


Liposomes in Cancer Therapy: How Did We Start and Where Are We Now


Abstract
:1. Introduction: Cancer and Chemotherapy
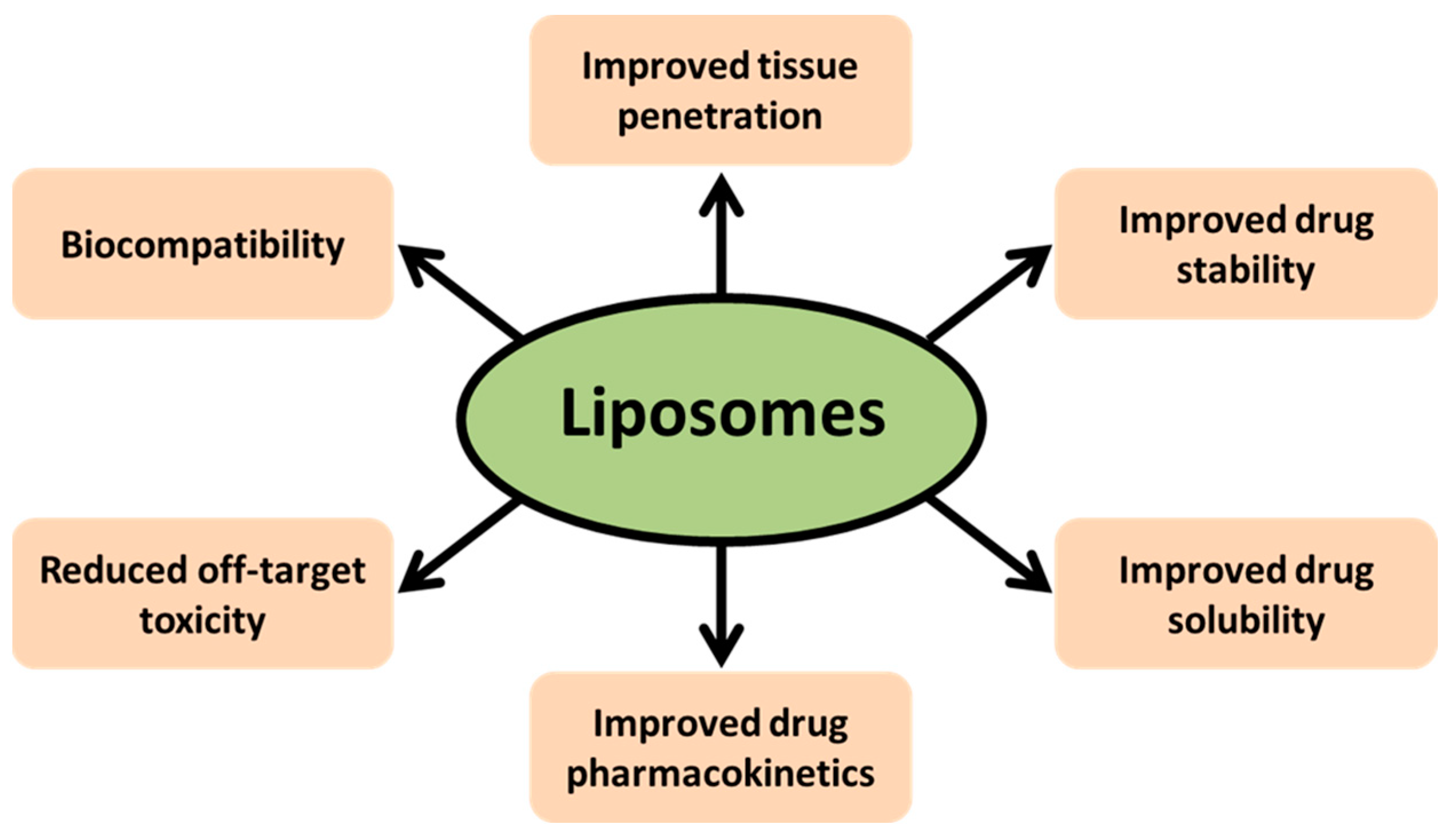
2. Nanoparticles and Liposome-Mediated Drug Delivery
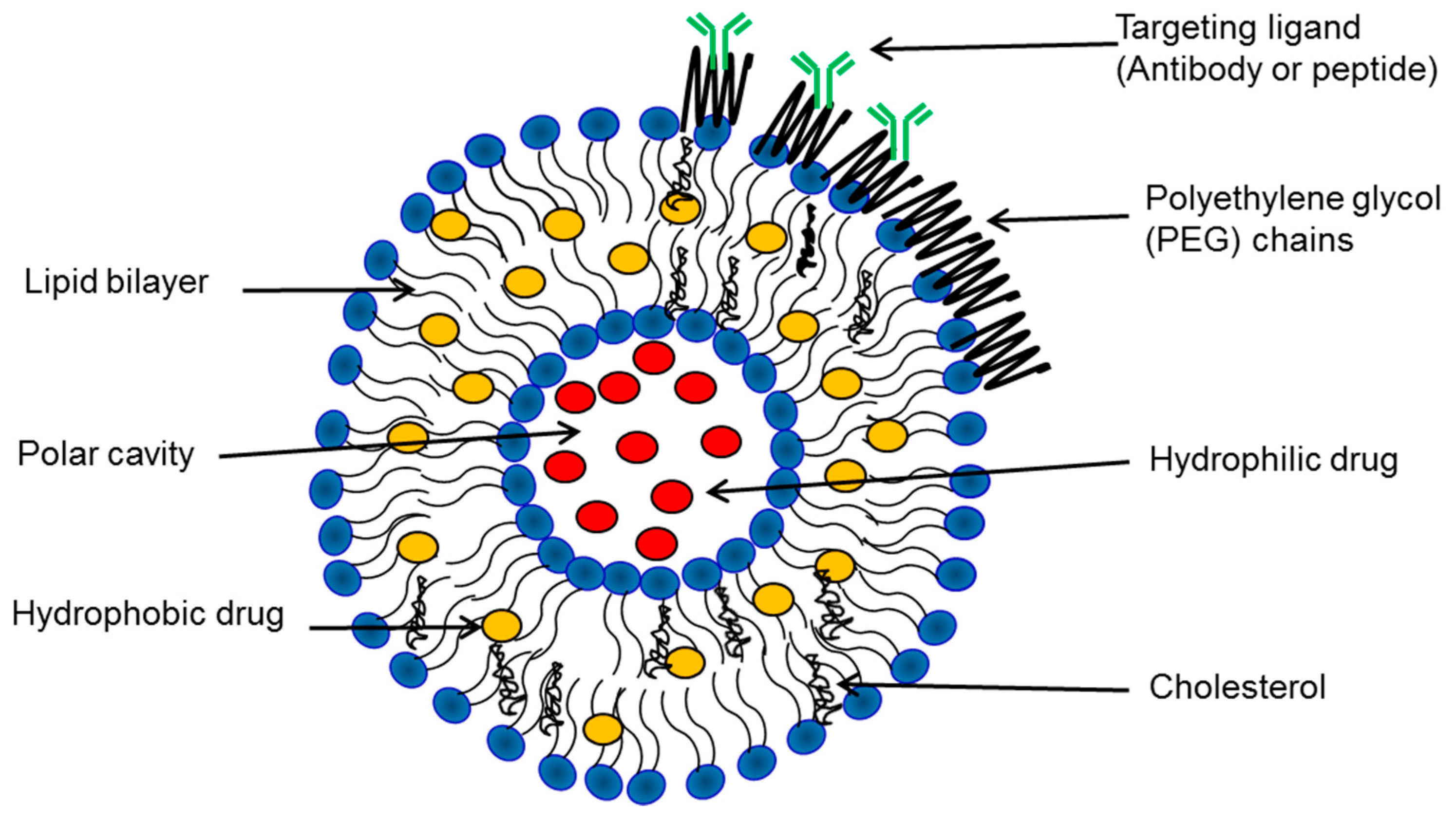
3. Biophysical Characterization of Liposomes
3.1. Transmission Electron Microscopy (TEM)
3.2. Dynamic Light Scattering (DLS)
3.3. Fluorescence Correlation Spectroscopy (FCS)
3.4. Encapsulation Efficiency (EE)
3.5. Phospholipid Content
4. Preparation and Properties of Liposomes
4.1. Thin Lipid Film Hydration
4.2. Reverse Phase Evaporation
4.3. Dehydration-Rehydration
4.4. Microfluidic Techniques
5. Active Loading vs. Passive Loading of Drugs in Liposomes
6. Strategies for Targeting Liposomes to Tumors
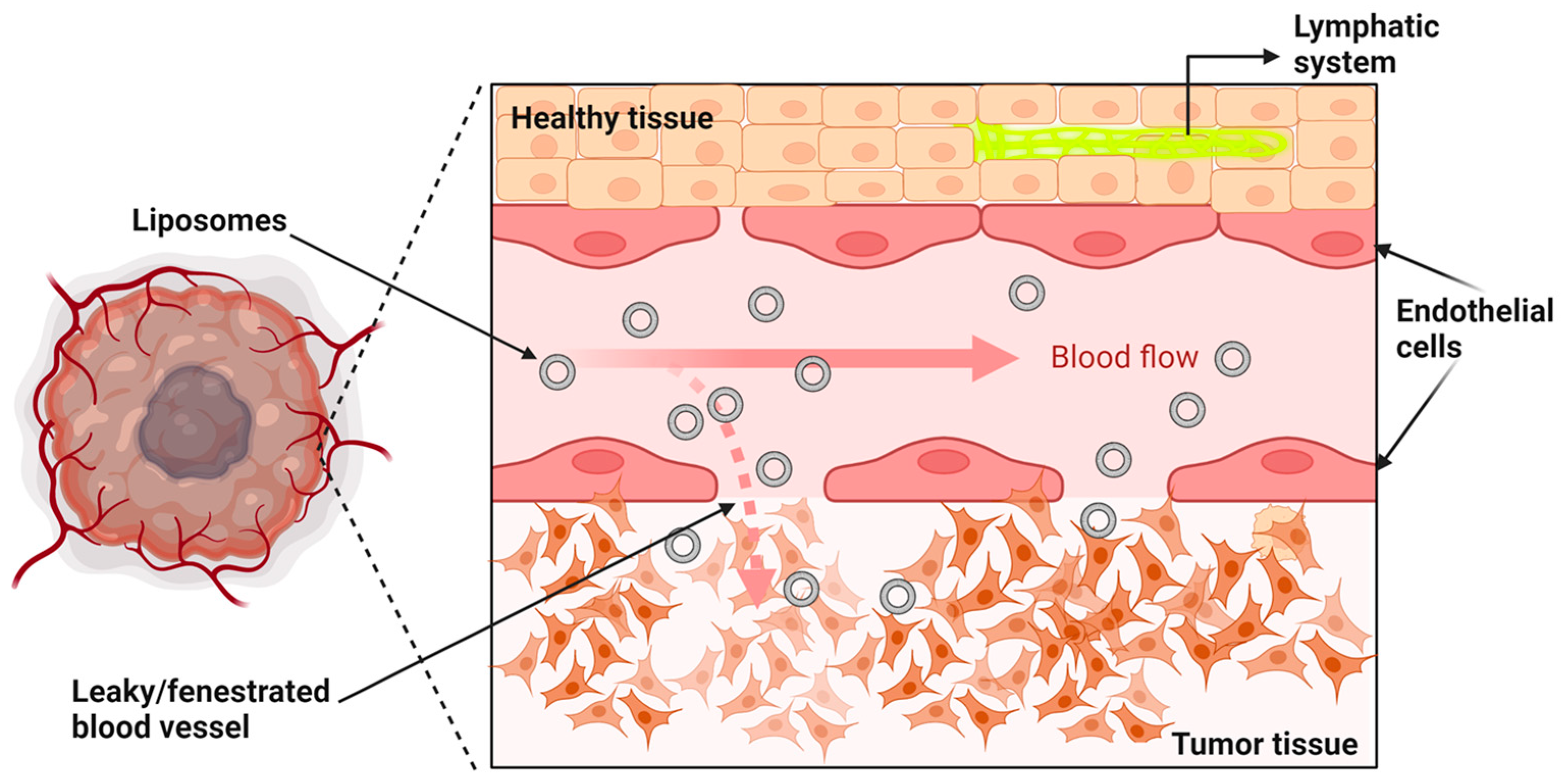
6.1. Liposomes and the EPR Effect (Passive Targeting)
6.2. Active Targeting of Liposomes
6.3. Local Stimuli to Trigger Drug Release from Liposomes
7. PEGylation of Liposomes
7.1. Accelerated Blood Clearance
7.2. Cell Uptake and Cargo Delivery of PEGylated Nanoparticles
7.3. Cleavable PEG Coatings
8. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.M.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 3 March 2023).
- Siegel, R.; DeSantis, C.; Virgo, K.; Stein, K.; Mariotto, A.; Smith, T.; Cooper, D.; Gansler, T.; Lerro, C.; Fedewa, S.; et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J. Clin. 2012, 62, 220–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowherd, S.M. Tumor staging and grading: A primer. Methods Mol. Biol. 2012, 823, 1–18. [Google Scholar] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Gamal, H.; Tawfik, W.; Fahmy, H.M.; El-Sayyad, H.H. Breakthroughs of using Photodynamic Therapy and Gold Nanoparticles in Cancer Treatment. In Proceedings of the IEEE International Conference on Nanoelectronics, Nanophotonics, Nanomaterials, Nanobioscience & Nanotechnology (5NANO), Kottayam, Kerala, India, 29–30 April 2021; pp. 1–4. [Google Scholar]
- Zhang, Q.; Li, L. Photodynamic combinational therapy in cancer treatment. Off. J. Balk. Union Oncol. 2018, 23, 561–567. [Google Scholar]
- Katz, A.; Ferrer, M.; Suarez, J.F. Comparison of quality of life after stereotactic body radiotherapy and surgery for early-stage prostate cancer. Radiat. Oncol. 2012, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- Kintzel, P.E.; Chase, S.L.; Schultz, L.M.; O’Rourke, T.J. Increased risk of metabolic syndrome, diabetes mellitus, and cardiovascular disease in men receiving androgen deprivation therapy for prostate cancer. Pharmacotherapy 2008, 28, 1511–1522. [Google Scholar] [CrossRef]
- Saraswathy, M.; Gong, S. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013, 31, 1397–1407. [Google Scholar] [CrossRef]
- Lackner, M.R.; Wilson, T.R.; Settleman, J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012, 8, 999–1014. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Kalscheuer, S.M.; Panyam, J. Exploiting nanotechnology to overcome tumor drug resistance: Challenges and opportunities. Adv. Drug Deliv. Rev. 2013, 65, 1731–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, J.J.; Sanchez de Medina, F.; Castano, B.; Bujanda, L.; Romero, M.R.; Martinez-Augustin, O.; Moral-Avila, R.D.; Briz, O. Chemoprevention, chemotherapy, and chemoresistance in colorectal cancer. Drug Metab. Rev. 2012, 44, 148–172. [Google Scholar] [CrossRef] [PubMed]
- England, C.G.; Ng, C.F.; van Berkel, V.; Frieboes, H.B. A Review of Pharmacological Treatment Options for Lung Cancer: Emphasis on Novel Nanotherapeutics and Associated Toxicity. Curr. Drug Targets 2015, 16, 1057–1087. [Google Scholar] [CrossRef]
- Soliman, G.M. Nanoparticles as safe and effective delivery systems of antifungal agents: Achievements and challenges. Int. J. Pharm. 2017, 523, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Straubinger, R.M.; Arnold, R.D.; Zhou, R.; Mazurchuk, R.; Slack, J.E. Antivascular and antitumor activities of liposome-associated drugs. Anticancer Res. 2004, 24, 397–404. [Google Scholar]
- Deamer, D.W. From “banghasomes” to liposomes: A memoir of Alec Bangham, 1921–2010. FASEB J. 2010, 24, 1308–1310. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Ryman, B.E. Fate of protein-containing liposomes injected into rats. An approach to the treatment of storage diseases. Eur. J. Biochem. 1972, 24, 485–491. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Buckland, R.A. Enzyme-containing liposomes alleviate a model for storage disease. Nature 1973, 244, 170–172. [Google Scholar] [CrossRef]
- Gregoriadis, G. The carrier potential of liposomes in biology and medicine (first of two parts). N. Engl. J. Med. 1976, 295, 704–710. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Neerunjun, E.D. Treatment of tumour bearing mice with liponsome-entrapped actinomycin D prolongs their survival. Res. Commun. Chem. Pathol. Pharmacol. 1975, 10, 351–362. [Google Scholar]
- Gregoriadis, G. Liposomes in Drug Delivery: How It All Happened. Pharmaceutics 2016, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forssen, E.A.; Tokes, Z.A. Use of anionic liposomes for the reduction of chronic doxorubicin-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 1981, 78, 1873–1877. [Google Scholar] [CrossRef] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetsugu, S.; Kurisu, S.; Takenawa, T. Dynamic shaping of cellular membranes by phospholipids and membrane-deforming proteins. Physiol. Rev. 2014, 94, 1219–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowhan, W. Understanding phospholipid function: Why are there so many lipids? J. Biol. Chem. 2017, 292, 10755–10766. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.H. Mixed-chain phospholipids: Structures and chain-melting behavior. Lipids 2001, 36, 1077–1097. [Google Scholar] [CrossRef]
- Feigenson, G.W. Phase behavior of lipid mixtures. Nat. Chem. Biol. 2006, 2, 560–563. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Nir, S.; Oki, S. Permeability properties of phospholipid membranes: Effect of cholesterol and temperature. Biochim. Biophys. Acta 1972, 266, 561–583. [Google Scholar] [CrossRef]
- Deniz, A.; Sade, A.; Severcan, F.; Keskin, D.; Tezcaner, A.; Banerjee, S. Celecoxib-loaded liposomes: Effect of cholesterol on encapsulation and in vitro release characteristics. Biosci. Rep. 2010, 30, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardania, H.; Tarvirdipour, S.; Dorkoosh, F. Liposome-targeted delivery for highly potent drugs. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1478–1489. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Morata, L.; Giannotti, M.I.; Sanz, F. Influence of cholesterol on the phase transition of lipid bilayers: A temperature-controlled force spectroscopy study. Langmuir 2012, 28, 12851–12860. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Henriksen-Lacey, M.; Wilkhu, J.; Devitt, A.; Christensen, D.; Perrie, Y. Effect of incorporating cholesterol into DDA:TDB liposomal adjuvants on bilayer properties, biodistribution, and immune responses. Mol. Pharm. 2014, 11, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Eloy, J.O.; Claro de Souza, M.; Petrilli, R.; Barcellos, J.P.; Lee, R.J.; Marchetti, J.M. Liposomes as carriers of hydrophilic small molecule drugs: Strategies to enhance encapsulation and delivery. Colloids Surf. B Biointerfaces 2014, 123, 345–363. [Google Scholar] [CrossRef]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharm. Rev. 1999, 51, 691–743. [Google Scholar] [PubMed]
- Langer, R. Drug delivery and targeting. Nature 1998, 392 (Suppl. 6679), 5–10. [Google Scholar] [PubMed]
- Missaoui, W.N.; Arnold, R.D.; Cummings, B.S. Toxicological status of nanoparticles: What we know and what we don’t know. Chem. Biol. Interact. 2018, 295, 1–12. [Google Scholar] [CrossRef]
- Kshirsagar, N.A.; Pandya, S.K.; Kirodian, G.B.; Sanath, S. Liposomal drug delivery system from laboratory to clinic. J. Postgrad. Med. 2005, 51 (Suppl. S1), S5–S15. [Google Scholar]
- Najahi-Missaoui, W.; Arnold, R.D.; Cummings, B.S. Safe Nanoparticles: Are We There Yet? Int. J. Mol. Sci. 2020, 22, 385. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, M.R. Liposomes: An overview of manufacturing techniques. Cell. Mol. Biol. Lett. 2005, 10, 711–719. [Google Scholar] [PubMed]
- Chen, C.; Han, D.; Cai, C.; Tang, X. An overview of liposome lyophilization and its future potential. J. Control. Release 2010, 142, 299–311. [Google Scholar] [CrossRef]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar]
- Shang, L.; Nienhaus, K.; Nienhaus, G.U. Engineered nanoparticles interacting with cells: Size matters. J. Nanobiotechnol. 2014, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Mahl, D.; Diendorf, J.; Meyer-Zaika, W.; Epple, M. Possibilities and limitations of different analytical methods for the size determination of a bimodal dispersion of metallic nanoparticles. Colloids Surfaces Physicochem. Eng. Asp. 2011, 377, 386–392. [Google Scholar] [CrossRef]
- Cho, E.J.; Holback, H.; Liu, K.C.; Abouelmagd, S.A.; Park, J.; Yeo, Y. Nanoparticle characterization: State of the art, challenges, and emerging technologies. Mol. Pharm. 2013, 10, 2093–2110. [Google Scholar] [CrossRef] [Green Version]
- Pietroiusti, A.; Magrini, A. Engineered nanoparticles at the workplace: Current knowledge about workers’ risk. Occup. Med. 2014, 64, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Rocker, C.; Potzl, M.; Zhang, F.; Parak, W.J.; Nienhaus, G.U. A quantitative fluorescence study of protein monolayer formation on colloidal nanoparticles. Nat. Nanotechnol. 2009, 4, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Zemanova, L.; Schenk, A.; Valler, M.J.; Nienhaus, G.U.; Heilker, R. Confocal optics microscopy for biochemical and cellular high-throughput screening. Drug Discov. Today 2003, 8, 1085–1093. [Google Scholar] [CrossRef]
- Arifin, D.R.; Palmer, A.F. Determination of size distribution and encapsulation efficiency of liposome-encapsulated hemoglobin blood substitutes using asymmetric flow field-flow fractionation coupled with multi-angle static light scattering. Biotechnol. Prog. 2003, 19, 1798–1811. [Google Scholar] [CrossRef] [PubMed]
- Grabielle-Madelmont, C.; Lesieur, S.; Ollivon, M. Characterization of loaded liposomes by size exclusion chromatography. J. Biochem. Biophys. Methods 2003, 56, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Desormeaux, A.; Bergeron, M.G. Lymphoid tissue targeting of anti-HIV drugs using liposomes. Methods Enzymol. 2005, 391, 330–351. [Google Scholar]
- Petkowicz, J.; Byra, A.; Szumilo, T. The hypoglycaemic response of diabetic rats to insulin-liposomes. Acta Physiol. Pol. 1990, 41, 97–103. [Google Scholar]
- Lundahl, P.; Yang, Q. Liposome chromatography: Liposomes immobilized in gel beads as a stationary phase for aqueous column chromatography. J. Chromatogr. 1991, 544, 283–304. [Google Scholar] [CrossRef]
- Bragagni, M.; Mennini, N.; Ghelardini, C.; Mura, P. Development and characterization of niosomal formulations of doxorubicin aimed at brain targeting. J. Pharm. Pharm. Sci. 2012, 15, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, C.; Fardella, G.; Chiappini, I.; Perioli, L.; Vescovi, C.; Ricci, M.; Giovagnoli, S.; Scuota, S. UV spectroscopy and reverse-phase HPLC as novel methods to determine Capreomycin of liposomal fomulations. J. Pharm. Biomed. Anal. 2004, 36, 249–255. [Google Scholar] [CrossRef]
- Bartlett, G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar] [CrossRef]
- Mayer, L.D.; Tai, L.C.; Ko, D.S.; Masin, D.; Ginsberg, R.S.; Cullis, P.R.; Bally, M.B. Influence of vesicle size, lipid composition, and drug-to-lipid ratio on the biological activity of liposomal doxorubicin in mice. Cancer Res. 1989, 49, 5922–5930. [Google Scholar]
- Kulkarni, S.B.; Betageri, G.V.; Singh, M. Factors affecting microencapsulation of drugs in liposomes. J. Microencapsul. 1995, 12, 229–246. [Google Scholar] [CrossRef]
- Tardi, P.G.; Boman, N.L.; Cullis, P.R. Liposomal doxorubicin. J. Drug Target. 1996, 4, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Fenske, D.B.; Cullis, P.R. Liposomal nanomedicines. Expert Opin. Drug Deliv. 2008, 5, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Demetzos, C. Differential Scanning Calorimetry (DSC): A tool to study the thermal behavior of lipid bilayers and liposomal stability. J. Liposome Res. 2008, 18, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Franzen, U.; Ostergaard, J. Physico-chemical characterization of liposomes and drug substance-liposome interactions in pharmaceutics using capillary electrophoresis and electrokinetic chromatography. J. Chromatogr. A 2012, 1267, 32–44. [Google Scholar] [CrossRef]
- Rohilla, S.; Dureja, H. Recent Patents, Formulation and Characterization of Nanoliposomes. Recent Pat. Drug Deliv. Formul. 2015, 9, 213–224. [Google Scholar] [CrossRef]
- Vemuri, S.; Rhodes, C.T. Preparation and characterization of liposomes as therapeutic delivery systems: A review. Pharm. Acta Helvetiae 1995, 70, 95–111. [Google Scholar] [CrossRef]
- Ahmed, K.S.; Hussein, S.A.; Ali, A.H.; Korma, S.A.; Qiu, L.; Chen, J. Liposome: Composition, characterisation, preparation, and recent innovation in clinical applications. J. Drug Target. 2019, 27, 742–761. [Google Scholar] [CrossRef]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and innovation in the manufacturing process. Adv. Drug Deliv. Rev. 2020, 154–155, 102–122. [Google Scholar] [CrossRef]
- Crosasso, P.; Ceruti, M.; Brusa, P.; Arpicco, S.; Dosio, F.; Cattel, L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J. Control. Release 2000, 63, 19–30. [Google Scholar] [CrossRef]
- Hua, H.; Zhang, N.; Liu, D.; Song, L.; Liu, T.; Li, S.; Zhao, Y. Multifunctional gold nanorods and docetaxel-encapsulated liposomes for combined thermo- and chemotherapy. Int. J. Nanomed. 2017, 12, 7869–7884. [Google Scholar] [CrossRef] [Green Version]
- Castile, J.D.; Taylor, K.M. Factors affecting the size distribution of liposomes produced by freeze-thaw extrusion. Int. J. Pharm. 1999, 188, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R.; Janoff, A.S. Solute distributions and trapping efficiencies observed in freeze-thawed multilamellar vesicles. Biochim. Biophys. Acta 1985, 817, 193–196. [Google Scholar] [CrossRef]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. Methods Mol. Biol. 2017, 1522, 17–22. [Google Scholar]
- Olson, F.; Hunt, C.A.; Szoka, F.C.; Vail, W.J.; Papahadjopoulos, D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta 1979, 557, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Al-Azayzih, A.; Missaoui, W.N.; Cummings, B.S.; Somanath, P.R. Liposome-mediated delivery of the p21 activated kinase-1 (PAK-1) inhibitor IPA-3 limits prostate tumor growth in vivo. Nanomedicine 2016, 12, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Najahi-Missaoui, W.; Quach, N.D.; Somanath, P.R.; Cummings, B.S. Liposomes Targeting P21 Activated Kinase-1 (PAK-1) and Selective for Secretory Phospholipase A(2) (sPLA(2)) Decrease Cell Viability and Induce Apoptosis in Metastatic Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9396. [Google Scholar] [CrossRef] [PubMed]
- Mendez, R.; Banerjee, S. Sonication-Based Basic Protocol for Liposome Synthesis. Methods Mol. Biol. 2017, 1609, 255–260. [Google Scholar] [PubMed]
- Hope, M.J.; Bally, M.B.; Webb, G.; Cullis, P.R. Production of large unilamellar vesicles by a rapid extrusion procedure: Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 1985, 812, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta 1993, 1151, 201–215. [Google Scholar] [CrossRef]
- Fritze, A.; Hens, F.; Kimpfler, A.; Schubert, R.; Peschka-Süss, R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim. Biophys. Acta 2006, 1758, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szoka, F., Jr.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corace, G.; Angeloni, C.; Malaguti, M.; Hrelia, S.; Stein, P.C.; Brandl, M.; Gotti, R.; Luppi, B. Multifunctional liposomes for nasal delivery of the anti-Alzheimer drug tacrine hydrochloride. J. Liposome Res. 2014, 24, 323–335. [Google Scholar] [CrossRef]
- Zhu, X.; Xie, Y.; Zhang, Y.; Huang, H.; Huang, S.; Hou, L.; Zhang, H.; Li, Z.; Shi, J.; Zhang, Z. Thermo-sensitive liposomes loaded with doxorubicin and lysine modified single-walled carbon nanotubes as tumor-targeting drug delivery system. J. Biomater. Appl. 2014, 29, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, I.; Reszka, R.; Goan, S.R.; Naundorf, H. Carboplatin-liposomes (CPL) in immunodeficient mice: Improved antitumor activity for breast carcinomas and stimulation of hematopoiesis. Med. Oncol. 1994, 11, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Grainger, D.W. Lyophilized liposome-based parenteral drug development: Reviewing complex product design strategies and current regulatory environments. Adv. Drug Deliv. Rev. 2019, 151–152, 56–71. [Google Scholar] [CrossRef]
- Franzé, S.; Selmin, F.; Samaritani, E.; Minghetti, P.; Cilurzo, F. Lyophilization of Liposomal Formulations: Still Necessary, Still Challenging. Pharmaceutics 2018, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, S.E.; Gregoriadis, G.; Dick, R. Evaluation of the dehydration-rehydration method for production of contrast-carrying liposomes. Investig. Radiol. 1988, 23, 131–138. [Google Scholar] [CrossRef]
- Monnard, P.A.; Oberholzer, T.; Luisi, P. Entrapment of nucleic acids in liposomes. Biochim. Biophys. Acta 1997, 1329, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Osouli-Bostanabad, K.; Puliga, S.; Serrano, D.R.; Bucchi, A.; Halbert, G.; Lalatsa, A. Microfluidic Manufacture of Lipid-Based Nanomedicines. Pharmaceutics 2022, 14, 1940. [Google Scholar] [CrossRef]
- Shah, V.M.; Nguyen, D.X.; Patel, P.; Cote, B.; Al-Fatease, A.; Pham, Y.; Huynh, M.G.; Woo, Y.; Alani, A.W. Liposomes produced by microfluidics and extrusion: A comparison for scale-up purposes. Nanomedicine 2019, 18, 146–156. [Google Scholar] [CrossRef]
- Mijajlovic, M.; Wright, D.; Zivkovic, V.; Bi, J.X.; Biggs, M.J. Microfluidic hydrodynamic focusing based synthesis of POPC liposomes for model biological systems. Colloids Surf. B Biointerfaces 2013, 104, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Saveyn, H.; De Baets, B.; Thas, O.; Hole, P.; Smith, J.; Van der Meeren, P. Accurate particle size distribution determination by nanoparticle tracking analysis based on 2-D Brownian dynamics simulation. J. Colloid Interface Sci. 2010, 352, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, T.; Holscher, H.; Schimmel, T.; Baykara, M.Z.; Schwarz, U.D.; Garcia, R. Advanced atomic force microscopy techniques. Beilstein J. Nanotechnol. 2012, 3, 893–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiss, W.; Thanh, N.T.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV-vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Planken, K.L.; Colfen, H. Analytical ultracentrifugation of colloids. Nanoscale 2010, 2, 1849–1869. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.W.; Prince, R.C.; Crofts, A.R. The response of fluorescent amines to pH gradients across liposome membranes. Biochim. Biophys. Acta 1972, 274, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Zucker, D.; Marcus, D.; Barenholz, Y.; Goldblum, A. Liposome drugs’ loading efficiency: A working model based on loading conditions and drug’s physicochemical properties. J. Control. Release 2009, 139, 73–80. [Google Scholar] [CrossRef]
- Li, X.; Hirsh, D.J.; Cabral-Lilly, D.; Zirkel, A.; Gruner, S.M.; Janoff, A.S.; Perkins, W.R. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim. Biophys. Acta 1998, 1415, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Safra, T.; Muggia, F.; Jeffers, S.; Tsao-Wei, D.D.; Groshen, S.; Lyass, O.; Henderson, R.; Berry, G.; Gabizon, A. Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Annal. Oncol. 2000, 11, 1029–1033. [Google Scholar] [CrossRef]
- Cancer Medications Enquiry Database (CanMED). Surveillance Research Program SEER Website Tool. National Drug Code, Version 1.14.0; Division of Cancer Control and Population Sciences, National Cancer Institute: Bethesda, MA, USA, 2023.
- HRPA. DAUNOXOME 2 mg/mL, Liposomal Dispersion for Injection; HRPA: Toronto, ON, Canada, 2014; pp. 1–14. [Google Scholar]
- BioSpace. Clinigen And Galen Enter Exclusive Global Access Agreement for Chemotherapy Drug Daunoxome; BioSpace: Des Moines, IA, USA, 2016. [Google Scholar]
- Van Hoogevest, P.; Wendel, A. The use of natural and synthetic phospholipids as pharmaceutical excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forssen, E. The design and development of DaunoXome® for solid tumor targeting in vivo. Adv. Drug Deliv. Rev. 1997, 24, 133–150. [Google Scholar] [CrossRef]
- European Medicines Agency. DepoCyt; European Medicines Agency: Amsterdam, The Netherlands, 2017.
- U.S. Food and Drug Administration. Depocyt (Cytarabine); U.S. Food and Drug Administration: Silver Spring, MD, USA, 2017.
- WCG FDANews. Pacira Shutters DepCyt Operations after Years of Manufacturing Problems; WCG FDANews: Falls Church, VA, USA, 2017. [Google Scholar]
- U.S. Food & Drug Administration. Doxil (Liposomal) [Doxorubicin Hydrochloride] Label; U.S. Food & Drug Administration: Silver Spring, MD, USA, 2022.
- European Medicines Agency. Caelyx Pegylated Liposomal; European Medicines Agency: Amsterdam, The Netherlands, 2023.
- Taiwan Liposome Company, Ltd. Form F-1 Registration Statement; Taiwan Liposome Company, Ltd.: Taipei, Taiwan, 2018. [Google Scholar]
- Hsieh, Y.J.; Chang, C.H.; Huang, S.P.; Lin, C.W.; Wang, M.N.; Wu, Y.T.; Chen, Y.J.; Tsai, T.H. Effect of cyclosporin A on the brain regional distribution of doxorubicin in rats. Int. J. Pharm. 2008, 350, 265–271. [Google Scholar] [CrossRef]
- Hospira Australia Pty Ltd. Marqibo® (vinCRIStine Sulfate LIPOSOME Injection) for Intravenous Infusion [Package Insert]; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2022.
- Mifamurtide: CGP 19835, CGP 19835A, L-MTP-PE, liposomal MTP-PE, MLV 19835A, MTP-PE, muramyltripeptide phosphatidylethanolamine. Drugs R D 2008, 9, 131–135. [CrossRef] [PubMed]
- Vail, D.M.; MacEwen, E.G.; Kurzman, I.D.; Dubielzig, R.R.; Helfand, S.C.; Kisseberth, W.C.; London, C.A.; Obradovich, J.E.; Madewell, B.R.; Rodriguez, C.O. Liposome-encapsulated muramyl tripeptide phosphatidylethanolamine adjuvant immunotherapy for splenic hemangiosarcoma in the dog: A randomized multi-institutional clinical trial. Clin. Cancer Res. 1995, 1, 1165–1170. [Google Scholar]
- European Medicines Agency. Mepact (mifamurtide); European Medicines Agency: Amsterdam, The Netherlands, 2020.
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K.; et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [CrossRef]
- European Medicines Agency. Myocet Liposomal (Previously Myocet); European Medicines Agency: Amsterdam, The Netherlands, 2021.
- Swenson, C.E.; Perkins, W.R.; Roberts, P.; Janoff, A.S. Liposome technology and the development of Myocet™ (liposomal doxorubicin citrate). Breast 2001, 10, 1–7. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Onivyde (Irinotecan Hydrochloride); U.S. Food and Drug Administration: Silver Spring, MD, USA, 2023.
- European Medicines Agency. Onivyde Pegylated Liposomal (Previously Known as Onivyde); European Medicines Agency: Amsterdam, The Netherlands, 2022.
- Kalra, A.V.; Kim, J.; Klinz, S.G.; Paz, N.; Cain, J.; Drummond, D.C.; Nielsen, U.B.; Fitzgerald, J.B. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014, 74, 7003–7013. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Vyxeos; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2023.
- European Medicines Agency. Vyxeos Liposomal (Previously Known as Vyxeos); European Medicines Agency: Amsterdam, The Netherlands, 2022.
- European Medicines Agency. Zolsketil Pegylated Liposomal; European Medicines Agency: Amsterdam, The Netherlands, 2022.
- Dragovich, T.; Mendelson, D.; Kurtin, S.; Richardson, K.; Von Hoff, D.; Hoos, A. A Phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-refractory advanced colorectal cancer. Cancer Chemother. Pharmacol. 2006, 58, 759–764. [Google Scholar] [CrossRef]
- Gutiérrez-Puente, Y.; Tari, A.M.; Stephens, C.; Rosenblum, M.; Guerra, R.T.; Lopez-Berestein, G. Safety, pharmacokinetics, and tissue distribution of liposomal P-ethoxy antisense oligonucleotides targeted to Bcl-2. J. Pharmacol. Exp. Ther. 1999, 291, 865–869. [Google Scholar]
- Fasol, U.; Frost, A.; Buchert, M.; Arends, J.; Fiedler, U.; Scharr, D.; Scheuenpflug, J.; Mross, K. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Annal. Oncol. 2012, 23, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Van, P.; Tiemessen, H.; Metselaar, J.M.; Drescher, S.; Fahr, A. The Use of Phospholipids to Make Pharmaceutical Form Line Extensions. Eur. J. Lipid Sci. Technol. 2021, 123, 2000297. [Google Scholar]
- Li, C.; Wang, J.; Wang, C.; Li, Y.; Shen, D.; Guo, W.; Li, J.; Zhang, L. Liposomal Pharmaceutical Preparation and Method for Manufacturing the Same. U.S. 10,028,913B2, 24 July 2018. [Google Scholar]
- Gao, Y.; Huang, H.; Wang, X.; Bai, B.; Huang, Y.; Yang, H.; Zhang, Q.; Li, Y.; Li, Y.; Zhou, M.; et al. Safety and Efficacy of Mitoxantrone Hydrochloride Liposome in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma and Extranodal NK/T-Cell Lymphoma: A Prospective, Single-Arm, Open-Label, Multi-Center, Phase II Clinical Trial. Blood 2020, 136, 36–37. [Google Scholar] [CrossRef]
- Yarmolenko, P.S.; Zhao, Y.; Landon, C.; Spasojevic, I.; Yuan, F.; Needham, D.; Viglianti, B.L.; Dewhirst, M.W. Comparative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumours. Int. J. Hyperth. 2010, 26, 485–498. [Google Scholar] [CrossRef]
- Lyon, P.C.; Griffiths, L.F.; Lee, J.; Chung, D.; Carlisle, R.; Wu, F.; Middleton, M.R.; Gleeson, F.V.; Coussios, C.C. Clinical trial protocol for TARDOX: A phase I study to investigate the feasibility of targeted release of lyso-thermosensitive liposomal doxorubicin (ThermoDox(R)) using focused ultrasound in patients with liver tumours. J. Ther. Ultrasound 2017, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.J.; Melander, F.; Vikbjerg, A.F.; Petersen, S.A.; Madsen, M.W. Medical Use of sPLA2 Hydrolysable Liposomes. U.S. Patent 11,207,269B2, 28 December 2021. [Google Scholar]
- Jehn, C.F.; Boulikas, T.; Kourvetaris, A.; Possinger, K.; Lüftner, D. Pharmacokinetics of liposomal cisplatin (lipoplatin) in combination with 5-FU in patients with advanced head and neck cancer: First results of a phase III study. AntiCancer Res. 2007, 27, 471–475. [Google Scholar] [PubMed]
- Newman, M.S.; Colbern, G.T.; Working, P.K.; Engbers, C.; Amantea, M.A. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother. Pharmacol. 1999, 43, 1–7. [Google Scholar] [CrossRef]
- Seetharamu, N.; Kim, E.; Hochster, H.; Martin, F.; Muggia, F. Phase II study of liposomal cisplatin (SPI-77) in platinum-sensitive recurrences of ovarian cancer. AntiCancer Res. 2010, 30, 541–545. [Google Scholar]
- Ogihara, I.; Kojima, S.; Jay, M. Tumor uptake of 67Ga-carrying liposomes. Eur. J. Nucl. Med. 1986, 11, 405–411. [Google Scholar] [CrossRef]
- Zhao, W.; Zhuang, S.; Qi, X.R. Comparative study of the in vitro and in vivo characteristics of cationic and neutral liposomes. Int. J. Nanomed. 2011, 6, 3087–3098. [Google Scholar]
- Krasnici, S.; Werner, A.; Eichhorn, M.E.; Schmitt-Sody, M.; Pahernik, S.A.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Naujoks, K.; et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int. J. Cancer 2003, 105, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine 2013, 8, 1509–1528. [Google Scholar] [CrossRef] [Green Version]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol. 2008, 26, 57–64. [Google Scholar] [CrossRef]
- Maeda, H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 53–71. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2004, 56, 1649–1659. [Google Scholar] [CrossRef]
- Sawant, R.R.; Torchilin, V.P. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012, 14, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Madni, A.; Sarfraz, M.; Rehman, M.; Ahmad, M.; Akhtar, N.; Ahmad, S.; Tahir, N.; Ijaz, S.; Al-Kassas, R.; Lobenberg, R. Liposomal drug delivery: A versatile platform for challenging clinical applications. J. Pharm. Pharm. Sci. 2014, 17, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol. Sci. 1994, 15, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Siegal, T.; Horowitz, A.; Gabizon, A. Doxorubicin encapsulated in sterically stabilized liposomes for the treatment of a brain tumor model: Biodistribution and therapeutic efficacy. J. Neurosurg. 1995, 83, 1029–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Working, P.K.; Newman, M.S.; Huang, K.S.; Mayhew, E.; Vaage, J.; Lasic, D.D. Pharmacokinetics, Biodistribution and Therapeutic Efficacy of Doxorubicin Encapsulated in Stealth® Liposomes (Doxil®). J. Liposome Res. 1994, 4, 667–687. [Google Scholar] [CrossRef]
- Phase Transition Temperatures for Glycerophospholipids. Available online: https://avantilipids.com/tech-support/physical-properties/phase-transition-temps (accessed on 26 February 2023).
- 18:0 PC (DSPC) 1,2-Distearoyl-sn-glycero-3-phosphocholine. Available online: https://avantilipids.com/product/850365 (accessed on 26 February 2023).
- Hydro Soy PC L-α-phosphatidylcholine, Hydrogenated (Soy). Available online: https://avantilipids.com/product/840058 (accessed on 26 February 2023).
- Hong, R.L.; Tseng, Y.L. Phase I and pharmacokinetic study of a stable, polyethylene-glycolated liposomal doxorubicin in patients with solid tumors: The relation between pharmacokinetic property and toxicity. Cancer 2001, 91, 1826–1833. [Google Scholar] [CrossRef]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Chou, H.H.; Wang, K.L.; Chen, C.A.; Wei, L.H.; Lai, C.H.; Hsieh, C.Y.; Yang, Y.C.; Twu, N.F.; Chang, T.C.; Yen, M.S.; et al. Pegylated liposomal doxorubicin (Lipo-Dox) for platinum-resistant or refractory epithelial ovarian carcinoma: A Taiwanese gynecologic oncology group study with long-term follow-up. Gynecol. Oncol. 2006, 101, 423–428. [Google Scholar] [CrossRef]
- Hsiao, S.M.; Chen, C.A.; Lin, H.H.; Hsieh, C.Y.; Wei, L.H. Phase II trial of carboplatin and distearoylphosphatidylcholine pegylated liposomal doxorubicin (Lipo-Dox) in recurrent platinum-sensitive ovarian cancer following front-line therapy with paclitaxel and platinum. Gynecol. Oncol. 2009, 112, 35–39. [Google Scholar] [CrossRef]
- Yao, N.; Kao, W.; Chao, T.; Hsieh, R.; Lin, J.; Su, C.; Lo, S. A phase II study of gemcitabine and liposomal doxorubicin (Lipo-Dox) as first line chemotherapy in the treatment of metastatic breast cancer. J. Clin. Oncol. 2006, 24, 10688. [Google Scholar] [CrossRef]
- White, S.C.; Lorigan, P.; Margison, G.P.; Margison, J.M.; Martin, F.; Thatcher, N.; Anderson, H.; Ranson, M. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br. J. Cancer 2006, 95, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Lu, C.; Khuri, F.R.; Tonda, M.; Glisson, B.S.; Liu, D.; Jung, M.; Hong, W.K.; Herbst, R.S. A phase II study of STEALTH cisplatin (SPI-77) in patients with advanced non-small cell lung cancer. Lung Cancer 2001, 34, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Kroll, A.; Pillukat, M.H.; Hahn, D.; Schnekenburger, J. Current in vitro methods in nanoparticle risk assessment: Limitations and challenges. Eur. J. Pharm. Biopharm. 2009, 72, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Raj, R.; Mongia, P.; Sahu, S.K.; Ram, A. Nanocarriers based anticancer drugs: Current scenario and future perceptions. Curr. Drug Targets 2015, 17, 206–228. [Google Scholar] [CrossRef]
- Perez-Herrero, E.; Fernandez-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Cao, H.; He, W.; Gao, F.; Liu, Y.; Yin, L. Novel drug delivery liposomes targeted with a fully human anti-VEGF165 monoclonal antibody show superior antitumor efficacy in vivo. Biomed. Pharmacother. 2015, 73, 48–57. [Google Scholar] [CrossRef]
- Gao, J.; Chen, H.; Song, H.; Su, X.; Niu, F.; Li, W.; Li, B.; Dai, J.; Wang, H.; Guo, Y. Antibody-targeted immunoliposomes for cancer treatment. Mini Rev. Med. Chem. 2013, 13, 2026–2035. [Google Scholar] [CrossRef]
- Fegan, A.; Kumarapperuma, S.C.; Wagner, C.R. Chemically Self-Assembled Antibody Nanostructures as Potential Drug Carriers. Mol. Pharm. 2012, 9, 3218–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007, 9, E128–E147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhier, F.; Feron, O.; Preat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-targeted liposome design: Challenges and fundamental considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.O.; Petrilli, R.; Trevizan, L.N.F.; Chorilli, M. Immunoliposomes: A review on functionalization strategies and targets for drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, C.; Lu, L.; Jiang, H.; Wang, F.; Zhang, X. Transcytosable Peptide-Paclitaxel Prodrug Nanoparticle for Targeted Treatment of Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 4646. [Google Scholar] [CrossRef]
- Li, J.; Kataoka, K. Chemo-physical Strategies to Advance the in Vivo Functionality of Targeted Nanomedicine: The Next Generation. J. Am. Chem. Soc. 2021, 143, 538–559. [Google Scholar] [CrossRef]
- Hussain, S.; Pluckthun, A.; Allen, T.M.; Zangemeister-Wittke, U. Antitumor activity of an epithelial cell adhesion molecule targeted nanovesicular drug delivery system. Mol. Cancer Ther. 2007, 6, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Koshkaryev, A.; Sawant, R.; Deshpande, M.; Torchilin, V. Immunoconjugates and long circulating systems: Origins, current state of the art and future directions. Adv. Drug Deliv. Rev. 2013, 65, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Hong, K.; Kirpotin, D.B.; Meyer, O.; Papahadjopoulos, D.; Benz, C.C. Anti-HER2 immunoliposomes for targeted therapy of human tumors. Cancer Lett. 1997, 118, 153–160. [Google Scholar] [CrossRef]
- Park, J.W.; Kirpotin, D.B.; Hong, K.; Shalaby, R.; Shao, Y.; Nielsen, U.B.; Marks, J.D.; Papahadjopoulos, D.; Benz, C.C. Tumor targeting using anti-her2 immunoliposomes. J. Control. Release 2001, 74, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Elbayoumi, T.A.; Torchilin, V.P. Enhanced accumulation of long-circulating liposomes modified with the nucleosome-specific monoclonal antibody 2C5 in various tumours in mice: Gamma-imaging studies. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- ElBayoumi, T.A.; Torchilin, V.P. Tumor-targeted nanomedicines: Enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin. Cancer Res. 2009, 15, 1973–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbayoumi, T.A.; Torchilin, V.P. Enhanced cytotoxicity of monoclonal anticancer antibody 2C5-modified doxorubicin-loaded PEGylated liposomes against various tumor cell lines. Eur. J. Pharm. Sci. 2007, 32, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Mumbengegwi, D.R.; Charrois, G.J. Anti-CD19-targeted liposomal doxorubicin improves the therapeutic efficacy in murine B-cell lymphoma and ameliorates the toxicity of liposomes with varying drug release rates. Clin. Cancer Res. 2005, 11, 3567–3573. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.W.; Allen, T.M. Targeted delivery of anti-CD19 liposomal doxorubicin in B-cell lymphoma: A comparison of whole monoclonal antibody, Fab’ fragments and single chain Fv. J. Control. Release 2008, 126, 50–58. [Google Scholar] [CrossRef]
- Arias, J.L. Drug targeting strategies in cancer treatment: An overview. Mini Rev. Med. Chem. 2011, 11, 1–17. [Google Scholar] [CrossRef]
- Ta, T.; Porter, T.M. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J. Control. Release 2013, 169, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: State of the art review. Int. J. Nanomed. 2014, 9, 4387–4398. [Google Scholar]
- May, J.P.; Li, S.D. Hyperthermia-induced drug targeting. Expert Opin. Drug Deliv. 2013, 10, 511–527. [Google Scholar] [CrossRef]
- Grull, H.; Langereis, S. Hyperthermia-triggered drug delivery from temperature-sensitive liposomes using MRI-guided high intensity focused ultrasound. J. Control. Release 2012, 161, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, F.; Steffen, K.J.; Mallik, S. Enzyme-Responsive Liposomes for the Delivery of Anticancer Drugs. Bioconjug. Chem. 2017, 28, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Mock, J.N.; Costyn, L.J.; Wilding, S.L.; Arnold, R.D.; Cummings, B.S. Evidence for distinct mechanisms of uptake and antitumor activity of secretory phospholipase A2 responsive liposome in prostate cancer. Integr. Biol. 2013, 5, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodle, M.C.; Matthay, K.K.; Newman, M.S.; Hidayat, J.E.; Collins, L.R.; Redemann, C.; Martin, F.J.; Papahadjopoulos, D. Versatility in lipid compositions showing prolonged circulation with sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1105, 193–200. [Google Scholar] [CrossRef]
- Du, H.; Chandaroy, P.; Hui, S.W. Grafted poly-(ethylene glycol) on lipid surfaces inhibits protein adsorption and cell adhesion. Biochim. Biophys. Acta 1997, 1326, 236–248. [Google Scholar] [CrossRef] [Green Version]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Dams, E.; Laverman, P.; Oyen, W.; Storm, G.; Scherphof, G.; Van der Meer, J.; Corstens, F.; Boerman, O. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharm. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar]
- Li, C.; Cao, J.; Wang, Y.; Zhao, X.; Deng, C.; Wei, N.; Yang, J.; Cui, J. Accelerated blood clearance of pegylated liposomal topotecan: Influence of polyethylene glycol grafting density and animal species. J. Pharm. Sci. 2012, 101, 3864–3876. [Google Scholar] [CrossRef]
- Xu, H.; Ye, F.; Hu, M.; Yin, P.; Zhang, W.; Li, Y.; Yu, X.; Deng, Y. Influence of phospholipid types and animal models on the accelerated blood clearance phenomenon of PEGylated liposomes upon repeated injection. Drug Deliv. 2015, 22, 598–607. [Google Scholar] [CrossRef]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef]
- Verhoef, J.J.F.; Carpenter, J.F.; Anchordoquy, T.J.; Schellekens, H. Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics. Drug Discov. Today 2014, 19, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gao, X.; Yan, X.; Deng, Y.; Ma, H. PEGylated nanoemulsions containing 1,2-distearoyl-sn-glycero-3-phosphoglycerol induced weakened accelerated blood clearance phenomenon. Drug Deliv. Transl. Res. 2022, 12, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, W.; Shen, Y.; Mu, H.; Zhang, Y.; Liang, R.; Wang, A.; Sun, K.; Fu, F. Effects of a novel pH-sensitive liposome with cleavable esterase-catalyzed and pH-responsive double smart mPEG lipid derivative on ABC phenomenon. Int. J. Nanomed. 2011, 6, 2053–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Wang, K.; Deng, Y.; Chen, D. Effects of cleavable PEG-cholesterol derivatives on the accelerated blood clearance of PEGylated liposomes. Biomaterials 2010, 31, 4757–4763. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, T.; Liu, M.Y.; Song, Y.Z.; Liu, X.R.; Deng, Y.H. Polysialic Acid Modified Liposomes for Improving Pharmacokinetics and Overcoming Accelerated Blood Clearance Phenomenon. Coatings 2020, 10, 834. [Google Scholar] [CrossRef]
- Sadzuka, Y.; Kishi, K.; Hirota, S.; Sonobe, T. Effect of polyethyleneglycol (PEG) chain on cell uptake of PEG-modified liposomes. J. Liposome Res. 2003, 13, 157–172. [Google Scholar] [CrossRef]
- Song, L.Y.; Ahkong, Q.F.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.J.; Mui, B. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim. Biophys. Acta 2002, 1558, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ghaferi, M.; Raza, A.; Koohi, M.; Zahra, W.; Akbarzadeh, A.; Ebrahimi Shahmabadi, H.; Alavi, S.E. Impact of PEGylated Liposomal Doxorubicin and Carboplatin Combination on Glioblastoma. Pharmaceutics 2022, 14, 2183. [Google Scholar] [CrossRef]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome-cell interactions in vitro: Effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef]
- Brandenberger, C.; Mühlfeld, C.; Ali, Z.; Lenz, A.G.; Schmid, O.; Parak, W.J.; Gehr, P.; Rothen-Rutishauser, B. Quantitative evaluation of cellular uptake and trafficking of plain and polyethylene glycol-coated gold nanoparticles. Small 2010, 6, 1669–1678. [Google Scholar] [CrossRef]
- Zhang, Y.; Kohler, N.; Zhang, M. Surface modification of superparamagnetic magnetite nanoparticles and their intracellular uptake. Biomaterials 2002, 23, 1553–1561. [Google Scholar] [CrossRef]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernández, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface Functionalization of Nanoparticles with Polyethylene Glycol: Effects on Protein Adsorption and Cellular Uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Webster, P.; Davis, M.E. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur. J. Cell Biol. 2004, 83, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.L.; Majzoub, R.N.; Shirazi, R.S.; Ewert, K.K.; Chen, Y.J.; Liang, K.S.; Safinya, C.R. Endosomal escape and transfection efficiency of PEGylated cationic liposome-DNA complexes prepared with an acid-labile PEG-lipid. Biomaterials 2012, 33, 4928–4935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Jiang, X.; Huang, Y.; Zhang, C.; Ping, Q. pH-Sensitive mPEG-Hz-Cholesterol Conjugates as a Liposome Delivery System. J. Bioact. Compat. Polym. 2010, 25, 527–542. [Google Scholar] [CrossRef]
- Parr, M.J.; Masin, D.; Cullis, P.R.; Bally, M.B. Accumulation of liposomal lipid and encapsulated doxorubicin in murine Lewis lung carcinoma: The lack of beneficial effects by coating liposomes with poly(ethylene glycol). J. Pharmacol. Exp. Ther. 1997, 280, 1319–1327. [Google Scholar]
- Zalba, S.; ten Hagen, T.L.M.; Burgui, C.; Garrido, M.J. Stealth nanoparticles in oncology: Facing the PEG dilemma. J. Controll. Release 2022, 351, 22–36. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.; Gao, S.; Lu, A.; Yang, D.; Jiang, H.; He, Y.; Shi, K. Cleavable PEGylation: A strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017, 24 (Suppl. S1), 22–32. [Google Scholar] [CrossRef] [Green Version]
- Holland, J.W.; Hui, C.; Cullis, P.R.; Madden, T.D. Poly(ethylene glycol)—Lipid conjugates regulate the calcium-induced fusion of liposomes composed of phosphatidylethanolamine and phosphatidylserine. Biochemistry 1996, 35, 2618–2624. [Google Scholar] [CrossRef]
- Hong, R.L.; Huang, C.J.; Tseng, Y.L.; Pang, V.F.; Chen, S.T.; Liu, J.J.; Chang, F.H. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: Is surface coating with polyethylene glycol beneficial? Clin. Cancer Res. 1999, 5, 3645–3652. [Google Scholar]
- Zhao, G.; Long, L.; Zhang, L.; Peng, M.; Cui, T.; Wen, X.; Zhou, X.; Sun, L.; Che, L. Smart pH-sensitive nanoassemblies with cleavable PEGylation for tumor targeted drug delivery. Sci. Rep. 2017, 7, 3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Shum, P.; Thompson, D.H. Acid-triggered release via dePEGylation of DOPE liposomes containing acid-labile vinyl ether PEG-lipids. J. Control. Release 2003, 91, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.R. Are cancer cells acidic? Br. J. Cancer 1991, 64, 425–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Hu, Y.B.; Dammer, E.B.; Ren, R.J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Kanamala, M.; Palmer, B.D.; Jamieson, S.M.; Wilson, W.R.; Wu, Z. Dual pH-sensitive liposomes with low pH-triggered sheddable PEG for enhanced tumor-targeted drug delivery. Nanomedicine 2019, 14, 1971–1989. [Google Scholar] [CrossRef]
- McNeeley, K.M.; Karathanasis, E.; Annapragada, A.V.; Bellamkonda, R.V. Masking and triggered unmasking of targeting ligands on nanocarriers to improve drug delivery to brain tumors. Biomaterials 2009, 30, 3986–3995. [Google Scholar] [CrossRef]
- Kale, A.A.; Torchilin, V.P. “Smart” drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J. Liposome Res. 2007, 17, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Kale, A.A.; Torchilin, V.P. Enhanced transfection of tumor cells in vivo using “Smart” pH-sensitive TAT-modified pegylated liposomes. J. Drug Target. 2007, 15, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Kuai, R.; Yuan, W.; Li, W.; Qin, Y.; Tang, J.; Yuan, M.; Fu, L.; Ran, R.; Zhang, Z.; He, Q. Targeted delivery of cargoes into a murine solid tumor by a cell-penetrating peptide and cleavable poly(ethylene glycol) comodified liposomal delivery system via systemic administration. Mol. Pharm. 2011, 8, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Hak, S.; Helgesen, E.; Hektoen, H.H.; Huuse, E.M.; Jarzyna, P.A.; Mulder, W.J.; Haraldseth, O.; Davies, C.e.L. The effect of nanoparticle polyethylene glycol surface density on ligand-directed tumor targeting studied in vivo by dual modality imaging. ACS Nano 2012, 6, 5648–5658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Guo, M.; Zhan, G.; Shi, D.; Shi, L.; Gan, L.; Zhao, Y.; Yang, X. NIR-triggered ligand-presenting nanocarriers for enhancing synergistic photothermal-chemotherapy. J. Control. Release 2023, 353, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yin, Z.; Chang, Y.; Wang, H.; Xu, J.-F.; Zhang, X. Tumor acidity-induced charge-reversal liposomal doxorubicin with enhanced cancer cell uptake and anticancer activity. Giant 2021, 6, 100052. [Google Scholar] [CrossRef]
- Savoy, E.A.O.; Yoon, F.P.; Mesbahi, H.; Knight, N.J.R.; Berkman, C.E. Chapter 6 Acid-labile Linkers. In Chemical Linkers in Antibody-Drug Conjugates (ADCs); The Royal Society of Chemistry: London, UK, 2022; pp. 213–231. [Google Scholar]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef]
- Olatunji, F.P.; Herman, J.W.; Kesic, B.N.; Olabode, D.; Berkman, C.E. A click-ready pH-triggered phosphoramidate-based linker for controlled release of monomethyl auristatin E. Tetrahedron Lett. 2020, 61, 152398. [Google Scholar] [CrossRef]
- Choy, C.J.; Ley, C.R.; Davis, A.L.; Backer, B.S.; Geruntho, J.J.; Clowers, B.H.; Berkman, C.E. Second-Generation Tunable pH-Sensitive Phosphoramidate-Based Linkers for Controlled Release. Bioconjug. Chem. 2016, 27, 2206–2213. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Product Name | Drug | Lipid Composition (Molar Ratio) | Indication | Approval Date | Marketing Status | Ref. |
|---|---|---|---|---|---|---|
| DaunoXome® (US) | Daunorubicin | DSPC:CL (2:1) | AIDS-related Kaposi’s sarcoma | 1995–1997 (EMA) 1996 (FDA) | US discontinued (2016), global on-demand access (2016, EU, UK, AU, NZ, HK) | [104,105,106,107,108] |
| DepoCyt® (US, EU) | Cytarabine | CL:TR:DOPC:DPPG (w/w/4.4:1.2:5.7:1) | Lymphomatous meningitis | 1999 (FDA) 2001 (EMA) | Withdrawn production by company (2017) | [109,110,111] |
| Doxil® (US)/ Caelyx® (EU) a | Doxorubicin | HSPC:CL:MPEG 2000-DSPE (calc. 3:2:0.9, w/w 3:1:1) | AIDS-related Kaposi’s sarcoma, recurrent ovarian cancer, multiple myeloma, metastatic breast cancer (EU only) | 1995 (FDA) 1996 (EMA) | Active (US, EU) | [102,112,113] |
| Lipo-Dox® (TW) | Doxorubicin | DSPC:CL:MPEG 2000-DSPE (3:2:0.3) | AIDS-related Kaposi’s sarcoma, ovarian cancer, breast cancer, multiple myeloma | 1998 (TW) | Active (TW) | [114,115] |
| Marqibo® (US) | Vincristine | SM:CL (60:40) | Acute lymphoblastic leukemia | 2012 (FDA) | US discontinued (2020) | [104,116] |
| Mepact® (EU) | Mifamurtide | DOPS:POPC (3:7) | Osteosarcoma | 2009 (EMA) | Active (EU) | [117,118,119] |
| Myocet® (EU) | Doxorubicin | PC:CL (55:45) | Metastatic breast cancer | 2000 (EMA) | Active (EU) | [120,121,122] |
| Onivyde®/Nal-IRI (EU, US) | Irinotecan | DSPC:CL:MPEG 2000-DSPE (3:2:0.015) | Pancreatic cancer | 1996 (FDA) 2016 (EMA) | Active (US, EU) | [123,124,125] |
| Vyxeos®/CPX-351 (EU, US) | Cytarabine: daunorubicin (5:1 mol. ratio) | DSPG:DSPC:CL (7:2:1) | Newly diagnosed therapy–related acute myeloid leukemia, acute myeloid leukemia with myelodysplasia-related changes | 2017 (FDA) 2018 (EMA) | Active (US, EU) | [126,127] |
| Zolsketil® (EU) a | Doxorubicin | HSPC:CL:MPEG 2000-DSPE | Metastatic breast cancer, advanced ovarian cancer, multiple myeloma, AIDS-related Kaposi’s sarcoma | 2022 (EMA) | Active (EU) | [128] |
| Product Name | Drug | Lipid Composition (Molar Ratio) | Conditions | Delivery Mechanism | Status | Ref. |
|---|---|---|---|---|---|---|
| L-NDDP/ Aroplatin™ | cis-bis-neodecanoato-trans-R,R-1,2- diaminocyclohexane platinum (II) | DMPC:DMPG | B-cell lymphoma, malignant mesothelioma, pancreatic cancer, colorectal cancer, solid tumors | EPR | Phase I/II | [129] |
| BP1002 (US) | Antisense oligonucleotide against BCl-2 | DOPC:ASO (20:1) | Acute myeloid leukemia, advanced lymphoid malignancies | EPR | Phase I | [130] |
| EndoTAG® (EU, US, TW, UA) | Paclitaxel | DOTAP:DOPC (53:47) | Breast cancer, pancreatic cancer, liver cancer | Electrostatic | Phase II/III | [131,132] |
| PLM60 (US, CN) | Mitoxantrone | HSPC:CL:MPEG 2000-DSPE (w/w 3:1:1) | Advanced hepatocellular carcinoma, small-cell lung cancer, non-Hodgkin’s lymphoma, recurrent/refractory lymphomas | EPR | Phase I/II | [133,134] |
| ThermoDox® (US) | Doxorubicin | DPPC:MSPC:MPEG2000-DSPE (90:10:4) | Hepatocellular carcinoma, colorectal cancer, pediatric cancer, liver neoplasms, pancreatic cancer, breast cancer | Temperature | Phase I/II/III | [135,136] |
| LiPlaCis (DK) | Cisplatin | DSPC:DSPG:MPEG 2000-DSPE (mol.% 70:25:5) | Adv./refractory solid tumors, metastatic breast cancer, prostate cancer, skin cancer | sPLA2 targeted | Phase I/II | [137] |
| Lipoplatin™ | Cisplatin | SPC-3: DPPG: CL: MPEG2000-DSPE | Malignant pleural effusions | Fusogenic | Phase I | [138] |
| SPI-077 | Cisplatin | HSPC:CL:MPEG2000-DSPE (51:44:5) | Ovarian cancer | EPR | Phase II | [139,140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fulton, M.D.; Najahi-Missaoui, W. Liposomes in Cancer Therapy: How Did We Start and Where Are We Now. Int. J. Mol. Sci. 2023, 24, 6615. https://doi.org/10.3390/ijms24076615
Fulton MD, Najahi-Missaoui W. Liposomes in Cancer Therapy: How Did We Start and Where Are We Now. International Journal of Molecular Sciences. 2023; 24(7):6615. https://doi.org/10.3390/ijms24076615
Chicago/Turabian StyleFulton, Melody D., and Wided Najahi-Missaoui. 2023. "Liposomes in Cancer Therapy: How Did We Start and Where Are We Now" International Journal of Molecular Sciences 24, no. 7: 6615. https://doi.org/10.3390/ijms24076615
APA StyleFulton, M. D., & Najahi-Missaoui, W. (2023). Liposomes in Cancer Therapy: How Did We Start and Where Are We Now. International Journal of Molecular Sciences, 24(7), 6615. https://doi.org/10.3390/ijms24076615