
Iron Deficiency and Iron Deficiency Anemia: Potential Risk Factors in Bone Loss


Abstract
:1. Introduction
2. Iron Deficiency and Iron Deficiency Anemia
2.1. Epidemiology
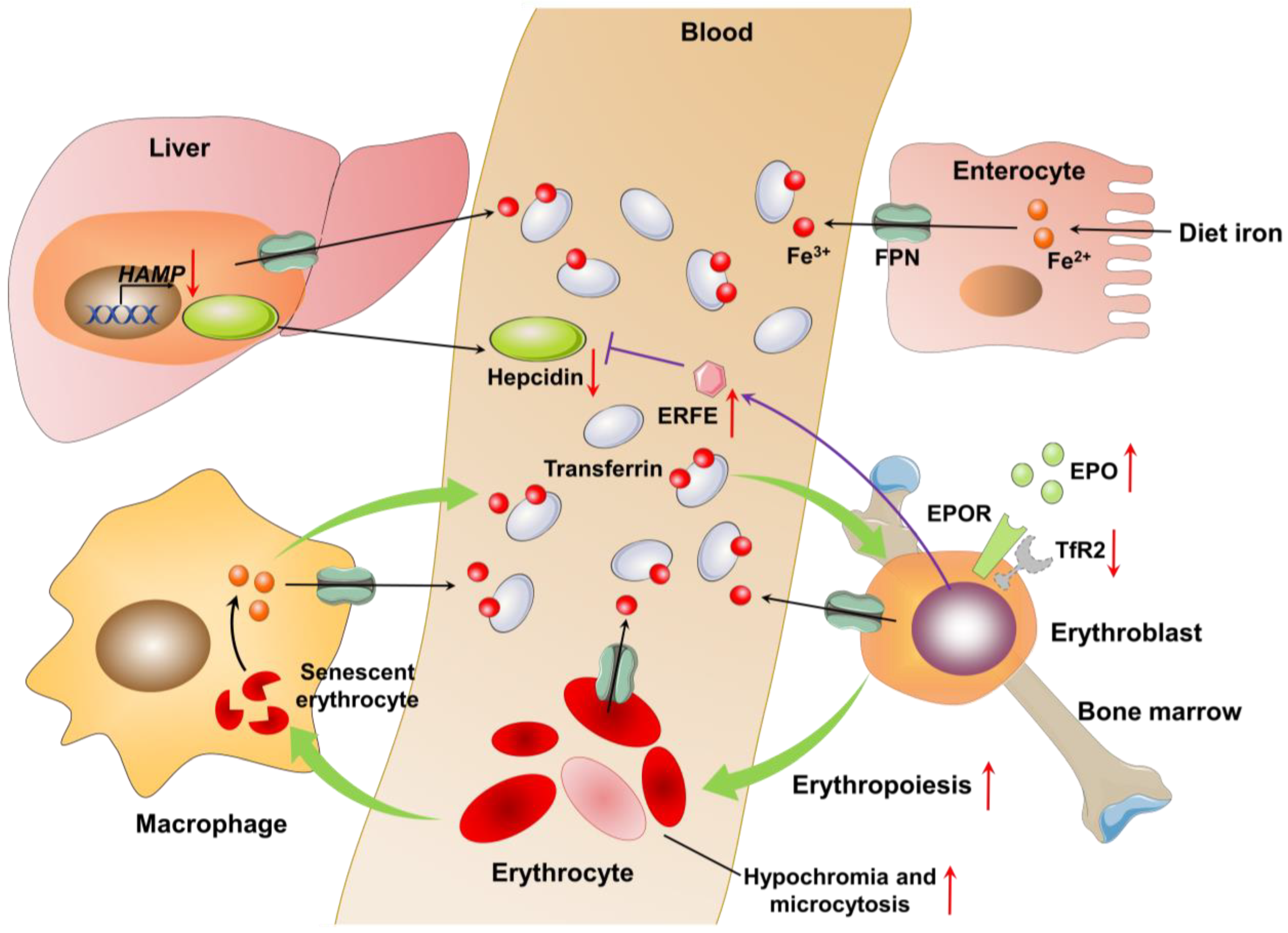
2.2. Pathophysiology
2.3. Diagnosis of Iron Deficiency and Iron Deficiency Anemia
3. Iron Deficiency and Bone Loss
3.1. Dietary Iron Deficiency and Bone Loss
3.2. Iron Deficiency Anemia and Bone Loss
3.3. Iron Deficiency Anemia in Chronic Kidney Disease Complicated by Osteoporosis
4. The Potential Mechanism of Bone Loss Induced by Iron Deficiency
4.1. Physiological Iron in Bone Homeostasis
4.2. Iron-Deficiency-Induced Hypoxia in Bone Homeostasis
4.3. Iron in Collagen Synthesis
4.4. Iron in Vitamin D Metabolism
5. Prevention of Iron Deficiency
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasricha, S.-R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.; Cacoub, P.; Macdougall, I.C.; Peyrin-Biroulet, L. Iron deficiency anaemia. Lancet 2016, 387, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Pinto, V.M.; Forni, G.L. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. Int. J. Mol. Sci. 2020, 21, 8771. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Violet, P.-C.; George, N.A.; Ali, F.; Bhanvadia, S.; Wong, R.; Tisdale, J.F.; Fitzhugh, C.; Levine, M.; Thein, S.L.; et al. Dietary iron restriction improves markers of disease severity in murine sickle cell anemia. Blood 2021, 137, 1553–1555. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.; Perapoch Amadó, M.; Moore, S.E. The Role of Iron in Brain Development: A Systematic Review. Nutrients 2020, 12, 2001. [Google Scholar] [CrossRef]
- Nairz, M.; Weiss, G. Iron in infection and immunity. Mol. Asp. Med. 2020, 75, 100864. [Google Scholar] [CrossRef]
- Houston, B.L.; Hurrie, D.; Graham, J.; Perija, B.; Rimmer, E.; Rabbani, R.; Bernstein, C.N.; Turgeon, A.F.; Fergusson, D.A.; Houston, D.S.; et al. Efficacy of iron supplementation on fatigue and physical capacity in non-anaemic iron-deficient adults: A systematic review of randomised controlled trials. BMJ Open 2018, 8, e019240. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.L.; Chen, L.R.; Tsao, H.M.; Chen, K.H. Iron Deficiency Anemia as a Risk Factor for Osteoporosis in Taiwan: A Nationwide Population-Based Study. Nutrients 2017, 9, 616. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.A.; Shin, D.W.; Yoo, J.H.; Ko, H.Y.; Jeong, S.M. Anemia and Risk of Fractures in Older Korean Adults: A Nationwide Population-Based Study. J. Bone Miner. Res. 2019, 34, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Walton, J.; Russell, L.; Wolman, R.; Wardley-Smith, B.; Green, J.R.; Mitchell, A.; Reeve, J. Dietary determinants of post-menopausal bone loss at the lumbar spine: A possible beneficial effect of iron. Osteoporos. Int. 2006, 17, 1165–1173. [Google Scholar] [CrossRef]
- WHO. Anaemia in Children. 2021. Available online: https://apps.who.int/gho/data/view.main.ANAEMIACHILDRENREGv?lang=en (accessed on 15 February 2023).
- WHO. Anaemia Women of Reproductive Age. 2021. Available online: https://apps.who.int/gho/data/view.main.ANAEMIAWOMENREPRODUCTIVEREGv?lang=en (accessed on 15 February 2023).
- WHO. Anaemia in Pregnant Women. 2021. Available online: https://apps.who.int/gho/data/view.main.ANAEMIAWOMENPWREGv?lang=en (accessed on 15 February 2023).
- WHO. The Global Prevalence of Anaemia in 2011. 2015. Available online: https://www.who.int/publications/i/item/9789241564960 (accessed on 15 February 2023).
- Petry, N.; Olofin, I.; Hurrell, R.F.; Boy, E.; Wirth, J.P.; Moursi, M.; Donahue Angel, M.; Rohner, F. The Proportion of Anemia Associated with Iron Deficiency in Low, Medium, and High Human Development Index Countries: A Systematic Analysis of National Surveys. Nutrients 2016, 8, 693. [Google Scholar] [CrossRef]
- Namaste, S.M.; Rohner, F.; Huang, J.; Bhushan, N.L.; Flores-Ayala, R.; Kupka, R.; Mei, Z.; Rawat, R.; Williams, A.M.; Raiten, D.J.; et al. Adjusting ferritin concentrations for inflammation: Biomarkers Reflecting Inflammation and Nutritional Determinants of Anemia (BRINDA) project. Am. J. Clin. Nutr. 2017, 106, 359s–371s. [Google Scholar] [CrossRef]
- Mei, Z.; Cogswell, M.E.; Looker, A.C.; Pfeiffer, C.M.; Cusick, S.E.; Lacher, D.A.; Grummer-Strawn, L.M. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am. J. Clin. Nutr. 2011, 93, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Pottie, K.; Greenaway, C.; Feightner, J.; Welch, V.; Swinkels, H.; Rashid, M.; Narasiah, L.; Kirmayer, L.J.; Ueffing, E.; MacDonald, N.E.; et al. Evidence-based clinical guidelines for immigrants and refugees. CMAJ 2011, 183, E824–E925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balarajan, Y.; Ramakrishnan, U.; Özaltin, E.; Shankar, A.H.; Subramanian, S.V. Anaemia in low-income and middle-income countries. Lancet 2011, 378, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tang, Q.; Zeng, Y. Melatonin: Potential avenue for treating iron overload disorders. Ageing Res. Rev. 2022, 81, 101717. [Google Scholar] [CrossRef] [PubMed]
- Pagani, A.; Nai, A.; Silvestri, L.; Camaschella, C. Hepcidin and Anemia: A Tight Relationship. Front. Physiol. 2019, 10, 1294. [Google Scholar] [CrossRef]
- Nai, A.; Lidonnici, M.R.; Rausa, M.; Mandelli, G.; Pagani, A.; Silvestri, L.; Ferrari, G.; Camaschella, C. The second transferrin receptor regulates red blood cell production in mice. Blood 2015, 125, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.L.; Senecal, T.; Ghosh, M.C.; Ollivierre-Wilson, H.; Tu, T.; Rouault, T.A. Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts. Blood 2011, 118, 2868–2877. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.C.; Lesperance, L.; Bernstein, H. Screening for iron deficiency. Pediatr. Rev. 2002, 23, 171–178. [Google Scholar] [CrossRef]
- Toxqui, L.; Vaquero, M.P. Chronic iron deficiency as an emerging risk factor for osteoporosis: A hypothesis. Nutrients 2015, 7, 2324–2344. [Google Scholar] [CrossRef] [Green Version]
- Turawa, E.; Awotiwon, O.; Dhansay, M.A.; Cois, A.; Labadarios, D.; Bradshaw, D.; Pillay-van Wyk, V. Prevalence of Anaemia, Iron Deficiency, and Iron Deficiency Anaemia in Women of Reproductive Age and Children under 5 Years of Age in South Africa (1997–2021): A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 12799. [Google Scholar] [CrossRef]
- Medeiros, D.M.; Plattner, A.; Jennings, D.; Stoecker, B. Bone morphology, strength and density are compromised in iron-deficient rats and exacerbated by calcium restriction. J. Nutr. 2002, 132, 3135–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, D.M.; Stoecker, B.; Plattner, A.; Jennings, D.; Haub, M. Iron deficiency negatively affects vertebrae and femurs of rats independently of energy intake and body weight. J. Nutr. 2004, 134, 3061–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parelman, M.; Stoecker, B.; Baker, A.; Medeiros, D. Iron restriction negatively affects bone in female rats and mineralization of hFOB osteoblast cells. Exp. Biol. Med. 2006, 231, 378–386. [Google Scholar] [CrossRef]
- Lobo, A.R.; Gaievski, E.H.S.; Colli, C. Hemoglobin Regeneration Efficiency in Anemic Rats: Effects on Bone Mineral Composition and Biomechanical Properties. Biol. Trace Elem. Res. 2011, 143, 403–411. [Google Scholar] [CrossRef]
- Harris, M.M.; Houtkooper, L.B.; Stanford, V.A.; Parkhill, C.; Weber, J.L.; Flint-Wagner, H.; Weiss, L.; Going, S.B.; Lohman, T. Dietary iron is associated with bone mineral density in healthy postmenopausal women. J. Nutr. 2003, 133, 3598–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, J.; Harris, M.M.; Stanford, V.A.; Lohman, T.G.; Cussler, E.; Going, S.B.; Houtkooper, L.B. Dietary iron positively influences bone mineral density in postmenopausal women on hormone replacement therapy. J. Nutr. 2005, 135, 863–869. [Google Scholar] [CrossRef] [Green Version]
- Toxqui, L.; Perez-Granados, A.M.; Blanco-Rojo, R.; Wright, I.; de la Piedra, C.; Vaquero, M.P. Low iron status as a factor of increased bone resorption and effects of an iron and vitamin D-fortified skimmed milk on bone remodelling in young Spanish women. Eur. J. Nutr. 2014, 53, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Jang, J.S.; Lee, D.R.; Kim, Y.H.; Nam, G.E.; Han, B.D.; Do Han, K.; Cho, K.H.; Kim, S.M.; Choi, Y.S.; et al. Serum ferritin levels are positively associated with bone mineral density in elderly Korean men: The 2008-2010 Korea National Health and Nutrition Examination Surveys. J. Bone Miner. Metab. 2014, 32, 683–690. [Google Scholar] [CrossRef]
- Cesari, M.; Pahor, M.; Lauretani, F.; Penninx, B.W.; Bartali, B.; Russo, R.; Cherubini, A.; Woodman, R.; Bandinelli, S.; Guralnik, J.M.; et al. Bone density and hemoglobin levels in older persons: Results from the InCHIANTI study. Osteoporos. Int. 2005, 16, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Laudisio, A.; Marzetti, E.; Pagano, F.; Bernabei, R.; Zuccala, G. Haemoglobin levels are associated with bone mineral density in the elderly: A population-based study. Clin. Rheumatol. 2009, 28, 145–151. [Google Scholar] [CrossRef]
- Korkmaz, U.; Korkmaz, N.; Yazici, S.; Erkan, M.; Baki, A.E.; Yazici, M.; Ozhan, H.; Ataoğlu, S. Anemia as a risk factor for low bone mineral density in postmenopausal Turkish women. Eur. J. Intern. Med. 2012, 23, 154–158. [Google Scholar] [CrossRef]
- Onal, E.D.; Usluogulları, A. Anemia and osteoporosis: Causal association or epiphenomenon? Eur. J. Intern. Med. 2012, 23, e117. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Teng, Z.; Xu, S.; Zhang, X.; Liu, J.; Yue, Q.; Zhu, Y.; Zeng, Y. The Analysis for Anemia Increasing Fracture Risk. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e925707. [Google Scholar] [CrossRef]
- Jørgensen, L.; Skjelbakken, T.; Løchen, M.L.; Ahmed, L.; Bjørnerem, A.; Joakimsen, R.; Jacobsen, B.K. Anemia and the risk of non-vertebral fractures: The Tromsø Study. Osteoporos. Int. 2010, 21, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Looker, A.C. Hemoglobin and hip fracture risk in older non-Hispanic white adults. Osteoporos. Int. 2014, 25, 2389–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliger, A.S.; Foley, R.N.; Goldfarb, D.S.; Goldstein, S.L.; Johansen, K.; Singh, A.; Szczech, L. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for Anemia in CKD. Am. J. Kidney Dis. 2013, 62, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarlane, S.I.; Chen, S.C.; Whaley-Connell, A.T.; Sowers, J.R.; Vassalotti, J.A.; Salifu, M.O.; Li, S.; Wang, C.; Bakris, G.; McCullough, P.A.; et al. Prevalence and associations of anemia of CKD: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am. J. Kidney Dis. 2008, 51, S46–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awan, A.A.; Walther, C.P.; Richardson, P.A.; Shah, M.; Winkelmayer, W.C.; Navaneethan, S.D. Prevalence, correlates and outcomes of absolute and functional iron deficiency anemia in nondialysis-dependent chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 129–136. [Google Scholar] [CrossRef]
- Batchelor, E.K.; Kapitsinou, P.; Pergola, P.E.; Kovesdy, C.P.; Jalal, D.I. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. J. Am. Soc. Nephrol. 2020, 31, 456–468. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Mechanisms of Anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Lee, T.C.; Montez-Rath, M.E.; Paik, J.; Chertow, G.M.; Desai, M.; Winkelmayer, W.C. Trends in acute nonvariceal upper gastrointestinal bleeding in dialysis patients. J. Am. Soc. Nephrol. 2012, 23, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Baaten, C.; Schröer, J.R.; Floege, J.; Marx, N.; Jankowski, J.; Berger, M.; Noels, H. Platelet Abnormalities in CKD and Their Implications for Antiplatelet Therapy. Clin. J. Am. Soc. Nephrol. 2022, 17, 155–170. [Google Scholar] [CrossRef]
- Liang, C.C.; Wang, S.M.; Kuo, H.L.; Chang, C.T.; Liu, J.H.; Lin, H.H.; Wang, I.K.; Yang, Y.F.; Lu, Y.J.; Chou, C.Y.; et al. Upper gastrointestinal bleeding in patients with CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Besarab, A.; Coyne, D.W. Iron supplementation to treat anemia in patients with chronic kidney disease. Nat. Rev. Nephrol. 2010, 6, 699–710. [Google Scholar] [CrossRef]
- Malyszko, J.; Malyszko, J.S.; Matuszkiewicz-Rowinska, J. Hepcidin as a therapeutic target for anemia and inflammation associated with chronic kidney disease. Expert Opin. Ther. Targets 2019, 23, 407–421. [Google Scholar] [CrossRef]
- van Swelm, R.P.L.; Wetzels, J.F.M.; Swinkels, D.W. The multifaceted role of iron in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef]
- Najar, M.S.; Mir, M.M.; Muzamil, M. Prevalence of osteoporosis in patients with chronic kidney disease (stages 3–5) in comparison with age- and sex-matched controls: A study from Kashmir Valley Tertiary Care Center. Saudi J. Kidney Dis. Transpl. 2017, 28, 538–544. [Google Scholar] [PubMed]
- Abdalbary, M.; Sobh, M.; Elnagar, S.; Elhadedy, M.A.; Elshabrawy, N.; Abdelsalam, M.; Asadipooya, K.; Sabry, A.; Halawa, A.; El-Husseini, A. Management of osteoporosis in patients with chronic kidney disease. Osteoporos. Int. 2022, 33, 2259–2274. [Google Scholar] [CrossRef]
- Sidibé, A.; Auguste, D.; Desbiens, L.C.; Fortier, C.; Wang, Y.P.; Jean, S.; Moore, L.; Mac-Way, F. Fracture Risk in Dialysis and Kidney Transplanted Patients: A Systematic Review. JBMR Plus 2019, 3, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-Y.; Chen, L.-R.; Chen, K.-H. Osteoporosis in Patients with Chronic Kidney Diseases: A Systemic Review. Int. J. Mol. Sci. 2020, 21, 6846. [Google Scholar] [CrossRef] [PubMed]
- Ensrud, K.E.; Lui, L.Y.; Taylor, B.C.; Ishani, A.; Shlipak, M.G.; Stone, K.L.; Cauley, J.A.; Jamal, S.A.; Antoniucci, D.M.; Cummings, S.R. Renal function and risk of hip and vertebral fractures in older women. Arch. Intern. Med. 2007, 167, 133–139. [Google Scholar] [CrossRef]
- Jadoul, M.; Albert, J.M.; Akiba, T.; Akizawa, T.; Arab, L.; Bragg-Gresham, J.L.; Mason, N.; Prutz, K.G.; Young, E.W.; Pisoni, R.L. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2006, 70, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- Mittalhenkle, A.; Gillen, D.L.; Stehman-Breen, C.O. Increased risk of mortality associated with hip fracture in the dialysis population. Am. J. Kidney Dis. 2004, 44, 672–679. [Google Scholar] [CrossRef]
- Pazianas, M.; Miller, P.D. Osteoporosis and Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD): Back to Basics. Am. J. Kidney Dis. 2021, 78, 582–589. [Google Scholar] [CrossRef]
- Patino, E.; Doty, S.B.; Bhatia, D.; Meza, K.; Zhu, Y.S.; Rivella, S.; Choi, M.E.; Akchurin, O. Carbonyl iron and iron dextran therapies cause adverse effects on bone health in juveniles with chronic kidney disease. Kidney Int. 2020, 98, 1210–1224. [Google Scholar] [CrossRef]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Rivella, S. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 2017, 16, 400–423. [Google Scholar] [CrossRef] [Green Version]
- Langdahl, B.; Ferrari, S.; Dempster, D.W. Bone modeling and remodeling: Potential as therapeutic targets for the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 2016, 8, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Kubatzky, K.F.; Uhle, F.; Eigenbrod, T. From macrophage to osteoclast - How metabolism determines function and activity. Cytokine 2018, 112, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [PubMed] [Green Version]
- McDonald, M.M.; Khoo, W.H.; Ng, P.Y.; Xiao, Y.; Zamerli, J.; Thatcher, P.; Kyaw, W.; Pathmanandavel, K.; Grootveld, A.K.; Moran, I.; et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 2021, 184, 1330–1347. [Google Scholar] [CrossRef]
- Matic, I.; Matthews, B.G.; Wang, X.; Dyment, N.A.; Worthley, D.L.; Rowe, D.W.; Grcevic, D.; Kalajzic, I. Quiescent Bone Lining Cells Are a Major Source of Osteoblasts During Adulthood. Stem Cells 2016, 34, 2930–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef] [PubMed]
- Eghbali-Fatourechi, G.Z.; Lamsam, J.; Fraser, D.; Nagel, D.; Riggs, B.L.; Khosla, S. Circulating osteoblast-lineage cells in humans. N. Engl. J. Med. 2005, 352, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wei, W.; Yang, M.; Du, Y.; Wan, Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014, 20, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.A.; Fumoto, T.; Iwai, K.; Takeshita, S.; Ito, M.; Shimohata, N.; Aburatani, H.; Taketani, S.; Lelliott, C.J.; Vidal-Puig, A.; et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat. Med. 2009, 15, 259–266. [Google Scholar] [CrossRef]
- Agidigbi, T.S.; Kim, C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int. J. Mol. Sci. 2019, 20, 3576. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and copper in mitochondrial diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, W.; Ding, C.; Yao, G.; Zhao, H.; Wu, S. Deferoxamine inhibits iron-uptake stimulated osteoclast differentiation by suppressing electron transport chain and MAPKs signaling. Toxicol. Lett. 2019, 313, 50–59. [Google Scholar] [CrossRef]
- Guo, J.P.; Pan, J.X.; Xiong, L.; Xia, W.F.; Cui, S.; Xiong, W.C. Iron Chelation Inhibits Osteoclastic Differentiation In Vitro and in Tg2576 Mouse Model of Alzheimer’s Disease. PLoS ONE 2015, 10, e0139395. [Google Scholar] [CrossRef]
- Eastell, R.; Szulc, P. Use of bone turnover markers in postmenopausal osteoporosis. Lancet Diabetes Endocrinol. 2017, 5, 908–923. [Google Scholar] [CrossRef]
- Diaz-Castro, J.; Lopez-Frias, M.R.; Campos, M.S.; Lopez-Frias, M.; Alferez, M.J.; Nestares, T.; Ojeda, M.L.; Lopez-Aliaga, I. Severe nutritional iron-deficiency anaemia has a negative effect on some bone turnover biomarkers in rats. Eur. J. Nutr. 2012, 51, 241–247. [Google Scholar] [CrossRef]
- Shum, L.C.; White, N.S.; Mills, B.N.; Bentley, K.L.; Eliseev, R.A. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells Dev. 2016, 25, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messer, J.G.; Cooney, P.T.; Kipp, D.E. Iron chelator deferoxamine alters iron-regulatory genes and proteins and suppresses osteoblast phenotype in fetal rat calvaria cells. Bone 2010, 46, 1408–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.Y.; Zhao, L.P.; He, Y.F.; Li, G.F.; Gao, C.; Li, K.; Xu, Y.J. A comparison of the biological activities of human osteoblast hFOB1.19 between iron excess and iron deficiency. Biol. Trace Elem. Res. 2012, 150, 487–495. [Google Scholar] [CrossRef]
- Edwards, D.F., 3rd; Miller, C.J.; Quintana-Martinez, A.; Wright, C.S.; Prideaux, M.; Atkins, G.J.; Thompson, W.R.; Clinkenbeard, E.L. Differential Iron Requirements for Osteoblast and Adipocyte Differentiation. JBMR Plus 2021, 5, e10529. [Google Scholar] [CrossRef]
- Bo, L.; Liu, Z.; Zhong, Y.; Huang, J.; Chen, B.; Wang, H.; Xu, Y. Iron deficiency anemia’s effect on bone formation in zebrafish mutant. Biochem. Biophys. Res. Commun. 2016, 475, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, S.; Tsuboi, R.; Uehara, M.; Suzuki, K. Dietary iron deficiency decreases serum osteocalcin concentration and bone mineral density in rats. Biosci. Biotechnol. Biochem. 2006, 70, 2547–2550. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Castro, J.; Ramirez Lopez-Frias, M.; Campos, M.S.; Lopez-Frias, M.; Alferez, M.J.; Nestares, T.; Ortega, E.; Lopez-Aliaga, I. Goat milk during iron repletion improves bone turnover impaired by severe iron deficiency. J. Dairy Sci. 2011, 94, 2752–2761. [Google Scholar] [CrossRef] [Green Version]
- Balogh, E.; Paragh, G.; Jeney, V. Influence of Iron on Bone Homeostasis. Pharmaceuticals 2018, 11, 107. [Google Scholar] [CrossRef] [Green Version]
- Ledesma-Colunga, M.G.; Weidner, H.; Vujic Spasic, M.; Hofbauer, L.C.; Baschant, U.; Rauner, M. Shaping the bone through iron and iron-related proteins. Semin. Hematol. 2021, 58, 188–200. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Liu, Y.; Yang, Z.; Aimaijiang, M.; Ma, R.; Yang, Y.; Zhang, Y.; Zhou, Y. Hypoxia-Inducible Factors Signaling in Osteogenesis and Skeletal Repair. Int. J. Mol. Sci. 2022, 23, 11201. [Google Scholar] [CrossRef]
- Mendoza, S.V.; Genetos, D.; Yellowley, C. Hypoxia inducible factor-2α signaling in the skeletal system. JBMR Plus 2023, e10733. [Google Scholar] [CrossRef]
- Hiraga, T. Hypoxic Microenvironment and Metastatic Bone Disease. Int. J. Mol. Sci. 2018, 19, 3523. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Gilbert, S.R.; Wang, Y.; Cao, X.; Shen, X.; Ramaswamy, G.; Jacobsen, K.A.; Alaql, Z.S.; Eberhardt, A.W.; Gerstenfeld, L.C.; et al. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc. Natl. Acad. Sci. USA 2008, 105, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 2007, 117, 1616–1626. [Google Scholar] [CrossRef]
- Shomento, S.H.; Wan, C.; Cao, X.; Faugere, M.C.; Bouxsein, M.L.; Clemens, T.L.; Riddle, R.C. Hypoxia-inducible factors 1alpha and 2alpha exert both distinct and overlapping functions in long bone development. J. Cell. Biochem. 2010, 109, 196–204. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Rankin, E.B.; Castellini, L.; Alcudia, J.F.; LaGory, E.L.; Andersen, R.; Rhodes, S.D.; Wilson, T.L.; Mohammad, K.S.; Castillo, A.B.; et al. Oxygen-sensing PHDs regulate bone homeostasis through the modulation of osteoprotegerin. Genes Dev. 2015, 29, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Mangiavini, L.; Merceron, C.; Araldi, E.; Khatri, R.; Gerard-O’Riley, R.; Wilson, T.L.; Rankin, E.B.; Giaccia, A.J.; Schipani, E. Loss of VHL in mesenchymal progenitors of the limb bud alters multiple steps of endochondral bone development. Dev. Biol. 2014, 393, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Xing, W.; Pourteymoor, S.; Mohan, S. Conditional disruption of the prolyl hydroxylase domain-containing protein 2 (Phd2) gene defines its key role in skeletal development. J. Bone Miner. Res. 2014, 29, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Kizaka-Kondoh, S.; Hirota, K.; Hiraoka, M.; Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007, 67, 4157–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, Y.; Sato, Y.; Kobayashi, T.; Yoshida, S.; Mori, T.; Kanagawa, H.; Katsuyama, E.; Fujie, A.; Hao, W.; Miyamoto, K.; et al. HIF1α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16568–16573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tando, T.; Sato, Y.; Miyamoto, K.; Morita, M.; Kobayashi, T.; Funayama, A.; Kanaji, A.; Hao, W.; Watanabe, R.; Oike, T.; et al. Hif1α is required for osteoclast activation and bone loss in male osteoporosis. Biochem. Biophys. Res. Commun. 2016, 470, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Merceron, C.; Ranganathan, K.; Wang, E.; Tata, Z.; Makkapati, S.; Khan, M.P.; Mangiavini, L.; Yao, A.Q.; Castellini, L.; Levi, B.; et al. Hypoxia-inducible factor 2α is a negative regulator of osteoblastogenesis and bone mass accrual. Bone Res. 2019, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Park, K.H.; Yu, H.G.; Kook, E.; Song, W.H.; Lee, G.; Koh, J.T.; Shin, H.I.; Choi, J.Y.; Huh, Y.H.; et al. Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice. Bone Res. 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Knowles, H.J. Distinct roles for the hypoxia-inducible transcription factors HIF-1α and HIF-2α in human osteoclast formation and function. Sci. Rep. 2020, 10, 21072. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, K.H.; Lee, G.; Kim, S.J.; Song, W.H.; Kwon, S.H.; Koh, J.T.; Huh, Y.H.; Ryu, J.H. Hypoxia-inducible factor-2α mediates senescence-associated intrinsic mechanisms of age-related bone loss. Exp. Mol. Med. 2021, 53, 591–604. [Google Scholar] [CrossRef]
- Gelse, K.; Poschl, E.; Aigner, T. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [Green Version]
- Currey, J.D. The structure and mechanics of bone. J. Mater. Sci. 2012, 47, 41–54. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Gentile, P.; Chiono, V.; Ciardelli, G. Collagen for bone tissue regeneration. Acta Biomater. 2012, 8, 3191–3200. [Google Scholar] [CrossRef] [PubMed]
- Viguet-Carrin, S.; Garnero, P.; Delmas, P. The role of collagen in bone strength. Osteoporos. Int. 2006, 17, 319–336. [Google Scholar] [CrossRef]
- Tuderman, L.; Myllylä, R.; Kivirikko, K.I. Mechanism of the prolyl hydroxylase reaction. Role of co-substrates. Eur. J. Biochem. 1977, 80, 341–348. [Google Scholar] [CrossRef] [PubMed]
- De Jong, L.; Kemp, A. Stoicheiometry and kinetics of the prolyl 4-hydroxylase partial reaction. Biochim. Biophys. Acta 1984, 787, 105–111. [Google Scholar] [CrossRef]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [Green Version]
- Wright, I.; Blanco-Rojo, R.; Fernández, M.C.; Toxqui, L.; Moreno, G.; Pérez-Granados, A.M.; de la Piedra, C.; Remacha, Á.F.; Vaquero, M.P. Bone remodelling is reduced by recovery from iron-deficiency anaemia in premenopausal women. J. Physiol. Biochem. 2013, 69, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.M.; Swallow, E.A.; Metzger, C.E.; Kohler, R.; Wallace, J.M.; Stacy, A.J.; Allen, M.R.; Gasier, H.G. Iron deficiency and high-intensity running interval training do not impact femoral or tibial bone in young female rats. Br. J. Nutr. 2022, 128, 1518–1525. [Google Scholar] [CrossRef]
- Al-Daghri, N.M.; Yakout, S.; Ghaleb, A.; Hussain, S.D.; Sabico, S. Iron and 25-hydroxyvitamin D in postmenopausal women with osteoporosis. Am. J. Transl. Res. 2022, 14, 1387–1405. [Google Scholar]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Cytochrome P450-mediated metabolism of vitamin D. J. Lipid Res. 2014, 55, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Burt, L.A.; Billington, E.O.; Rose, M.S.; Raymond, D.A.; Hanley, D.A.; Boyd, S.K. Effect of High-Dose Vitamin D Supplementation on Volumetric Bone Density and Bone Strength: A Randomized Clinical Trial. JAMA 2019, 322, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Goltzman, D. Functions of vitamin D in bone. Histochem. Cell Biol. 2018, 149, 305–312. [Google Scholar] [CrossRef] [PubMed]
- van Driel, M.; Pols, H.A.; van Leeuwen, J.P. Osteoblast differentiation and control by vitamin D and vitamin D metabolites. Curr. Pharm. Des. 2004, 10, 2535–2555. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, J.; Kawamura, S.; Okiji, F.; Shirazaki, M.; Sakai, S.; Saito, H.; Ishii, M. Sphingosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proc. Natl. Acad. Sci. USA 2013, 110, 7009–7013. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Cong, Q.; Xia, X.; Leong, W.F.; Yeh, J.; Miao, D.; Mishina, Y.; Liu, H.; Li, B. Pharmacologic Calcitriol Inhibits Osteoclast Lineage Commitment via the BMP-Smad1 and IκB-NF-κB Pathways. J. Bone Miner. Res. 2017, 32, 1406–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polzonetti, V.; Pucciarelli, S.; Vincenzetti, S.; Polidori, P. Dietary Intake of Vitamin D from Dairy Products Reduces the Risk of Osteoporosis. Nutrients 2020, 12, 1743. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Wu, C.-C.; Liao, M.-T.; Shyu, J.-F.; Hung, C.-F.; Yen, T.-H.; Lu, C.-L.; Lu, K.-C. Role of nutritional vitamin D in osteoporosis treatment. Clin. Chim. Acta 2018, 484, 179–191. [Google Scholar] [CrossRef]
- Cheng, J.B.; Levine, M.A.; Bell, N.H.; Mangelsdorf, D.J.; Russell, D.W. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc. Natl. Acad. Sci. USA 2004, 101, 7711–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, N.; Sakaki, T.; Ohta, M.; Inouye, K. Metabolism of Vitamin D3 by Human CYP27A1. Biochem. Biophys. Res. Commun. 2000, 273, 977–984. [Google Scholar] [CrossRef]
- Roizen, J.D.; Li, D.; O’Lear, L.; Javaid, M.K.; Shaw, N.J.; Ebeling, P.R.; Nguyen, H.H.; Rodda, C.P.; Thummel, K.E.; Thacher, T.D.; et al. CYP3A4 mutation causes vitamin D-dependent rickets type. J. Clin. Investig. 2018, 128, 1913–1918. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Kaufmann, M.; Weber, S.; Irwin, A.; Goos, C.; John, U.; Misselwitz, J.; Klaus, G.; Kuwertz-Bröking, E.; Fehrenbach, H.; et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N. Engl. J. Med. 2011, 365, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Galan, P.; Hercberg, S.; Touitou, Y. The activity of tissue enzymes in iron-deficient rat and man: An overview. Comp. Biochem. Physiol. B 1984, 77, 647–653. [Google Scholar] [CrossRef]
- Blanco-Rojo, R.; Perez-Granados, A.M.; Toxqui, L.; Zazo, P.; de la Piedra, C.; Vaquero, M.P. Relationship between vitamin D deficiency, bone remodelling and iron status in iron-deficient young women consuming an iron-fortified food. Eur. J. Nutr. 2013, 52, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Grindulis, H.; Scott, P.H.; Belton, N.R.; Wharton, B.A. Combined deficiency of iron and vitamin D in Asian toddlers. Arch. Dis. Child. 1986, 61, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Qader, E.A.; Alkhateeb, N.E. Vitamin D Status in Children with Iron Deficiency and/or Anemia. Int. J. Pediatr. 2016, 4, 3571–3577. [Google Scholar]
- El-Adawy, E.H.; Zahran, F.E.; Shaker, G.A.; Seleem, A. Vitamin D Status in Egyptian Adolescent Females with Iron Deficiency Anemia and Its Correlation with Serum Iron Indices. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 519–525. [Google Scholar] [CrossRef]
- Jin, H.J.; Lee, J.H.; Kim, M.K. The prevalence of vitamin D deficiency in iron-deficient and normal children under the age of 24 months. Blood Res. 2013, 48, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Casal, M.N.; Pasricha, S.R.; Martinez, R.X.; Lopez-Perez, L.; Peña-Rosas, J.P. Serum or plasma ferritin concentration as an index of iron deficiency and overload. Cochrane Database Syst. Rev. 2021, 5, Cd011817. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Kim, J.H.; Ahn, E.H.; Yoo, E.G.; Kim, M.K. Iron and vitamin D status in breastfed infants and their mothers. Korean J. Pediatr. 2015, 58, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Li, R.; Gu, S.; Zhao, Y.; Yang, L. The effect of iron dextran on vitamin D3 metabolism in SD rats. Nutr. Metab. 2022, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, S.; Katsumata, R.; Matsumoto, N.; Inoue, H.; Takahashi, N.; Uehara, M. Iron deficiency decreases renal 25-hydroxyvitamin D3-1α-hydroxylase activity and bone formation in rats. BMC Nutr. 2016, 2, 33. [Google Scholar] [CrossRef] [Green Version]
- Noonan, M.L.; Ni, P.; Solis, E.; Marambio, Y.G.; Agoro, R.; Chu, X.; Wang, Y.; Gao, H.; Xuei, X.; Clinkenbeard, E.L.; et al. Osteocyte Egln1/Phd2 links oxygen sensing and biomineralization via FGF23. Bone Res. 2023, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Latic, N.; Erben, R.G. FGF23 and Vitamin D Metabolism. JBMR Plus 2021, 5, e10558. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Araya, K.; Fukumoto, S.; Backenroth, R.; Takeuchi, Y.; Nakayama, K.; Ito, N.; Yoshii, N.; Yamazaki, Y.; Yamashita, T.; Silver, J.; et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005, 90, 5523–5527. [Google Scholar] [CrossRef] [Green Version]
- Farrow, E.G.; Yu, X.; Summers, L.J.; Davis, S.I.; Fleet, J.C.; Allen, M.R.; Robling, A.G.; Stayrook, K.R.; Jideonwo, V.; Magers, M.J.; et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef] [Green Version]
- Clinkenbeard, E.L.; Farrow, E.G.; Summers, L.J.; Cass, T.A.; Roberts, J.L.; Bayt, C.A.; Lahm, T.; Albrecht, M.; Allen, M.R.; Peacock, M.; et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J. Bone Miner. Res. 2014, 29, 361–369. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Koch, T.A.; Bregman, D.B. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J. Bone Miner. Res. 2013, 28, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A.; Peacock, M.; Gray, A.K.; Padgett, L.R.; Hui, S.L.; Econs, M.J. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J. Clin. Endocrinol. Metab. 2011, 96, 3541–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Katz, R.; Ginsberg, C.; Bullen, A.; Vallon, V.; Thomson, S.; Moe, O.W.; Hoofnagle, A.N.; de Leeuw, P.W.; Kroon, A.A.; et al. Renal Clearance of Fibroblast Growth Factor-23 (FGF23) and its Fragments in Humans. J. Bone Miner. Res. 2022, 37, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, R. How to ensure adequate iron absorption from iron-fortified food. Nutr. Rev. 2002, 60, S7–S15; discussion S43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrell, R.F.; Reddy, M.B.; Juillerat, M.A.; Cook, J.D. Degradation of phytic acid in cereal porridges improves iron absorption by human subjects. Am. J. Clin. Nutr. 2003, 77, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Schlemmer, U.; Frølich, W.; Prieto, R.M.; Grases, F. Phytate in foods and significance for humans: Food sources, intake, processing, bioavailability, protective role and analysis. Mol. Nutr. Food. Res. 2009, 53 (Suppl. 2), S330–S375. [Google Scholar] [CrossRef]
- Liang, J.; Han, B.Z.; Nout, M.J.; Hamer, R.J. Effects of soaking, germination and fermentation on phytic acid, total and in vitro soluble zinc in brown rice. Food Chem. 2008, 110, 821–828. [Google Scholar] [CrossRef]
- Stoffel, N.U.; von Siebenthal, H.K.; Moretti, D.; Zimmermann, M.B. Oral iron supplementation in iron-deficient women: How much and how often? Mol. Asp. Med. 2020, 75, 100865. [Google Scholar] [CrossRef]
- Gautam, C.S.; Saha, L.; Sekhri, K.; Saha, P.K. Iron deficiency in pregnancy and the rationality of iron supplements prescribed during pregnancy. Medscape J. Med. 2008, 10, 283. [Google Scholar]
- Tolkien, Z.; Stecher, L.; Mander, A.P.; Pereira, D.I.; Powell, J.J. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0117383. [Google Scholar] [CrossRef] [Green Version]
- Macdougall, I.C.; Bock, A.H.; Carrera, F.; Eckardt, K.U.; Gaillard, C.; Van Wyck, D.; Roubert, B.; Nolen, J.G.; Roger, S.D. FIND-CKD: A randomized trial of intravenous ferric carboxymaltose versus oral iron in patients with chronic kidney disease and iron deficiency anaemia. Nephrol. Dial. Transplant. 2014, 29, 2075–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, O.H.; Ainsworth, M.; Coskun, M.; Weiss, G. Management of Iron-Deficiency Anemia in Inflammatory Bowel Disease: A Systematic Review. Medicine 2015, 94, e963. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hsu, J.C.; Koo, H.; Cormode, D.P. Repurposing ferumoxytol: Diagnostic and therapeutic applications of an FDA-approved nanoparticle. Theranostics 2022, 12, 796–816. [Google Scholar] [CrossRef]
- Trujillo-Alonso, V.; Pratt, E.C.; Zong, H.; Lara-Martinez, A.; Kaittanis, C.; Rabie, M.O.; Longo, V.; Becker, M.W.; Roboz, G.J.; Grimm, J.; et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat. Nanotechnol. 2019, 14, 616–622. [Google Scholar] [CrossRef]
- Zhang, G.; Zhen, C.; Yang, J.; Zhang, Z.; Wu, Y.; Che, J.; Shang, P. 1-2T static magnetic field combined with Ferumoxytol prevent unloading-induced bone loss by regulating iron metabolism in osteoclastogenesis. J. Orthop. Transl. 2023, 38, 126–140. [Google Scholar]
- Liu, L.; Jin, R.; Duan, J.; Yang, L.; Cai, Z.; Zhu, W.; Nie, Y.; He, J.; Xia, C.; Gong, Q.; et al. Bioactive iron oxide nanoparticles suppress osteoclastogenesis and ovariectomy-induced bone loss through regulating the TRAF6-p62-CYLD signaling complex. Acta Biomater. 2020, 103, 281–292. [Google Scholar] [CrossRef]
- Lynch, S. Food iron absorption and its importance for the design of food fortification strategies. Nutr. Rev. 2002, 60, S3–S6; discussion S42–S43. [Google Scholar] [CrossRef]
- Dary, O.; Freire, W.; Kim, S. Iron compounds for food fortification: Guidelines for Latin America and the Caribbean 2002. Nutr. Rev. 2002, 60, S50–S61. [Google Scholar]
- Allen, L.; De Benoist, B.; Dary, O.; Hurrell, R. Guidelines on Food Fortification with Micronutrients; WHO/FAO: Geneva, Switzerland, 2006. [Google Scholar]
- Nayak, S.N.; Aravind, B.; Malavalli, S.S.; Sukanth, B.S.; Poornima, R.; Bharati, P.; Hefferon, K.; Kole, C.; Puppala, N. Omics Technologies to Enhance Plant Based Functional Foods: An Overview. Front. Genet. 2021, 12, 742095. [Google Scholar] [CrossRef]
- Lucca, P.; Hurrell, R.; Potrykus, I. Fighting iron deficiency anemia with iron-rich rice. J. Am. Coll. Nutr. 2002, 21, 184s–190s. [Google Scholar] [CrossRef]
- Masuda, H.; Ishimaru, Y.; Aung, M.S.; Kobayashi, T.; Kakei, Y.; Takahashi, M.; Higuchi, K.; Nakanishi, H.; Nishizawa, N.K. Iron biofortification in rice by the introduction of multiple genes involved in iron nutrition. Sci. Rep. 2012, 2, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Markers | Normal | Iron Depletion | Iron Deficiency without Anemia | Iron Deficiency Anemia |
|---|---|---|---|---|
| Current | ||||
| Hemoglobin (g/dL) | N | N | N | L |
| Mean corpuscular volume (fL) | N | N | N | L |
| Mean corpuscular hemoglobin (pg) | N | N | N/L | L |
| Ferritin (μg/L) | N | L | L | L |
| Serum iron (μg/dL) | N | N | L | L |
| Transferrin saturation (%) | N | N | L | L |
| Proposed | ||||
| Reticulocyte hemoglobin content (pg) | N | N | L | L |
| Soluble transferrin receptor (mg/L) | N | N | H | H |
| Total iron-binding capacity | N | N | H | H |
| Hepcidin | N | N/L | L | L |
| Zinc protoporphyrin | N | N | N | H |
| Population, Age | No Anemia | Anemia | ||
|---|---|---|---|---|
| Mild | Moderate | Severe | ||
| Children, 6–59 months | ≥110 | 100–109 | 70–99 | <70 |
| Children, 5–11 years | ≥115 | 110–114 | 80–109 | <80 |
| Children, 12–14 years | ≥120 | 110–119 | 80–109 | <80 |
| Non-pregnant women, 15 years and above | ≥120 | 110–119 | 80–109 | <80 |
| Pregnant women | ≥110 | 100–109 | 70–99 | <70 |
| Men, 15 years and above | ≥130 | 110–129 | 80–109 | <80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Li, Q.; Feng, Y.; Zeng, Y. Iron Deficiency and Iron Deficiency Anemia: Potential Risk Factors in Bone Loss. Int. J. Mol. Sci. 2023, 24, 6891. https://doi.org/10.3390/ijms24086891
Yang J, Li Q, Feng Y, Zeng Y. Iron Deficiency and Iron Deficiency Anemia: Potential Risk Factors in Bone Loss. International Journal of Molecular Sciences. 2023; 24(8):6891. https://doi.org/10.3390/ijms24086891
Chicago/Turabian StyleYang, Jiancheng, Qingmei Li, Yan Feng, and Yuhong Zeng. 2023. "Iron Deficiency and Iron Deficiency Anemia: Potential Risk Factors in Bone Loss" International Journal of Molecular Sciences 24, no. 8: 6891. https://doi.org/10.3390/ijms24086891
APA StyleYang, J., Li, Q., Feng, Y., & Zeng, Y. (2023). Iron Deficiency and Iron Deficiency Anemia: Potential Risk Factors in Bone Loss. International Journal of Molecular Sciences, 24(8), 6891. https://doi.org/10.3390/ijms24086891