

Characterizing the Interplay of Lymphocytes in Graves’ Disease


Abstract
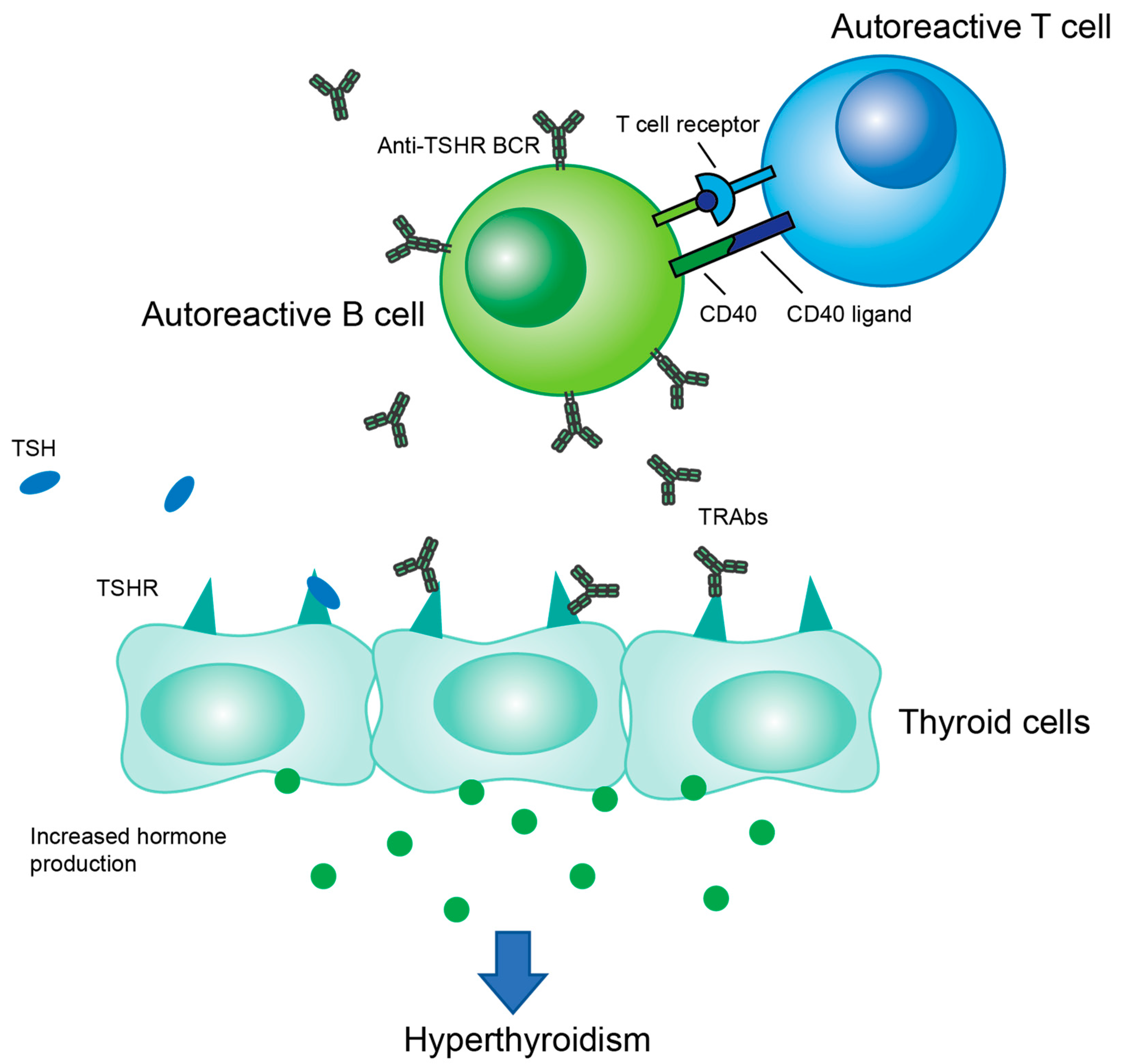
:1. Introduction
2. Autoantibodies
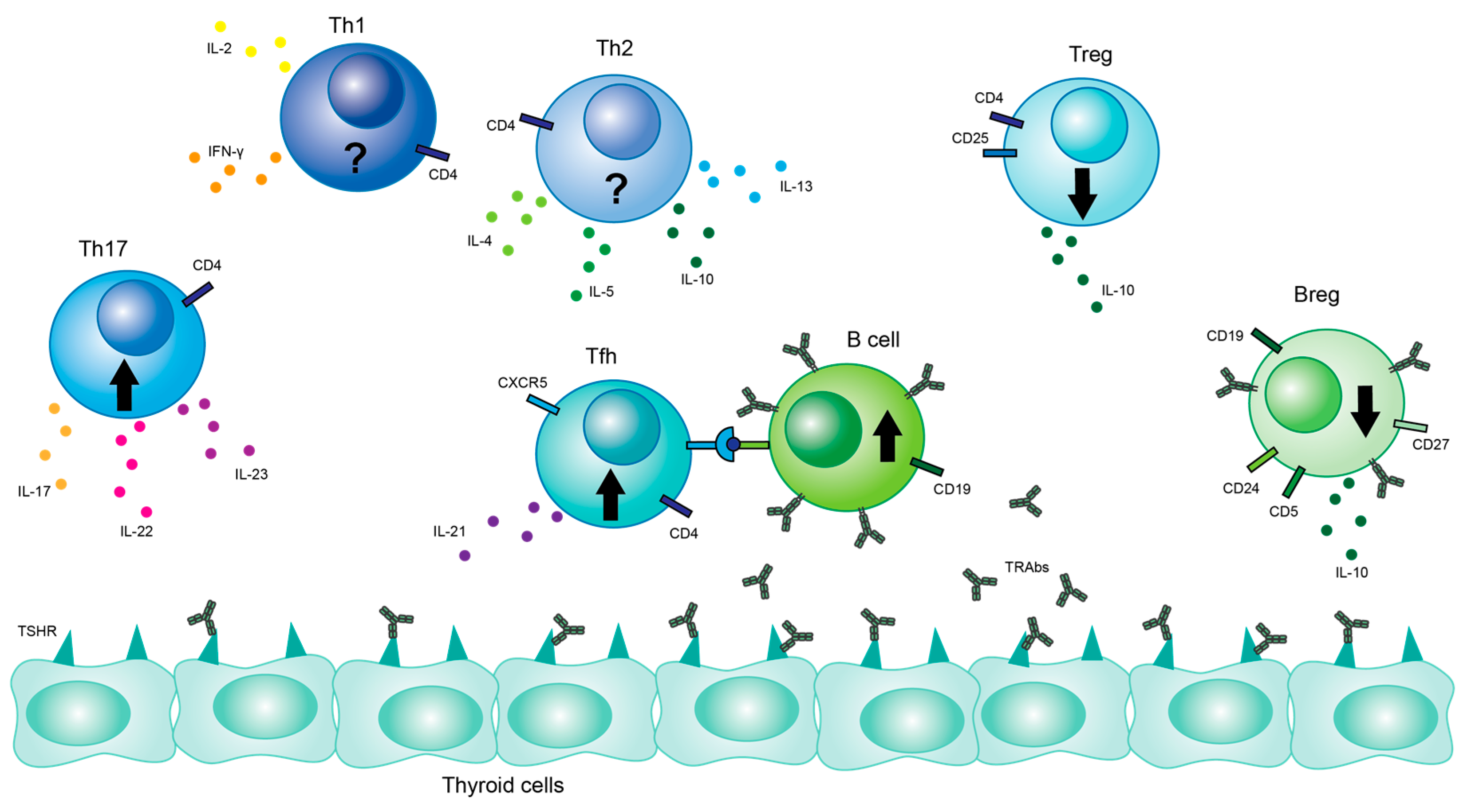
3. T Cells
3.1. Th17 Cells
3.2. Treg Cells
3.3. Th1/Th2 Cells
3.4. Tfh Cells
4. B Cells
Breg Cells
5. Immunophenotype-Based Treatments
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Smith, T.J.; Hegedüs, L. Graves’ Disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef]
- Ross, D.S.; Burch, H.B.; Cooper, D.S.; Greenlee, M.C.; Laurberg, P.; Maia, A.L.; Rivkees, S.A.; Samuels, M.; Sosa, J.A.; Stan, M.N.; et al. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid 2016, 26, 1343–1421. [Google Scholar] [CrossRef] [PubMed]
- Bartalena, L. Graves’ Disease: Complications. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bartalena, L.; Piantanida, E.; Gallo, D.; Ippolito, S.; Tanda, M.L. Management of Graves’ hyperthyroidism: Present and future. Expert Rev. Endocrinol. Metab. 2022, 17, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Subekti, I.; Pramono, L.A. Current Diagnosis and Management of Graves’ Disease. Acta Med. Indones 2018, 50, 177–182. [Google Scholar]
- Kahaly, G.J.; Bartalena, L.; Hegedüs, L.; Leenhardt, L.; Poppe, K.; Pearce, S.H. 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. Eur. Thyroid J. 2018, 7, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wang, X.; Wang, L.; Sun, X.; Tan, G.; Wei, W.; Zheng, G.; Ma, X.; Tian, D.; Yu, H. Genetics, Epigenetics, Cellular Immunology, and Gut Microbiota: Emerging Links With Graves’ Disease. Front. Cell Dev. Biol. 2021, 9, 794912. [Google Scholar] [CrossRef]
- Chu, X.; Pan, C.M.; Zhao, S.X.; Liang, J.; Gao, G.Q.; Zhang, X.M.; Yuan, G.Y.; Li, C.G.; Xue, L.Q.; Shen, M.; et al. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat. Genet. 2011, 43, 897–901. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, X.M.; Wang, X.; Sun, X.; Wang, L.J.; Li, X.Q.; Liu, X.Y.; Yu, H.S. Emerging Insights Into the Role of Epigenetics and Gut Microbiome in the Pathogenesis of Graves’ Ophthalmopathy. Front. Endocrinol. 2021, 12, 788535. [Google Scholar] [CrossRef]
- Sawicka-Gutaj, N.; Gruszczyński, D.; Zawalna, N.; Nijakowski, K.; Muller, I.; Karpiński, T.; Salvi, M.; Ruchała, M. Microbiota Alterations in Patients with Autoimmune Thyroid Diseases: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 3450. [Google Scholar] [CrossRef]
- Biscarini, F.; Masetti, G.; Muller, I.; Verhasselt, H.L.; Covelli, D.; Colucci, G.; Zhang, L.; Draman, M.S.; Okosieme, O.; Taylor, P.; et al. Gut microbiome associated with Graves’ disease and Graves’ orbitopathy: The INDIGO* multi-centre European study. J. Clin. Endocrinol. Metab. 2023. [Google Scholar] [CrossRef]
- Su, X.; Yin, X.; Liu, Y.; Yan, X.; Zhang, S.; Wang, X.; Lin, Z.; Zhou, X.; Gao, J.; Wang, Z.; et al. Gut Dysbiosis Contributes to the Imbalance of Treg and Th17 Cells in Graves’ Disease Patients by Propionic Acid. J. Clin. Endocrinol. Metab. 2020, 105, 3526–3547. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, E.; Shimokawa, C.; Steimle, A.; Desai, M.S.; Ohno, H. The impact of the gut microbiome on extra-intestinal autoimmune diseases. Nat. Rev. Immunol. 2023, 23, 9–23. [Google Scholar] [CrossRef]
- Hou, J.; Tang, Y.; Chen, Y.; Chen, D. The Role of the Microbiota in Graves’ Disease and Graves’ Orbitopathy. Front. Cell Infect. Microbiol. 2021, 11, 739707. [Google Scholar] [CrossRef] [PubMed]
- Furmaniak, J.; Sanders, J.; Núñez Miguel, R.; Rees Smith, B. Mechanisms of Action of TSHR Autoantibodies. Horm. Metab. Res. 2015, 47, 735–752. [Google Scholar] [CrossRef] [PubMed]
- Michalek, K.; Morshed, S.A.; Latif, R.; Davies, T.F. TSH receptor autoantibodies. Autoimmun. Rev. 2009, 9, 113–116. [Google Scholar] [CrossRef]
- Neumann, S.; Geras-Raaka, E.; Marcus-Samuels, B.; Gershengorn, M.C. Persistent cAMP signaling by thyrotropin (TSH) receptors is not dependent on internalization. FASEB J. 2010, 24, 3992–3999. [Google Scholar] [CrossRef]
- Morshed, S.A.; Davies, T.F. Graves’ Disease Mechanisms: The Role of Stimulating, Blocking, and Cleavage Region TSH Receptor Antibodies. Horm. Metab. Res. 2015, 47, 727–734. [Google Scholar] [CrossRef]
- Akamizu, T.; Kosugi, S.; Kohn, L.D.; Mori, T. Anti-thyrotropin (TSH) receptor antibody binding epitopes of TSH receptor: Site-directed mutagenesis approach. Nihon. Rinsho. 1994, 52, 1024–1030. [Google Scholar]
- Akamizu, T.; Ueda, Y.; Hua, L.; Okuda, J.; Mori, T. Establishment and characterization of an antihuman thyrotropin (TSH) receptor-specific CD4+ T cell line from a patient with Graves’ disease: Evidence for multiple T cell epitopes on the TSH receptor including the transmembrane domain. Thyroid 1995, 5, 259–264. [Google Scholar] [CrossRef]
- Inaba, H.; De Groot, L.J.; Akamizu, T. Thyrotropin Receptor Epitope and Human Leukocyte Antigen in Graves’ Disease. Front. Endocrinol. 2016, 7, 120. [Google Scholar] [CrossRef]
- Latif, R.; Teixeira, A.; Michalek, K.; Ali, M.R.; Schlesinger, M.; Baliram, R.; Morshed, S.A.; Davies, T.F. Antibody protection reveals extended epitopes on the human TSH receptor. PLoS ONE 2012, 7, e44669. [Google Scholar] [CrossRef]
- Janyga, S.; Marek, B.; Kajdaniuk, D.; Ogrodowczyk-Bobik, M.; Urbanek, A.; Bułdak, Ł. CD4+ cells in autoimmune thyroid disease. Endokrynol. Pol. 2021, 72, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Brummelman, J.; Pilipow, K.; Lugli, E. The Single-Cell Phenotypic Identity of Human CD8(+) and CD4(+) T Cells. Int. Rev. Cell Mol. Biol. 2018, 341, 63–124. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, A.; Kuchroo, V.K. Th17 cells: From precursors to players in inflammation and infection. Int. Immunol. 2009, 21, 489–498. [Google Scholar] [CrossRef]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef]
- Lee, Y.; Collins, M.; Kuchroo, V.K. Unexpected targets and triggers of autoimmunity. J. Clin. Immunol. 2014, 34 (Suppl. S1), S56–S60. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Zhou, J.; Fan, C.; Zhao, N.; Liu, Y.; Wang, S.; Cui, X.; Huang, M.; Guan, H.; Li, Y.; et al. Increased Circulating Th17 but Decreased CD4(+)Foxp3(+) Treg and CD19(+)CD1d(hi)CD5(+) Breg Subsets in New-Onset Graves’ Disease. Biomed. Res. Int. 2017, 2017, 8431838. [Google Scholar] [CrossRef]
- Li, C.; Yuan, J.; Zhu, Y.F.; Yang, X.J.; Wang, Q.; Xu, J.; He, S.T.; Zhang, J.A. Imbalance of Th17/Treg in Different Subtypes of Autoimmune Thyroid Diseases. Cell Physiol. Biochem. 2016, 40, 245–252. [Google Scholar] [CrossRef]
- Peng, D.; Xu, B.; Wang, Y.; Guo, H.; Jiang, Y. A high frequency of circulating th22 and th17 cells in patients with new onset graves’ disease. PLoS ONE 2013, 8, e68446. [Google Scholar] [CrossRef]
- Nanba, T.; Watanabe, M.; Inoue, N.; Iwatani, Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid 2009, 19, 495–501. [Google Scholar] [CrossRef]
- Torimoto, K.; Okada, Y.; Nakayamada, S.; Kubo, S.; Kurozumi, A.; Narisawa, M.; Tanaka, Y. Comprehensive immunophenotypic analysis reveals the pathological involvement of Th17 cells in Graves’ disease. Sci. Rep. 2022, 12, 16880. [Google Scholar] [CrossRef]
- Klatka, M.; Grywalska, E.; Partyka, M.; Charytanowicz, M.; Kiszczak-Bochynska, E.; Rolinski, J. Th17 and Treg cells in adolescents with Graves’ disease. Impact of treatment with methimazole on these cell subsets. Autoimmunity 2014, 47, 201–211. [Google Scholar] [CrossRef]
- Bossowski, A.; Moniuszko, M.; Idźkowska, E.; Grubczak, K.; Singh, P.; Bossowska, A.; Diana, T.; Kahaly, G.J. Decreased proportions of CD4 + IL17+/CD4 + CD25 + CD127- and CD4 + IL17+/CD4 + CD25 + CD127 - FoxP3+ T cells in children with autoimmune thyroid diseases. Autoimmunity 2016, 49, 320–328. [Google Scholar] [CrossRef]
- Liu, H.Y.; Shi, Z.Y.; Fan, D.; Zhang, S.X.; Wu, L.X.; Lu, K.Y.; Yang, S.Y.; Li, W.T.; Kang, J.F.; Li, C.H.; et al. Absolute reduction in peripheral regulatory T cells in patients with Graves’ disease and post-treatment recovery. Mol. Immunol. 2022, 144, 49–57. [Google Scholar] [CrossRef]
- Qin, Q.; Liu, P.; Liu, L.; Wang, R.; Yan, N.; Yang, J.; Wang, X.; Pandey, M.; Zhang, J.A. The increased but non-predominant expression of Th17- and Th1-specific cytokines in Hashimoto’s thyroiditis but not in Graves’ disease. Braz. J. Med. Biol. Res. 2012, 45, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- González-Amaro, R.; Marazuela, M. T regulatory (Treg) and T helper 17 (Th17) lymphocytes in thyroid autoimmunity. Endocrine 2016, 52, 30–38. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- Bossowski, A.; Moniuszko, M.; Dąbrowska, M.; Sawicka, B.; Rusak, M.; Jeznach, M.; Wójtowicz, J.; Bodzenta-Lukaszyk, A.; Bossowska, A. Lower proportions of CD4+CD25(high) and CD4+FoxP3, but not CD4+CD25+CD127(low) FoxP3+ T cell levels in children with autoimmune thyroid diseases. Autoimmunity 2013, 46, 222–230. [Google Scholar] [CrossRef]
- Mao, C.; Wang, S.; Xiao, Y.; Xu, J.; Jiang, Q.; Jin, M.; Jiang, X.; Guo, H.; Ning, G.; Zhang, Y. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves’ disease. J. Immunol. 2011, 186, 4734–4743. [Google Scholar] [CrossRef]
- Ji, X.; Wan, J.; Chen, R.; Wang, H.; Huang, L.; Wang, S.; Su, Z.; Xu, H. Low frequency of IL-10-producing B cells and high density of ILC2s contribute to the pathological process in Graves’ disease, which may be related to elevated-TRAb levels. Autoimmunity 2020, 53, 78–85. [Google Scholar] [CrossRef]
- Pan, D.; Shin, Y.H.; Gopalakrishnan, G.; Hennessey, J.; De Groot, L.J. Regulatory T cells in Graves’ disease. Clin. Endocrinol. 2009, 71, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, S.; Tang, X.; Li, J.; Zou, P. Changes of regulatory T cells in Graves’ disease. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Wodzinski, A.; Fu, P.; Levine, A.D.; Wald, D.N. Impairment of regulatory T-cell function in autoimmune thyroid disease. Thyroid 2013, 23, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Watanabe, M.; Iida, T.; Kuroda, S.; Matsuzuka, F.; Miyauchi, A.; Iwatani, Y. Apoptosis-induced decrease of intrathyroidal CD4(+)CD25(+) regulatory T cells in autoimmune thyroid diseases. Thyroid 2007, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, B.; Mu, K.; Zhang, J.-A. The pathogenesis of thyroid autoimmune diseases: New T lymphocytes—Cytokines circuits beyond the Th1−Th2 paradigm. J. Cell. Physiol. 2019, 234, 2204–2216. [Google Scholar] [CrossRef]
- Kocjan, T.; Wraber, B.; Repnik, U.; Hojker, S. Changes in Th1/Th2 cytokine balance in Graves’ desease. Pflügers Archiv. Eur. J. Physiol. 2000, 440, R094–R095. [Google Scholar] [CrossRef]
- Mintz, M.A.; Cyster, J.G. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 2020, 296, 48–61. [Google Scholar] [CrossRef]
- Gitlin, A.D.; Mayer, C.T.; Oliveira, T.Y.; Shulman, Z.; Jones, M.J.; Koren, A.; Nussenzweig, M.C. T cell help controls the speed of the cell cycle in germinal center B cells. Science 2015, 349, 643–646. [Google Scholar] [CrossRef]
- Zhu, C.; Ma, J.; Liu, Y.; Tong, J.; Tian, J.; Chen, J.; Tang, X.; Xu, H.; Lu, L.; Wang, S. Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J. Clin. Endocrinol. Metab. 2012, 97, 943–950. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, M.; Zeng, H.; Guo, Y.; Zhuang, Z.; Feng, Z.; Yan, H.; Xu, M.; Liang, W.; Yang, C.; et al. Elevated follicular helper T cells and expression of IL-21 in thyroid tissues are involved in the pathogenesis of Graves’ disease. Immunol. Res. 2015, 62, 163–174. [Google Scholar] [CrossRef]
- Schmitt, N.; Bentebibel, S.E.; Ueno, H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol. 2014, 35, 436–442. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, X.; Li, X.; Cui, D.; Xie, J. Constitutive Changes in Circulating Follicular Helper T Cells and Their Subsets in Patients with Graves’ Disease. J. Immunol. Res. 2018, 2018, 8972572. [Google Scholar] [CrossRef]
- Chen, J.; Tian, J.; Tang, X.; Rui, K.; Ma, J.; Mao, C.; Liu, Y.; Lu, L.; Xu, H.; Wang, S. MiR-346 regulates CD4⁺CXCR5⁺ T cells in the pathogenesis of Graves’ disease. Endocrine 2015, 49, 752–760. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, Z.; Liu, X.; Wei, L.; Li, S.; Zheng, X.; Yang, T.; Xu, X. The Frequency of Intrathyroidal Follicular Helper T Cells Varies with the Progression of Graves’ Disease and Hashimoto’s Thyroiditis. J. Immunol. Res. 2022, 2022, 4075522. [Google Scholar] [CrossRef]
- Aust, G.; Sittig, D.; Becherer, L.; Anderegg, U.; Schütz, A.; Lamesch, P.; Schmücking, E. The role of CXCR5 and its ligand CXCL13 in the compartmentalization of lymphocytes in thyroids affected by autoimmune thyroid diseases. Eur. J. Endocrinol. 2004, 150, 225–234. [Google Scholar] [CrossRef]
- Zhang, J.; Zeng, H.; Ren, M.; Yan, H.; Xu, M.; Feng, Z.; Liang, W.; Yang, C.; Cheng, H.; Ding, H.; et al. Interleukin-21 is associated with disease activity in patients with Graves’ disease. Endocrine 2014, 46, 539–548. [Google Scholar] [CrossRef]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, Y.; Tang, S.; Zhang, L.; Huang, Z.; Shi, X.; Fang, Y.; Yang, J.; Deng, X.; Wang, L.; et al. Aberrant expression of inhibitory receptors on B cells in patients with Graves’ disease. Hum. Immunol. 2022, 83, 144–152. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Hu, F. Double-negative (DN) B cells: An under-recognized effector memory B cell subset in autoimmunity. Clin. Exp. Immunol. 2021, 205, 119–127. [Google Scholar] [CrossRef]
- Racine, R.; Chatterjee, M.; Winslow, G.M. CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J. Immunol. 2008, 181, 1375–1385. [Google Scholar] [CrossRef]
- Rincon-Arevalo, H.; Wiedemann, A.; Stefanski, A.L.; Lettau, M.; Szelinski, F.; Fuchs, S.; Frei, A.P.; Steinberg, M.; Kam-Thong, T.; Hatje, K.; et al. Deep Phenotyping of CD11c(+) B Cells in Systemic Autoimmunity and Controls. Front. Immunol. 2021, 12, 635615. [Google Scholar] [CrossRef]
- Cao, Y.; Zhao, X.; You, R.; Zhang, Y.; Qu, C.; Huang, Y.; Yu, Y.; Gong, Y.; Cong, T.; Zhao, E.; et al. CD11c+ B Cells Participate in the Pathogenesis of Graves’ Disease by Secreting Thyroid Autoantibodies and Cytokines. Front. Immunol. 2022, 13, 836347. [Google Scholar] [CrossRef]
- Bossowski, A.; Urban, M.; Stasiak-Barmuta, A. Analysis of Changes in the Percentage of Β (CD19) and Τ (CD3) Lymphocytes, Subsets CD4, CD8 and their Memory (CD45RO), and Naive (CD45RA) Τ Cells in Children with Immune and Non-immune Thyroid Diseases. J. Pediatr. Endocrinol. Metab. 2003, 16, 63–70. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Grubczak, K.; Starosz, A.; Stożek, K.; Bossowski, F.; Moniuszko, M.; Bossowski, A. Regulatory B Cells Involvement in Autoimmune Phenomena Occurring in Pediatric Graves’ Disease Patients. Int. J. Mol. Sci. 2021, 22, 926. [Google Scholar] [CrossRef]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef]
- Burgueño-Bucio, E.; Mier-Aguilar, C.A.; Soldevila, G. The multiple faces of CD5. J. Leukoc. Biol. 2019, 105, 891–904. [Google Scholar] [CrossRef]
- Stożek, K.; Grubczak, K.; Marolda, V.; Eljaszewicz, A.; Moniuszko, M.; Bossowski, A. Lower proportion of CD19(+)IL-10(+) and CD19(+)CD24(+)CD27(+) but not CD1d(+)CD5(+)CD19(+)CD24(+)CD27(+) IL-10(+) B cells in children with autoimmune thyroid diseases. Autoimmunity 2020, 53, 46–55. [Google Scholar] [CrossRef]
- Zha, B.; Wang, L.; Liu, X.; Liu, J.; Chen, Z.; Xu, J.; Sheng, L.; Li, Y.; Chu, Y. Decrease in proportion of CD19+ CD24(hi) CD27+ B cells and impairment of their suppressive function in Graves’ disease. PLoS ONE 2012, 7, e49835. [Google Scholar] [CrossRef]
- Morandi, F.; Airoldi, I.; Marimpietri, D.; Bracci, C.; Faini, A.C.; Gramignoli, R. CD38, a Receptor with Multifunctional Activities: From Modulatory Functions on Regulatory Cell Subsets and Extracellular Vesicles, to a Target for Therapeutic Strategies. Cells 2019, 8, 1527. [Google Scholar] [CrossRef]
- Rydzewska, M.; Jaromin, M.; Pasierowska, I.E.; Stożek, K.; Bossowski, A. Role of the T and B lymphocytes in pathogenesis of autoimmune thyroid diseases. Thyroid Res. 2018, 11, 2. [Google Scholar] [CrossRef]
- El Fassi, D.; Nielsen, C.H.; Bonnema, S.J.; Hasselbalch, H.C.; Hegedüs, L. B lymphocyte depletion with the monoclonal antibody rituximab in Graves’ disease: A controlled pilot study. J. Clin. Endocrinol. Metab. 2007, 92, 1769–1772. [Google Scholar] [CrossRef]
- Heemstra, K.A.; Toes, R.E.; Sepers, J.; Pereira, A.M.; Corssmit, E.P.; Huizinga, T.W.J.; Romijn, J.A.; Smit, J.W. Rituximab in relapsing Graves’ disease, a phase II study. Eur. J. Endocrinol. 2008, 159, 609–615. [Google Scholar] [CrossRef]
- Ristov, J.; Espie, P.; Ulrich, P.; Sickert, D.; Flandre, T.; Dimitrova, M.; Müller-Ristig, D.; Weider, D.; Robert, G.; Schmutz, P.; et al. Characterization of the in vitro and in vivo properties of CFZ533, a blocking and non-depleting anti-CD40 monoclonal antibody. Am. J. Transplant. 2018, 18, 2895–2904. [Google Scholar] [CrossRef]
- Cheetham, T.D.; Cole, M.; Abinun, M.; Allahabadia, A.; Barratt, T.; Davies, J.H.; Dimitri, P.; Drake, A.; Mohamed, Z.; Murray, R.D.; et al. Adjuvant Rituximab—Exploratory Trial in Young People with Graves Disease. J. Clin. Endocrinol. Metab. 2021, 107, 743–754. [Google Scholar] [CrossRef]
- Furmaniak, J.; Sanders, J.; Sanders, P.; Miller-Gallacher, J.; Ryder, M.M.; Rees Smith, B. Practical applications of studies on the TSH receptor and TSH receptor autoantibodies. Endocrine 2020, 68, 261–264. [Google Scholar] [CrossRef]
- Jansson, L.; Vrolix, K.; Jahraus, A.; Martin, K.F.; Wraith, D.C. Immunotherapy with Apitopes Blocks the Immune Response to TSH Receptor in HLA-DR Transgenic Mice. Endocrinology 2018, 159, 3446–3457. [Google Scholar] [CrossRef]
- Pearce, S.H.S.; Dayan, C.; Wraith, D.C.; Barrell, K.; Olive, N.; Jansson, L.; Walker-Smith, T.; Carnegie, C.; Martin, K.F.; Boelaert, K.; et al. Antigen-Specific Immunotherapy with Thyrotropin Receptor Peptides in Graves’ Hyperthyroidism: A Phase I Study. Thyroid 2019, 29, 1003–1011. [Google Scholar] [CrossRef]
- Furmaniak, J.; Sanders, J.; Young, S.; Kabelis, K.; Sanders, P.; Evans, M.; Clark, J.; Wilmot, J.; Rees Smith, B. In vivo effects of a human thyroid-stimulating monoclonal autoantibody (M22) and a human thyroid-blocking autoantibody (K1-70). Auto Immun. Highlights 2012, 3, 19–25. [Google Scholar] [CrossRef]
- Furmaniak, J.; Sanders, J.; Clark, J.; Wilmot, J.; Sanders, P.; Li, Y.; Rees Smith, B. Preclinical studies on the toxicology, pharmacokinetics and safety of K1-70TM a human monoclonal autoantibody to the TSH receptor with TSH antagonist activity. Auto Immun. Highlights 2019, 10, 11. [Google Scholar] [CrossRef]
- Furmaniak, J.; Sanders, J.; Sanders, P.; Li, Y.; Rees Smith, B. TSH receptor specific monoclonal autoantibody K1-70(TM) targeting of the TSH receptor in subjects with Graves’ disease and Graves’ orbitopathy-Results from a phase I clinical trial. Clin. Endocrinol. 2022, 96, 878–887. [Google Scholar] [CrossRef]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytom. A 2014, 85, 36–42. [Google Scholar] [CrossRef]
- Andersson, A.K.; Feldmann, M.; Brennan, F.M. Neutralizing IL-21 and IL-15 inhibits pro-inflammatory cytokine production in rheumatoid arthritis. Scand. J. Immunol. 2008, 68, 103–111. [Google Scholar] [CrossRef]
- Bubier, J.A.; Bennett, S.M.; Sproule, T.J.; Lyons, B.L.; Olland, S.; Young, D.A.; Roopenian, D.C. Treatment of BXSB-Yaa mice with IL-21R-Fc fusion protein minimally attenuates systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2007, 1110, 590–601. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | Target | Status | NCT Number |
|---|---|---|---|
| Rituximab | CD20+ B cells | Phase II Trials | NCT00150111 |
| Iscalimab | CD40-CD154 B cell signaling pathway | Phase II Trials | NCT02713256 |
| ATX-GD-59 | Activation of APCs | Phase I Trials | NCT02973802 |
| K1-70™ | TSHR | Phase I Trials | NCT02904330 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, M.; Cheever, A.; Weber, K.S.; O’Neill, K.L. Characterizing the Interplay of Lymphocytes in Graves’ Disease. Int. J. Mol. Sci. 2023, 24, 6835. https://doi.org/10.3390/ijms24076835
Hansen M, Cheever A, Weber KS, O’Neill KL. Characterizing the Interplay of Lymphocytes in Graves’ Disease. International Journal of Molecular Sciences. 2023; 24(7):6835. https://doi.org/10.3390/ijms24076835
Chicago/Turabian StyleHansen, Mackenzie, Abigail Cheever, K. Scott Weber, and Kim L. O’Neill. 2023. "Characterizing the Interplay of Lymphocytes in Graves’ Disease" International Journal of Molecular Sciences 24, no. 7: 6835. https://doi.org/10.3390/ijms24076835
APA StyleHansen, M., Cheever, A., Weber, K. S., & O’Neill, K. L. (2023). Characterizing the Interplay of Lymphocytes in Graves’ Disease. International Journal of Molecular Sciences, 24(7), 6835. https://doi.org/10.3390/ijms24076835