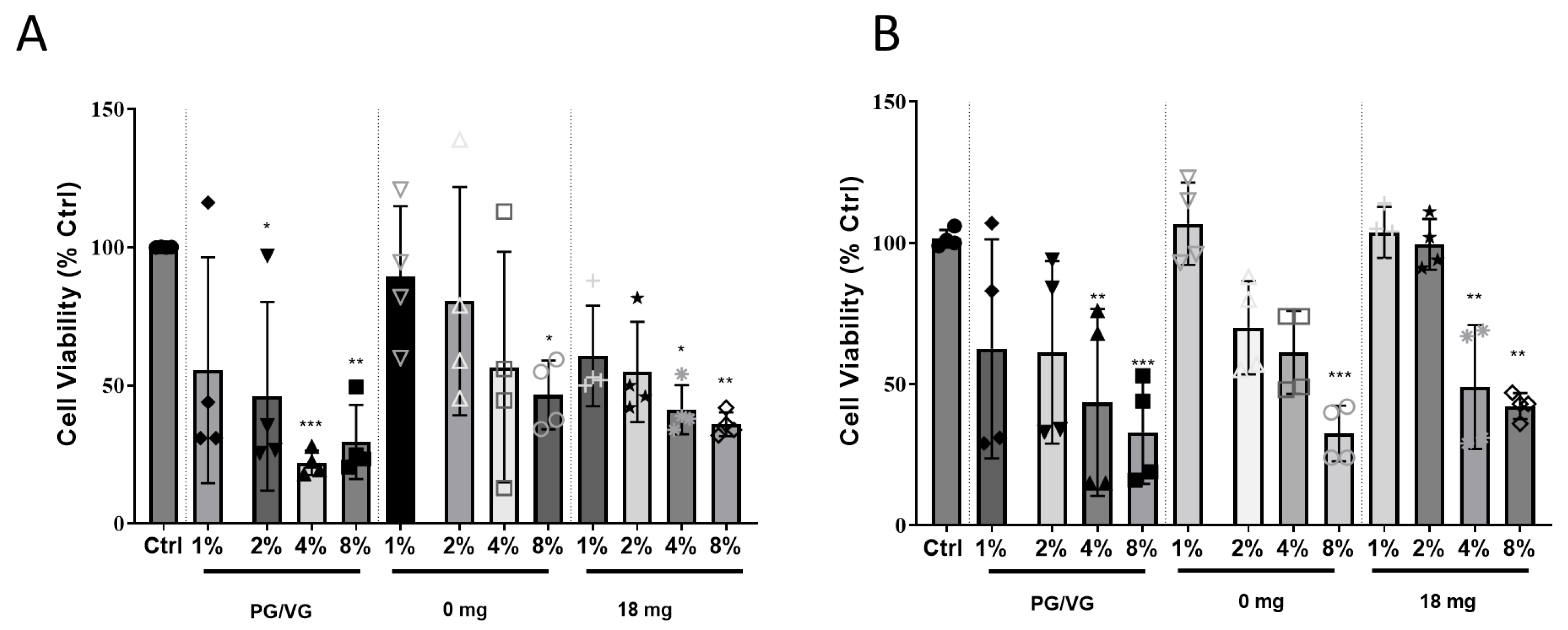
2.1. Exposure of HCAEC to EAC-Treated Lung Cell Conditioned Media Results in Cytotoxicity
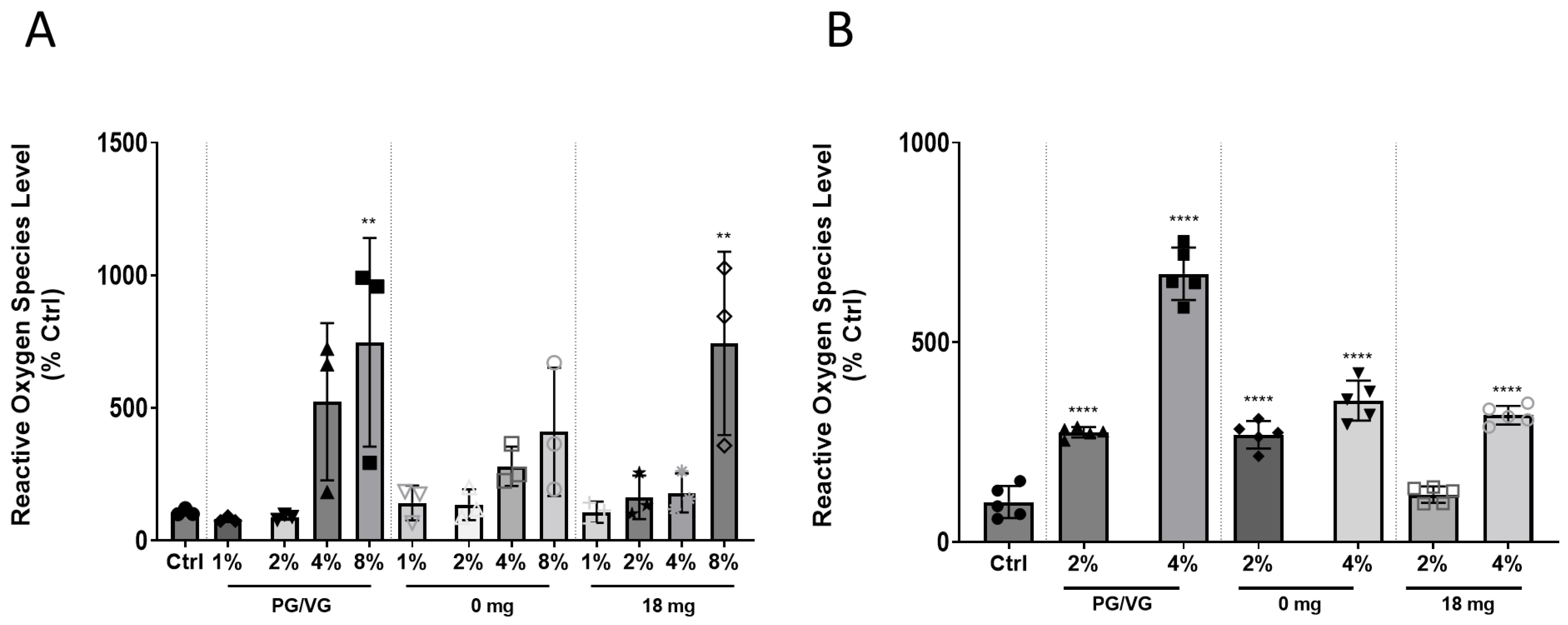
2.2. Direct Exposure to EAC or Indirectly to EAC-Lung Cell Conditioned Media Induces ROS Levels
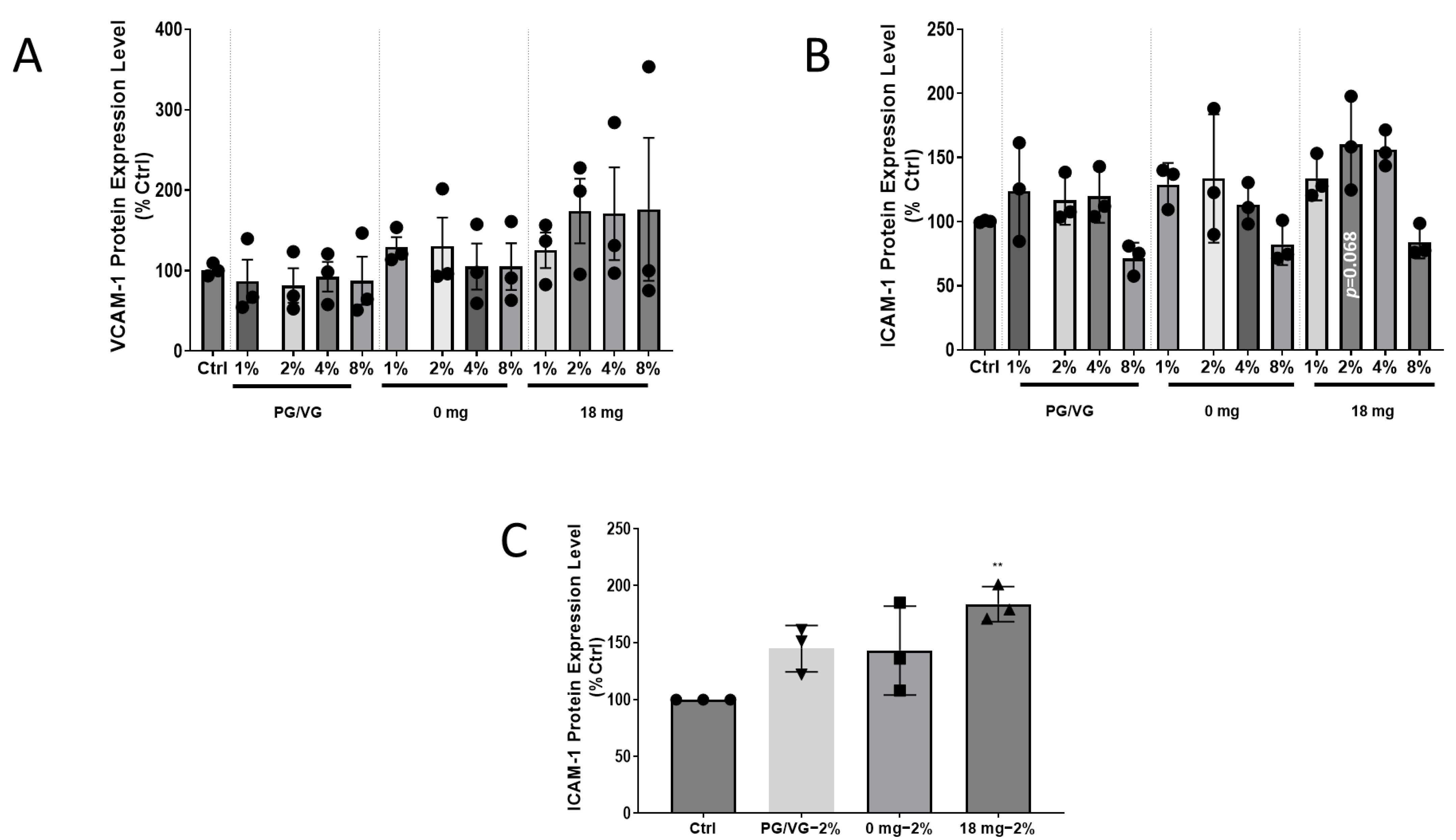
2.3. Adhesion Molecule Expression Increases in HCAEC after EAC Exposure for ICAM-1, but Not VCAM-1
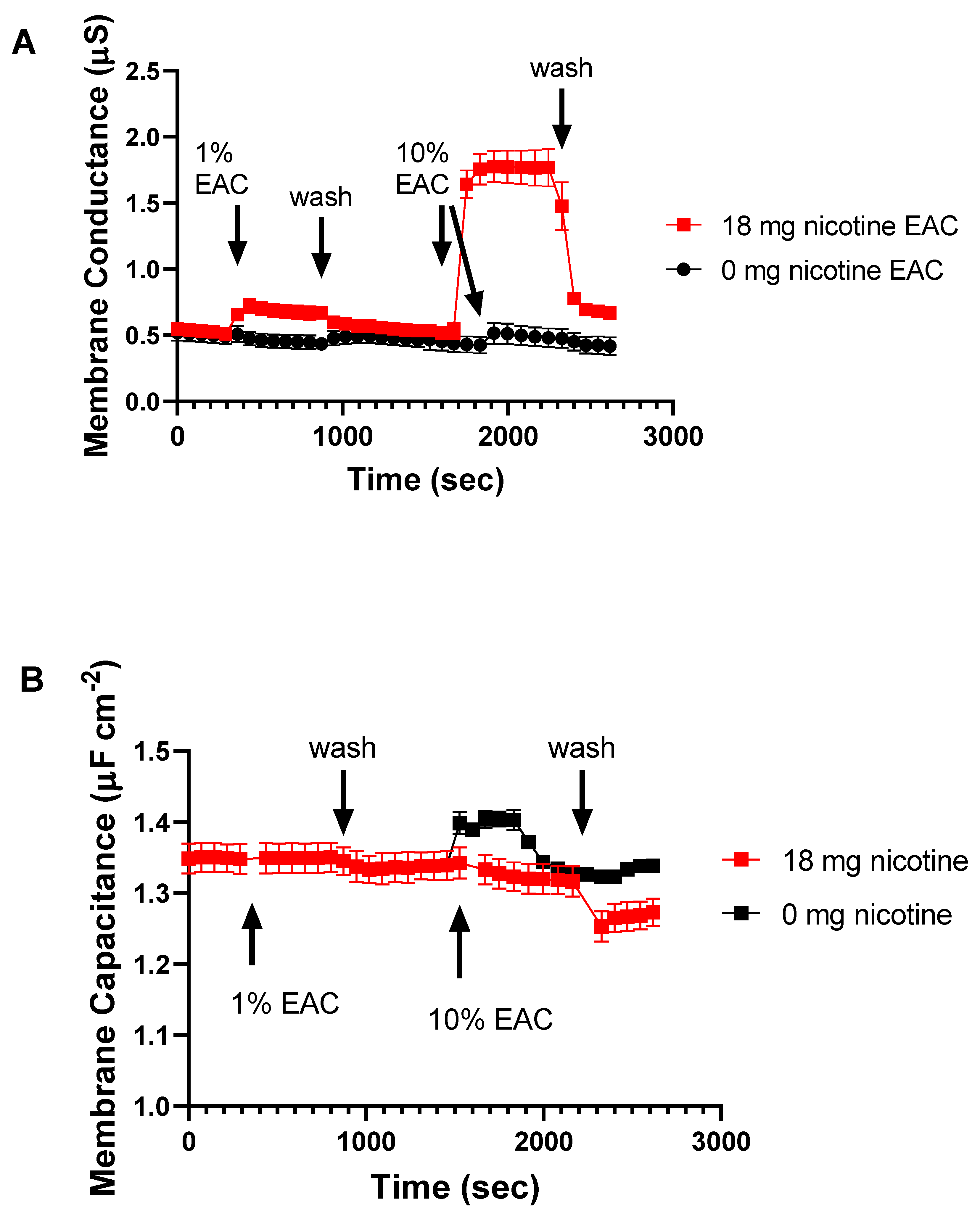
2.4. EAC from Nicotine Containing e-Liquid Alters Membrane Permeability
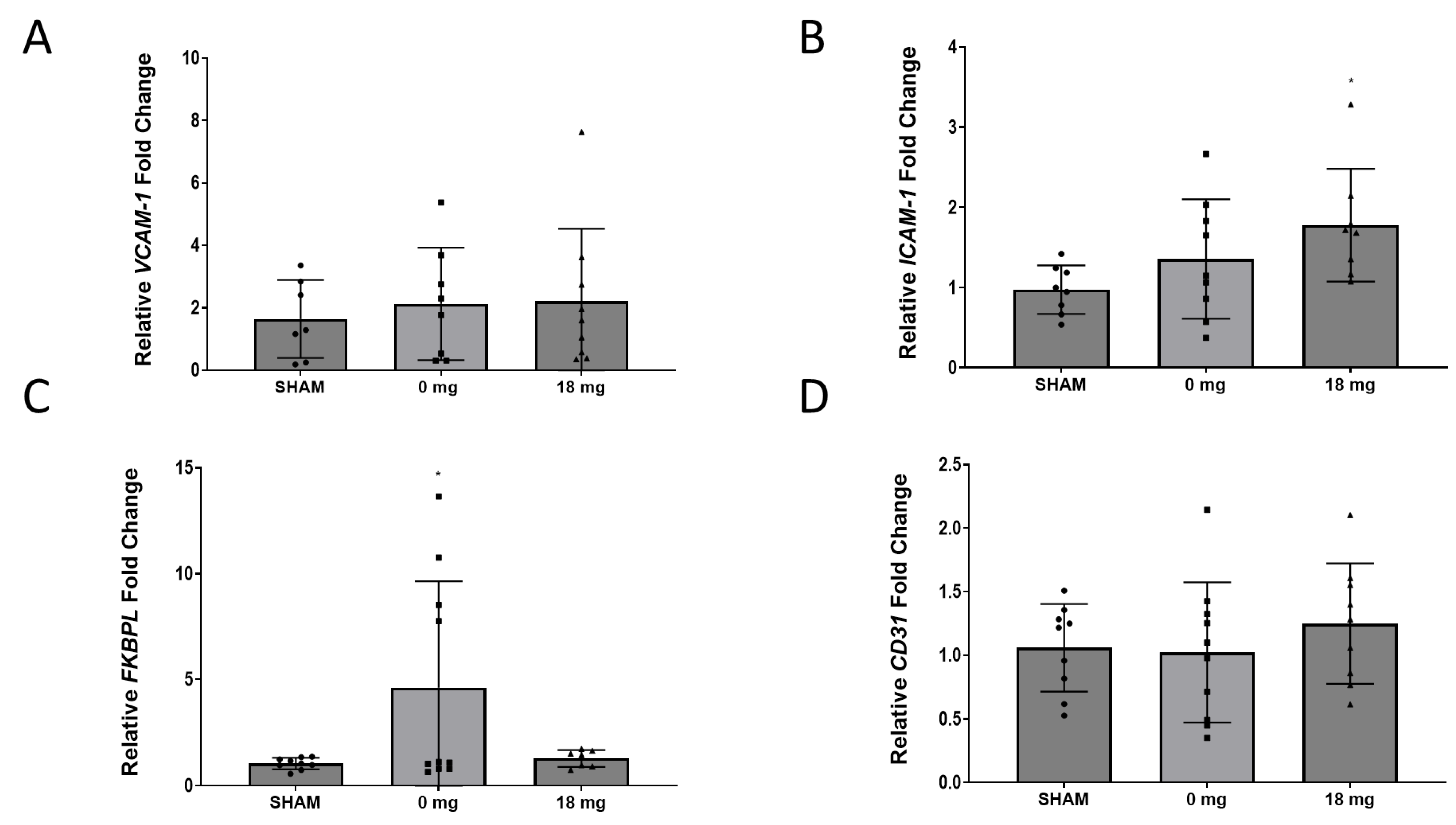
2.5. E-Cigarette Aerosol Increases ICAM-1 mRNA Expression in Murine Hearts
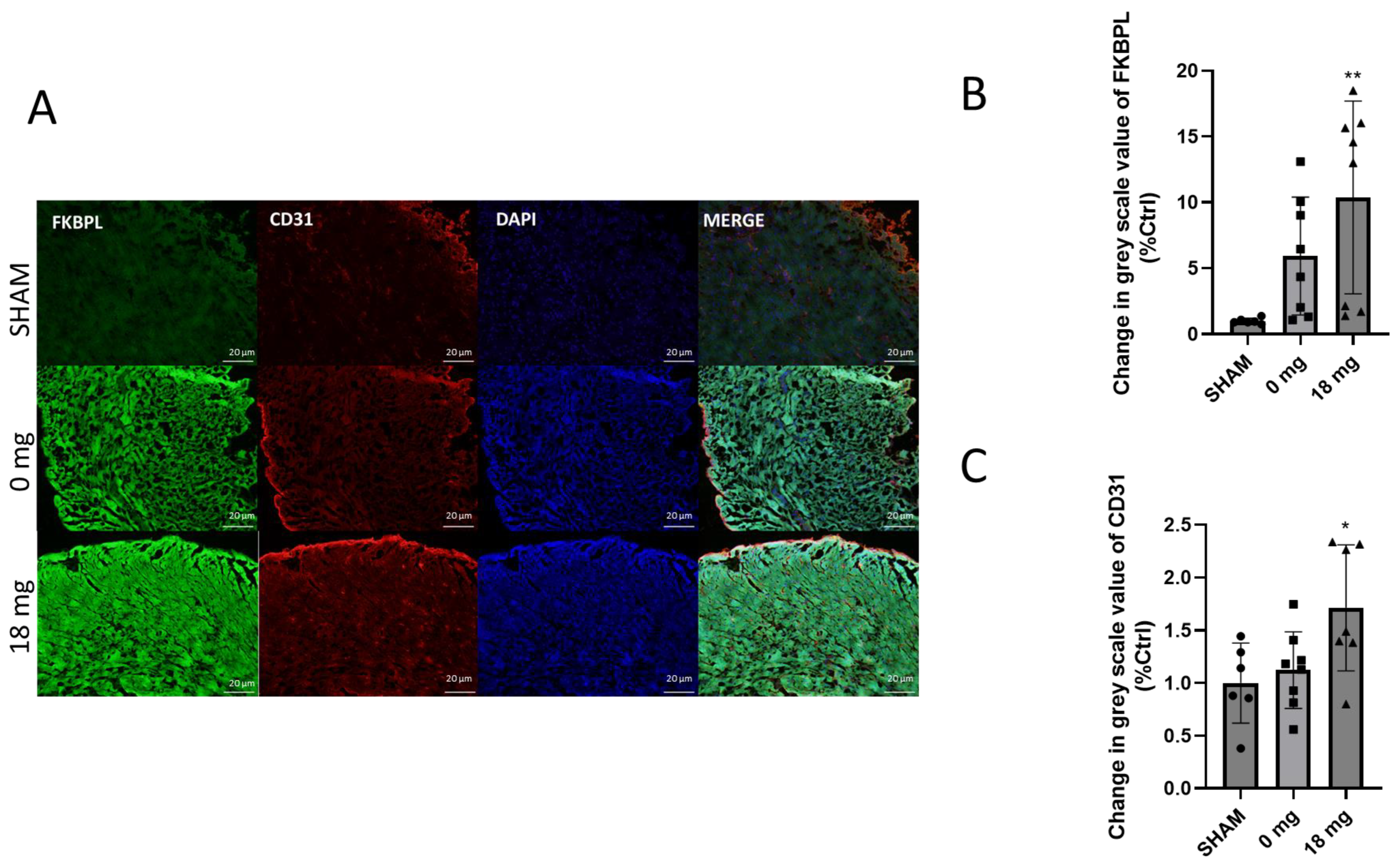
2.6. Cardiac Angiogenesis Markers Are Dysregulated by E-Cig Aerosol Exposure
3. Discussion
4. Materials and Methods
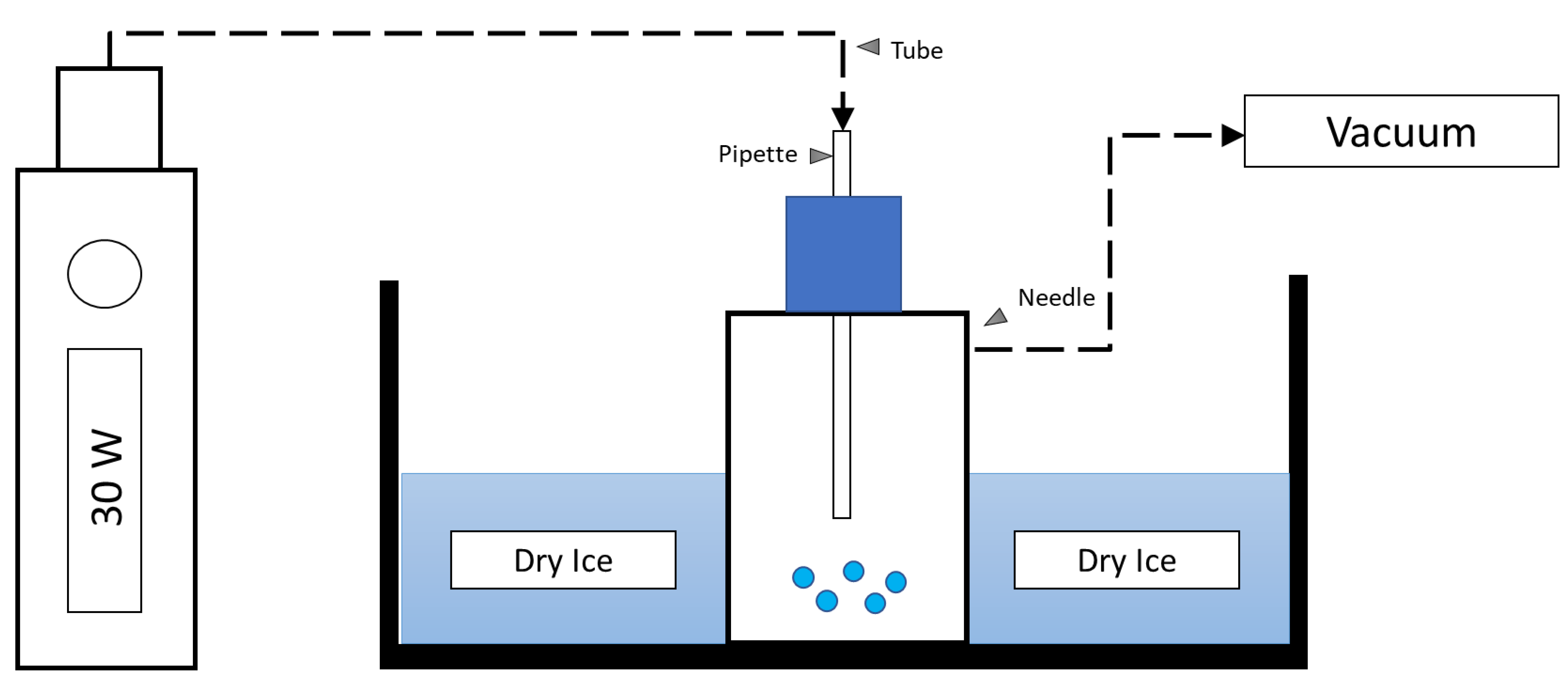
4.1. Generation of EAC
4.2. Cell Culture and Treatment Models
4.3. Cytotoxicity Assay
4.4. Intracellular Reactive Oxygen Species (ROS) Assay
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Animal Exposure
4.7. Immunohistochemistry of the Heart Tissue
4.8. Reverse Transcription-Polymerase Chain Reaction (RT-qPCR)
4.9. Tethered Bilayer Lipid Membrane (tBLMs) Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stewart, J.; Manmathan, G.; Wilkinson, P. Primary prevention of cardiovascular disease: A review of contemporary guidance and literature. JRSM Cardiovasc. Dis. 2017, 6, 204800401668721. [Google Scholar] [CrossRef] [Green Version]
- Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases/#tab=tab_1 (accessed on 22 April 2020).
- ABS. Heart, Stroke and Vascular Disease. Available online: https://www.abs.gov.au/statistics/health/health-conditions-and-risks/heart-stroke-and-vascular-disease/latest-release. (accessed on 22 April 2020).
- WHO. WHO Global Report: Mortality Attributable to Tobacco; WHO: Geneva, Switzerland, 2014.
- Banks, E.; Joshy, G.; Korda, R.; Stavreski, B.; Soga, K.; Egger, S.; Day, C.; Clarke, N.; Lewington, S.; Lopez, A. Tobacco smoking and risk of 36 cardiovascular disease subtypes: Fatal and non-fatal outcomes in a large prospective Australian study. BMC Med. 2019, 17, 128. [Google Scholar] [CrossRef] [Green Version]
- Abbot, N.C.; Stead, L.F.; White, A.R.; Barnes, J. Hypnotherapy for smoking cessation. Cochrane Database Syst. Rev. 1998, CD001008. [Google Scholar] [CrossRef]
- Anderson, C.; Majeste, A.; Hanus, J.; Wang, S. E-Cigarette Aerosol Exposure Induces Reactive Oxygen Species, DNA Damage, and Cell Death in Vascular Endothelial Cells. Toxicol. Sci. 2016, 154, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Tayyarah, R.; Long, G.A. Comparison of select analytes in aerosol from e-cigarettes with smoke from conventional cigarettes and with ambient air. Regul. Toxicol. Pharmacol. 2014, 70, 704–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, N.K.; Byron, M.J.; Abrams, D.B.; Shields, P.G. Novel nicotine delivery systems and public health: The rise of the ‘E-Cigarette’. Am. J. Public Health 2010, 100, 2340–2342. [Google Scholar] [CrossRef] [PubMed]
- Skotsimara, G.; Antonopoulos, A.; Oikonomou, E.; Siasos, G.; Ioakeimidis, N.; Tsalamandris, S.; Charalambous, G.; Galiatsatos, N.; Vlachopoulos, C.; Tousoulis, D. Cardiovascular effects of electronic cigarettes: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2019, 26, 1219–1228. [Google Scholar] [CrossRef]
- Tan, A.S.L.; Bigman, C.A. E-Cigarette Awareness and Perceived Harmfulness Prevalence and Associations with Smoking-Cessation Outcomes. Am. J. Prev. Med. 2014, 47, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.; Taudte, R.; Nizio, K.; Herok, G.; Cranfield, C.; Shimmon, R. Headspace analysis of E-cigarette fluids using comprehensive two dimensional GC×GC-TOF-MS reveals the presence of volatile and toxic compounds. J. Pharm. Biomed. Anal. 2021, 196, 113930. [Google Scholar] [CrossRef]
- Whitehead, A.K.; Erwin, A.P.; Yue, X. Nicotine and vascular dysfunction. Acta Physiol. 2021, 231, e13631. [Google Scholar] [CrossRef]
- Cheng, T. Chemical evaluation of electronic cigarettes. Tob. Control 2014, 23, ii11. [Google Scholar] [CrossRef]
- George, J.; Hussain, M.; Vadiveloo, T.; Ireland, S.; Hopkinson, P.; Struthers, A.; Donnan, P.; Khan, F.; Lang, C. Cardiovascular Effects of Switching from Tobacco Cigarettes to Electronic Cigarettes. J. Am. Coll. Cardiol. 2019, 74, 3112. [Google Scholar] [CrossRef]
- Barber, K.E.; Ghebrehiwet, B.; Yin, W.; Rubenstein, D.A. Endothelial Cell Inflammatory Reactions Are Altered in the Presence of E-Cigarette Extracts of Variable Nicotine. Cell. Mol. Bioeng. 2017, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Sorensen, K.; Gooch, V.; Miller, O.; Sullivan, I.; Lloyd, J.; Deanfield, J.; Spiehelhalter, D. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Bernhard, D.; Csordas, A.; Henderson, B.; Rossmann, A.; Kind, M.; Wick, G. Cigarette smoke metal-catalyzed protein oxidation leads to vascular endothelial cell contraction by depolymerization of microtubules. FASEB J. 2005, 19, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Januszewski, A.S.; Watson, C.; O’Niell, V.; McDonald, L.; Ledwidge, M.; Robson, T.; Jenkins, A.; Keech, A.; McClements, L. FKBPL is associated with metabolic parameters and is a novel determinant of cardiovascular disease. Sci. Rep. 2020, 10, 21655. [Google Scholar] [CrossRef] [PubMed]
- Yakkundi, A.; Bennett, R.; Herenandez-Negrete, I.; Delalande, J.; Hanna, M.; Lyubomska, O.; Arthur, K.; Short, A.; McKeen, H.; Nelson, L.; et al. FKBPL is a critical antiangiogenic regulator of developmental and pathological angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 845–854. [Google Scholar] [CrossRef] [Green Version]
- DeLisser, H.M.; Christofidou-Solomidou, M.; Strieter, R.; Burdick, M.; Robinson, C.; Wexler, R.; Kerr, J.; Garlanda, C.; Merwin, J.; Madri, J.; et al. Involvement of endothelial PECAM-1/CD31 in angiogenesis. Am. J. Pathol. 1997, 151, 671. [Google Scholar]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of PECAM-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.; Gudi, K.; Krause, A.; Sackrowitz, R.; Harvey, B.; Strulovici-Barel, Y.; Mezey, J.; Crystal, R. Circulating Endothelial Microparticles as a Measure of Early Lung Destruction in Cigarette Smokers. Am. J. Respir. Crit. Care Med. 2012, 184, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Kato, R.; Mizuno, S.; Kadowaki, M.; Shiozaki, K.; Akai, M.; Nakagawa, K.; Oikawa, T.; Iguchi, M.; Osanai, K.; Ishizaki, T.; et al. Sirt1 expression is associated with CD31 expression in blood cells from patients with chronic obstructive pulmonary disease. Respir. Res. 2016, 17, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, M.; Turnbull, T.; Sebastian, S.; Kempson, I. The mtt assay: Utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis. Int. J. Mol. Sci. 2021, 22, 12827. [Google Scholar] [CrossRef]
- Van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Squier, C.A. Penetration of nicotine and nitrosonornicotine across porcine oral mucosa. J. Appl. Toxicol. 1986, 6, 123–128. [Google Scholar] [CrossRef]
- Alghalayini, A.; Garcia, A.; Berry, T.; Cranfield, C.G. The Use of Tethered Bilayer Lipid Membranes to Identify the Mechanisms of Antimicrobial Peptide Interactions with Lipid Bilayers. Antibiotics 2019, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Galkina, E.; Ley, K. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef]
- Patel, R.B.; Colangelo, L.; Bielinski, S.; Larson, N.; Ding, J.; Allen, N.; Michos, E.; Shah, S.; Lloyd-Jones, D. Circulating vascular cell adhesion molecule-1 and incident heart failure: The multi-ethnic study of atherosclerosis (MESA). J. Am. Heart Assoc. 2020, 9, e019390. [Google Scholar] [CrossRef]
- Qasim, H.; Karim, Z.A.; Rivera, J.O.; Khasawneh, F.T.; Alshbool, F.Z. Impact of Electronic Cigarettes on the Cardiovascular System. J. Am. Heart Assoc. 2017, 6, e006353. [Google Scholar] [CrossRef]
- Romijnders, K.A.G.J.; Krusemann, E.; Boesveldt, S.; de Graaf, K.; de Vries, H.; Talhout, R. E-Liquid Flavor Preferences and Individual Factors Related to Vaping: A Survey among Dutch Never-Users, Smokers, Dual Users, and Exclusive Vapers. Int. J. Environ. Res. Public Health 2019, 16, 4661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Wu, D.; Ma, Y.; Ma, X.; Wang, S.; Li, F.; Li, M.; Zhang, T. Toxicity of electronic cigarettes: A general review of the origins, health hazards, and toxicity mechanisms. Sci. Total Environ. 2021, 772, 145475. [Google Scholar] [CrossRef] [PubMed]
- FDA. Premarket Tobacco Product Marketing Granted Orders. Available online: https://www.fda.gov/tobacco-products/premarket-tobacco-product-applications/premarket-tobacco-product-marketing-granted-orders (accessed on 22 April 2020).
- Fischman, J.S.; Sista, S.; Lee, D.K.; Cuadra, G.A.; Palazzolo, D.L. Flavorless vs. Flavored Electronic Cigarette-Generated Aerosol and E-Liquid on the Growth of Common Oral Commensal Streptococci. Front. Physiol. 2020, 11, 1513. [Google Scholar] [CrossRef] [PubMed]
- Smets, J.; Baeyens, F.; Chaumont, M.; Adriaens, K.; van Gucht, D. When Less is More: Vaping Low-Nicotine vs. High-Nicotine E-Liquid is Compensated by Increased Wattage and Higher Liquid Consumption. Int. J. Environ. Res. Public Health 2019, 16, 723. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, G.; Chan, Y.; Chapman, D.; Sukjamnong, S.; Nguyen, T.; Annissa, T.; McGrath, K.; Sharma, P.; Oliver, B. Maternal E-cigarette exposure in mice alters DNA methylation and lung cytokine expression in offspring. Am. J. Respir. Cell Mol. Biol. 2018, 58, 366–377. [Google Scholar] [CrossRef]
- Li, G.; Chan, Y.; Wang, B.; Saad, S.; George, J.; Oliver, B.; Chen, H. E-cigarettes damage the liver and alter nutrient metabolism in pregnant mice and their offspring. Ann. N. Y. Acad. Sci. 2020, 1475, 64–77. [Google Scholar] [CrossRef]
- Li, G.; Chan, Y.; Nguyen, L.; Mak, C.; Zaky, A.; Answer, A.; Shi, Y.; Nguyen, T.; Pollock, C.; Oliver, B.; et al. Impact of maternal e-cigarette vapor exposure on renal health in the offspring. Ann. N. Y. Acad. Sci. 2019, 1452, 65–77. [Google Scholar] [CrossRef]
- Behar, R.Z.; Wang, Y.; Talbot, P. Comparing the cytotoxicity of electronic cigarette fluids, aerosols and solvents. Tob. Control 2018, 27, 325. [Google Scholar] [CrossRef]
- Kosmider, L.; Sobczak, A.; Fik, M.; Knysak, J.; Zaciera, M.; Kurek, J.; Goniewicz, M. Carbonyl Compounds in Electronic Cigarette Vapors: Effects of Nicotine Solvent and Battery Output Voltage. Nicotine Tob. Res. 2014, 16, 1319. [Google Scholar] [CrossRef]
- Farsalinos, K.E.; Voudris, V.; Poulas, K. E-cigarettes generate high levels of aldehydes only in ‘dry puff’ conditions. Addiction 2015, 110, 1352–1356. [Google Scholar] [CrossRef] [PubMed]
- Putzhammer, R.; Doppler, C.; Jakschitz, T.; Heinz, K.; Förste, J.; Danzl, K.; Messner, B.; Bernhard, D. Vapours of US and EU Market Leader Electronic Cigarette Brands and Liquids Are Cytotoxic for Human Vascular Endothelial Cells. PLoS ONE 2016, 11, e0157337. [Google Scholar]
- Carnevale, R.; Sciarretta, S.; Violi, F.; Nocella, C.; Loffredo, L.; Perri, L.; Peruzzi, M.; Marullo, A.; De Falco, E.; Chimenti, I.; et al. Acute Impact of Tobacco vs. Electronic Cigarette Smoking on Oxidative Stress and Vascular Function. Chest 2016, 150, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R.; Keaney, J.F. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Rao, P.S.S.; Ande, A.; Sinha, N.; Kumar, A.; Kumar, S. Effects of Cigarette Smoke Condensate on Oxidative Stress, Apoptotic Cell Death, and HIV Replication in Human Monocytic Cells. PLoS ONE 2016, 11, e0155791. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, Y.; Sisler, J.D.; Shaffer, J.; Leonard, S.S.; Morris, A.M.; Qian, Y.; Bello, D.; Demokritou, P. Assessment of reactive oxygen species generated by electronic cigarettes using acellular and cellular approaches. J. Hazard. Mater. 2018, 344, 549–557. [Google Scholar] [CrossRef]
- Münzel, T.; Hahad, O.; Kuntic, M.; Keaney, J.; Deanfield, J.; Daiber, A. Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes. Eur. Heart J. 2020, 41, 4057–4070. [Google Scholar] [CrossRef]
- El-Mahdy, M.A.; Ewees, M.; Eid, M.; Mahgoup, E.; Khaleel, S.; Zweier, J. Electronic cigarette exposure causes vascular endothelial dysfunction due to NADPH oxidase activation and eNOS uncoupling. Am. J. Physiol.-Heart Circ. Physiol. 2022, 322, H549–H567. [Google Scholar] [CrossRef]
- Steyers, C.M.; Miller, F.J. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [Green Version]
- Makwana, O.; Smith, G.; Flockton, H.; Watters, G.; Lowe, F.; Breheny, D. Impact of cigarette versus electronic cigarette aerosol conditioned media on aortic endothelial cells in a microfluidic cardiovascular model. Sci. Rep. 2021, 11, 4747. [Google Scholar] [CrossRef]
- Muthumalage, T.; Prinz, M.; Ansah, K.; Gerloff, J.; Sundar, I.; Rahman, I. Inflammatory and Oxidative Responses Induced by Exposure to Commonly Used e-Cigarette Flavoring Chemicals and Flavored e-Liquids without Nicotine. Front. Physiol. 2018, 8, 1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: Differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review). Int. J. Mol. Med. 1999, 4, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Gerloff, J.; Sundar, I.; Freter, R.; Sekera, E.; Friedman, A.; Robinson, R.; Pagano, T.; Rahman, I. Inflammatory Response and Barrier Dysfunction by Different e-Cigarette Flavoring Chemicals Identified by Gas Chromatography–Mass Spectrometry in e-Liquids and e-Vapors on Human Lung Epithelial Cells and Fibroblasts. Appl. Vitr. Toxicol. 2017, 3, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.; Sesperez, K.; Chhor, M.; Ghorpandour, S.; Rennie, C.; Chung Ming, C.; Evenhuis, C.; Nikolic, V.; Orlic, N.K.; Mikovic, Z.; et al. Characterisation of Cardiac Health in the Reduced Uterine Perfusion Pressure Model and a 3D Cardiac Spheroid Model, of Preeclampsia. Biol. Sex Differ. 2021, 12, 31. [Google Scholar] [CrossRef]
- Cooke, J.P. Angiogenesis and the role of the endothelial nicotinic acetylcholine receptor. Life Sci. 2007, 80, 2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Cooke, J.P. Nicotine and Pathological Angiogenesis. Life Sci. 2012, 91, 1058. [Google Scholar] [CrossRef] [Green Version]
- Alqudah, A.; Eastwood, K.; Jerotic, D.; Todd, N.; Hoch, D.; McNally, R.; Obradovic, D.; Dugalic, S.; Hunter, A.; Holmes, V.; et al. FKBPL and SIRT-1 Are Downregulated by Diabetes in Pregnancy Impacting on Angiogenesis and Endothelial Function. Front. Endocrinol. 2021, 12, 459. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.; McNally, R.; Alqudah, A.; Jerotic, D.; Suvakov, S.; Obradovic, D.; Hoch, D.; Hombrebueno, J.; Campos, G.; Watson, C.; et al. Role of A Novel Angiogenesis FKBPL-CD44 Pathway in Preeclampsia Risk Stratification and Mesenchymal Stem Cell Treatment. J. Clin Endocrinol. Metab. 2021, 106, 26. [Google Scholar] [CrossRef]
- Santi, P. Partition and transport of verapamil and nicotine through artificial membranes. Int. J. Pharm. 1991, 68, 43–49. [Google Scholar] [CrossRef]
- Vis, M.A.M.; Ito, K.; Hofmann, S. Impact of Culture Medium on Cellular Interactions in in vitro Co-culture Systems. Front. Bioeng. Biotechnol. 2020, 8, 911. [Google Scholar] [CrossRef]
- McGrath, K.C.Y.; Li, X.H.; McRobb, L.S.; Heather, A.K. Inhibitory Effect of a French Maritime Pine Bark Extract-Based Nutritional Supplement on TNF-α-Induced Inflammation and Oxidative Stress in Human Coronary Artery Endothelial Cells. Evid.-Based Complement. Altern. Med. 2015, 2015, 260530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Li, G.E.; Chen, E.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Maternal E-Cigarette Exposure Results in Cognitive and Epigenetic Alterations in Offspring in a Mouse Model. Chem. Res. Toxicol. 2018, 31, 601–611. [Google Scholar] [CrossRef]
- StHelen, G.; Havel, C.; Dempsey, D.A.; Jacob, P.; Benowitz, N.L. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction 2016, 111, 535–544. [Google Scholar] [CrossRef]
- Bustin, S.A. Absolute quantification of mrna using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cranfield, C.; Carne, S.; Martinac, B.; Cornell, B. The assembly and use of tethered bilayer lipid membranes (tBLMs). Methods Mol. Biol. 2015, 1232, 45–53. [Google Scholar]
- Berry, T.; Dutta, D.; Chen, R.; Leong, A.; Wang, H.; Donald, W.; Parviz, M.; Cornell, B.; Willcox, M.; Kumar, N.; et al. Lipid Membrane Interactions of the Cationic Antimicrobial Peptide Chimeras Melimine and Cys-Melimine. Langmuir 2018, 34, 11586–11592. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) |
|---|---|
| β-actin (sense) | GATGTATGAAGGCTTTGGTC |
| β-actin (anti-sense) | TGTGCACTTTTATTGGTCTC |
| ICAM-1 (sense) | CAGTCTACAACTTTTCAGCTC |
| ICAM-1 (anti-sense) | CACACTTCACAGTTACTTGG |
| VCAM-1 (sense) | ACTGATTATCCAAGTCTCTCC |
| VCAM-1 (anti-sense) | CCATCCACAGACTTTAATACC |
| CD31 (sense) | CATCGCCACCTTAATAGTTG |
| CD31 (anti-sense) | CCAGAAACATCATCATAACCG |
| FKBPL (sense) | TCTCTCAGGGATCAGGAG |
| FKBPL (anti-sense) | TATTTAAGATTTGCTGGGCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhor, M.; Tulpar, E.; Nguyen, T.; Cranfield, C.G.; Gorrie, C.A.; Chan, Y.L.; Chen, H.; Oliver, B.G.; McClements, L.; McGrath, K.C. E-Cigarette Aerosol Condensate Leads to Impaired Coronary Endothelial Cell Health and Restricted Angiogenesis. Int. J. Mol. Sci. 2023, 24, 6378. https://doi.org/10.3390/ijms24076378
Chhor M, Tulpar E, Nguyen T, Cranfield CG, Gorrie CA, Chan YL, Chen H, Oliver BG, McClements L, McGrath KC. E-Cigarette Aerosol Condensate Leads to Impaired Coronary Endothelial Cell Health and Restricted Angiogenesis. International Journal of Molecular Sciences. 2023; 24(7):6378. https://doi.org/10.3390/ijms24076378
Chicago/Turabian StyleChhor, Michael, Esra Tulpar, Tara Nguyen, Charles G. Cranfield, Catherine A. Gorrie, Yik Lung Chan, Hui Chen, Brian G. Oliver, Lana McClements, and Kristine C. McGrath. 2023. "E-Cigarette Aerosol Condensate Leads to Impaired Coronary Endothelial Cell Health and Restricted Angiogenesis" International Journal of Molecular Sciences 24, no. 7: 6378. https://doi.org/10.3390/ijms24076378
APA StyleChhor, M., Tulpar, E., Nguyen, T., Cranfield, C. G., Gorrie, C. A., Chan, Y. L., Chen, H., Oliver, B. G., McClements, L., & McGrath, K. C. (2023). E-Cigarette Aerosol Condensate Leads to Impaired Coronary Endothelial Cell Health and Restricted Angiogenesis. International Journal of Molecular Sciences, 24(7), 6378. https://doi.org/10.3390/ijms24076378