
Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor

Abstract
:1. Introduction
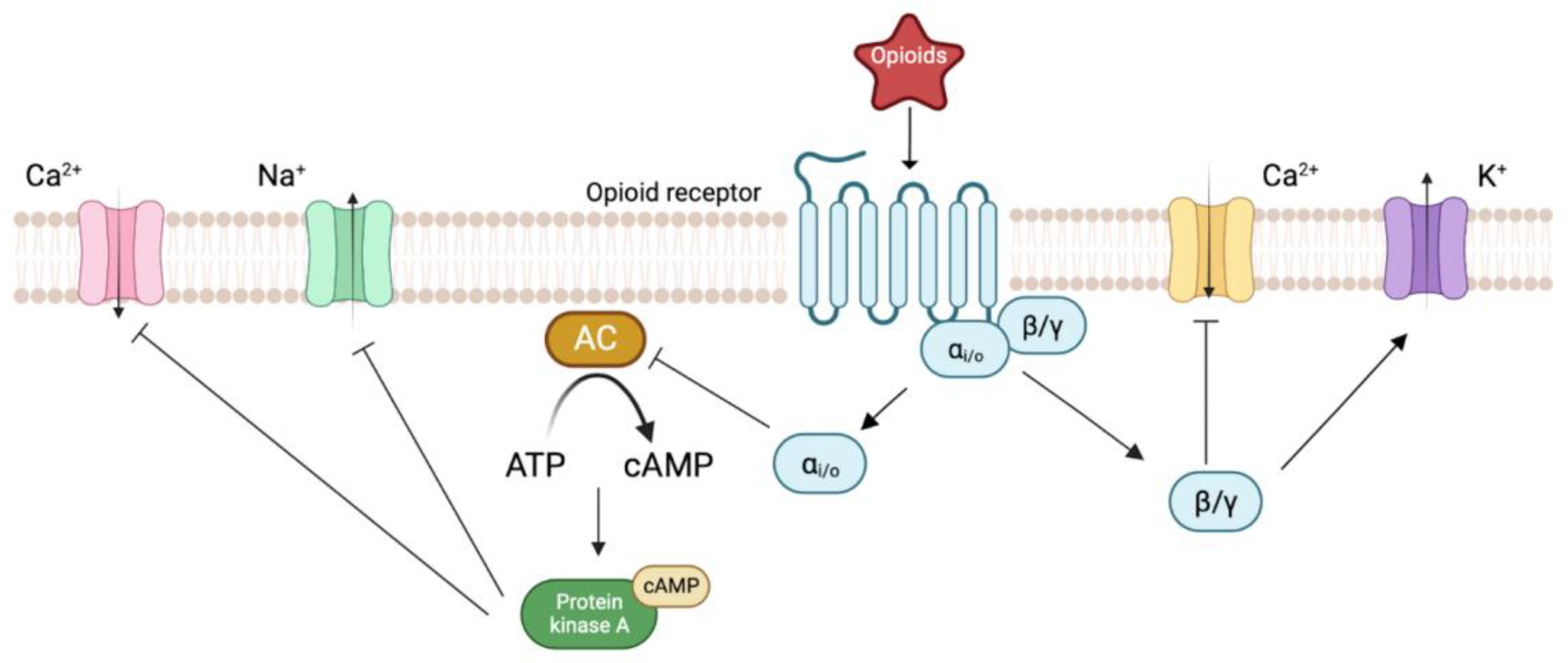
2. Biology of Opioid Receptors
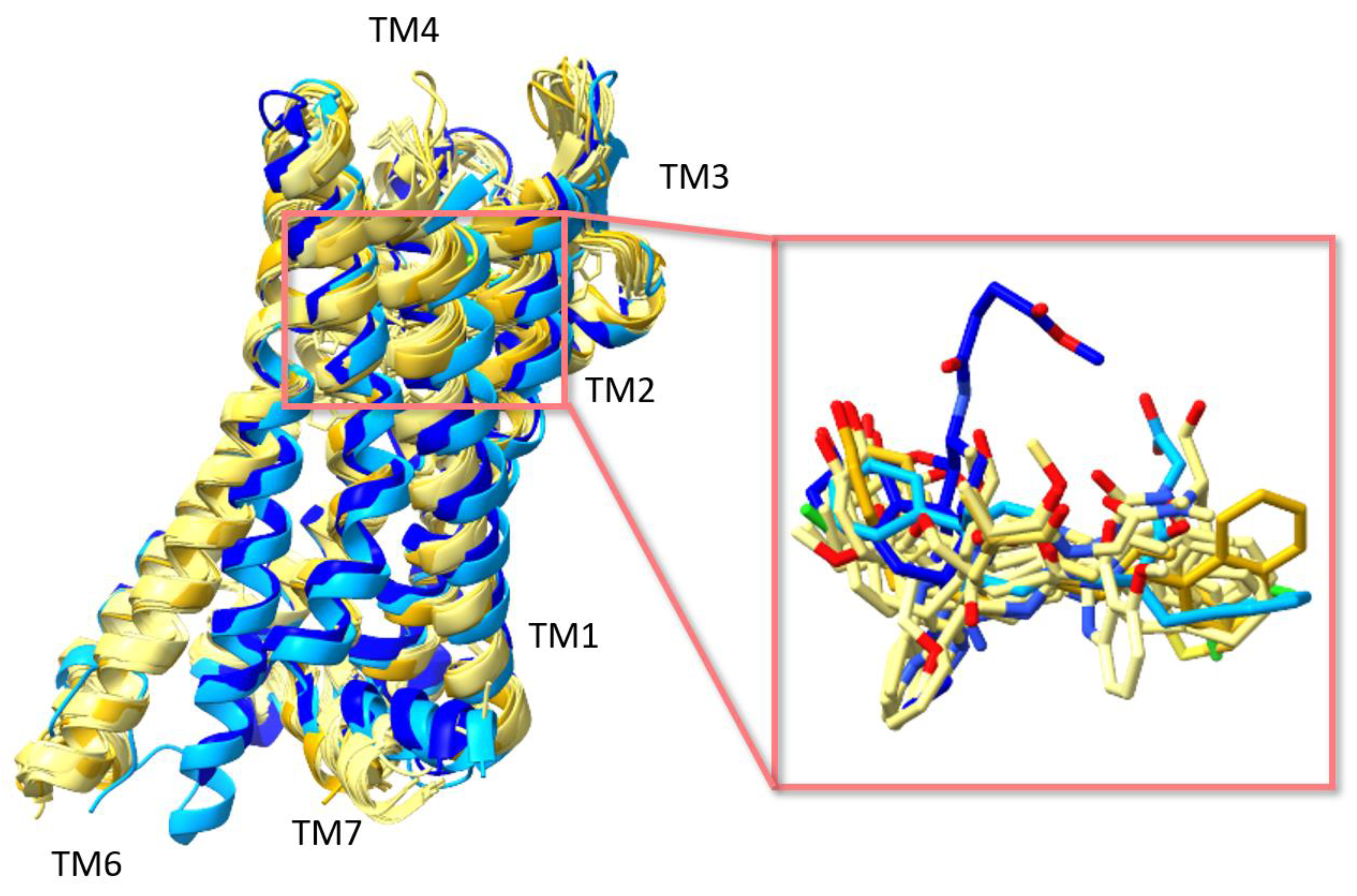
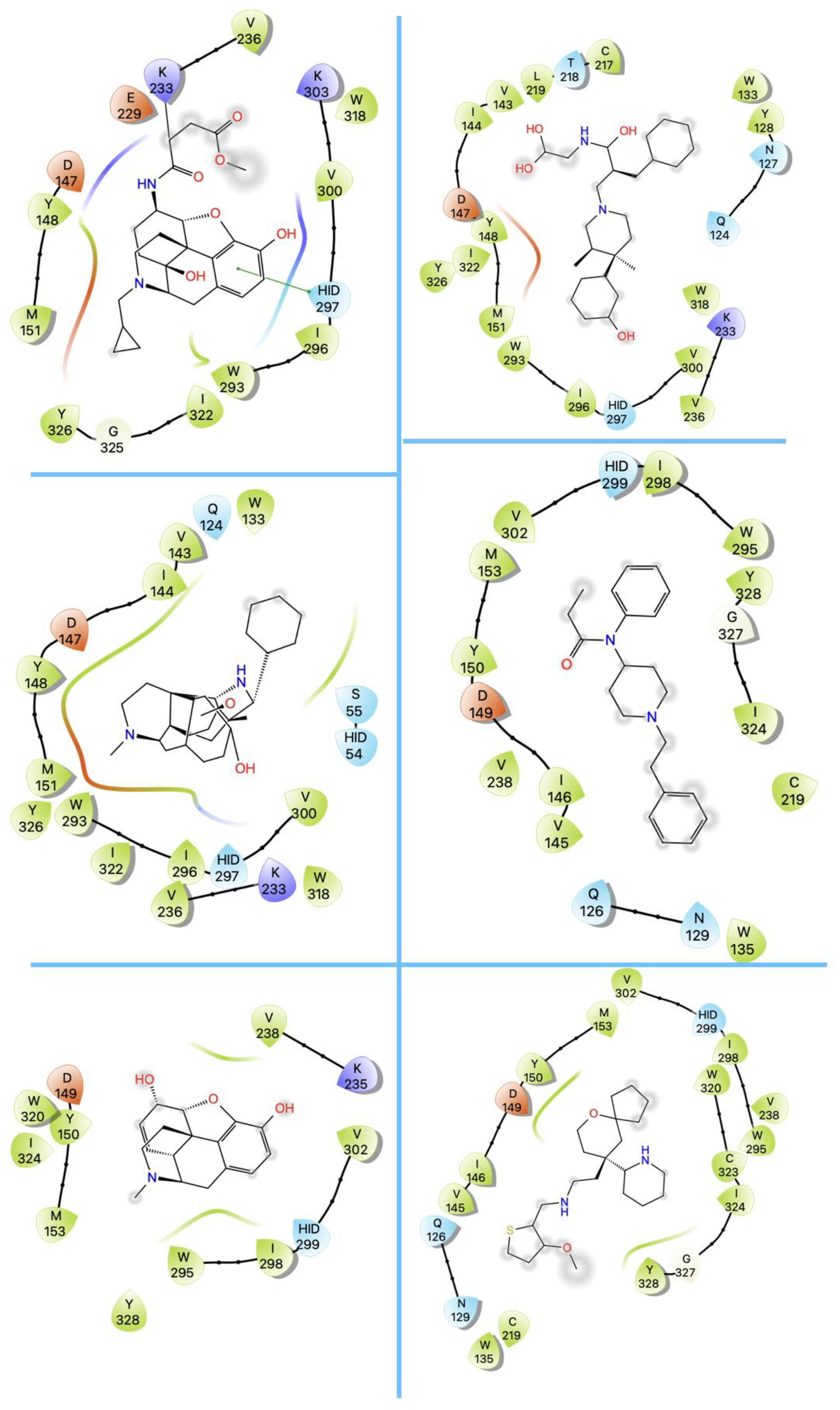
3. Structures of μ Opioid Receptor (MOR)
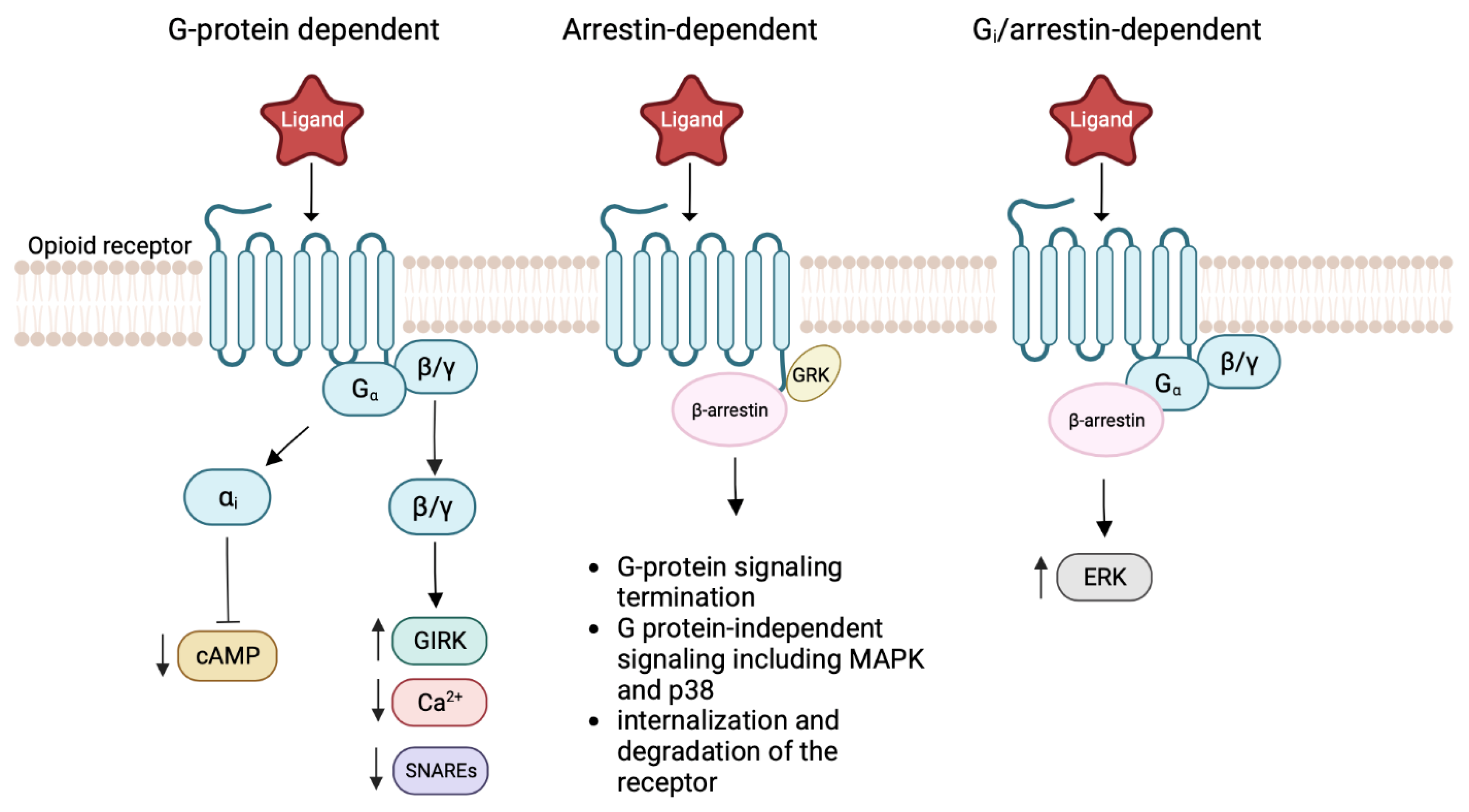
4. Allosteric Modulation of the MOR

5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ropero-Miller, J.D.; Speaker, P.J. The Hidden Costs of the Opioid Crisis and the Implications for Financial Management in the Public Sector. Forensic Sci. Int. Synerg. 2019, 1, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Florence, C.; Luo, F.; Rice, K. The Economic Burden of Opioid Use Disorder and Fatal Opioid Overdose in the United States, 2017. Drug Alcohol. Depend. 2021, 218, 108350. [Google Scholar] [CrossRef] [PubMed]
- Darcq, E.; Kieffer, B.L. Opioid Receptors: Drivers to Addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharm. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Huang, X.-P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Stevens, R.C.; Katritch, V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar] [CrossRef] [Green Version]
- Poli, G.; Dimmito, M.P.; Mollica, A.; Zengin, G.; Benyhe, S.; Zador, F.; Stefanucci, A. Discovery of Novel Μ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules 2019, 24, 3872. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, A.; Wang, H.; Zaidi, S.A.; DiBerto, J.F.; Che, T.; Qu, Q.; Robertson, M.J.; Madasu, M.K.; El Daibani, A.; Varga, B.R.; et al. Structure-Based Design of Bitopic Ligands for the µ-Opioid Receptor. Nature 2023, 613, 767–774. [Google Scholar] [CrossRef]
- Portoghese, P.S. Relationships Between Stereostructure and Pharmacological Activities. Annu. Rev. Pharmacol. 1970, 10, 51–76. [Google Scholar] [CrossRef]
- Portoghese, P.S. A New Concept on the Mode of Interaction of Narcotic Analgesics with Receptors. J. Med. Chem. 1965, 8, 609–616. [Google Scholar] [CrossRef]
- Beckett, A.H.; Casy, A.F. 5 Analgesics and Their Antagonists: Biochemical Aspects and Structure-Activity Relationships. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 1965; Volume 4, pp. 171–218. ISBN 978-0-444-53323-4. [Google Scholar]
- Pert, C.B.; Snyder, S.H. Opiate Receptor: Demonstration in Nervous Tissue. Science 1973, 179, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Terenius, L. Stereospecific Interaction Between Narcotic Analgesics and a Synaptic Plasma Membrane Fraction of Rat Cerebral Cortex. Acta Pharmacol. Toxicol. 2009, 32, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific Binding of the Potent Narcotic Analgesic [ 3 H]Etorphine to Rat-Brain Homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gropper, M.A.; Miller, R.D.; Cohen, N.H. (Eds.) Miller’s Anesthesia, 9th ed.; Elsevier: Philadelphia, PA, USA, 2020; ISBN 978-0-323-59604-6. [Google Scholar]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.-L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.-C. ORL1, a Novel Member of the Opioid Receptor Family: Cloning, Functional Expression and Localization. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-Dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [Green Version]
- Le Merrer, J.; Becker, J.A.J.; Befort, K.; Kieffer, B.L. Reward Processing by the Opioid System in the Brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef]
- Pathan, H.; Williams, J. Basic Opioid Pharmacology: An Update. Br. J. Pain 2012, 6, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Snyder, S.H.; Pasternak, G.W. Historical Review: Opioid Receptors. Trends Pharmacol. Sci. 2003, 24, 198–205. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal Structure of the Μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, X.; Yang, Y.; Chao, D.; H Lazarus, L.; Xia, Y. Current Research on Opioid Receptor Function. CDT 2012, 13, 230–246. [Google Scholar] [CrossRef] [Green Version]
- Wiesenfeld-Hallin, Z.; de Araúja Lucas, G.; Alster, P.; Xu, X.-J.; Hökfelt, T. Cholecystokinin/Opioid Interactions. Brain Res. 1999, 848, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W. Multiple Opiate Receptors: Déjà vu All over Again. Neuropharmacology 2004, 47, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid Receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone-Corsi, P. The Cyclic AMP Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.-T.; Hang, L.; Liu, T. Mu Opioid Receptor Heterodimers Emerge as Novel Therapeutic Targets: Recent Progress and Future Perspective. Front. Pharmacol. 2020, 11, 1078. [Google Scholar] [CrossRef]
- Tan, L.; Yan, W.; McCorvy, J.D.; Cheng, J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure–Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018, 61, 9841–9878. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Cong, X.; Maurel, D.; Déméné, H.; Vasiliauskaité-Brooks, I.; Hagelberger, J.; Peysson, F.; Saint-Paul, J.; Golebiowski, J.; Granier, S.; Sounier, R. Molecular Insights into the Biased Signaling Mechanism of the μ-Opioid Receptor. Mol. Cell 2021, 81, 4165–4175.e6. [Google Scholar] [CrossRef]
- Che, T.; Dwivedi-Agnihotri, H.; Shukla, A.K.; Roth, B.L. Biased Ligands at Opioid Receptors: Current Status and Future Directions. Sci. Signal. 2021, 14, eaav0320. [Google Scholar] [CrossRef]
- Kelly, B.; Hollingsworth, S.A.; Blakemore, D.C.; Owen, R.M.; Storer, R.I.; Swain, N.A.; Aydin, D.; Torella, R.; Warmus, J.S.; Dror, R.O. Delineating the Ligand–Receptor Interactions That Lead to Biased Signaling at the μ-Opioid Receptor. J. Chem. Inf. Model. 2021, 61, 3696–3707. [Google Scholar] [CrossRef]
- Nagi, K.; Pineyro, G. Kir3 Channel Signaling Complexes: Focus on Opioid Receptor Signaling. Front. Cell Neurosci. 2014, 8, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafi, A.; Kim, S.-K.; Goddard, W.A. Mechanism of β-Arrestin Recruitment by the μ-Opioid G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 16346–16355. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Patel, C.B.; Rockman, H.A. β-Arrestin: A Signaling Molecule and Potential Therapeutic Target for Heart Failure. J. Mol. Cell. Cardiol. 2011, 51, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Macé, G.; Miaczynska, M.; Zerial, M.; Nebreda, A.R. Phosphorylation of EEA1 by P38 MAP Kinase Regulates μ Opioid Receptor Endocytosis. EMBO J. 2005, 24, 3235–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, M.M.; Clark, A.L.; Haas, P.D.; Serna, J.S.; Hahn, J.W.; Kiss, A.; Coscia, C.J. μ and κ Opioid Receptors Activate ERK/MAPK via Different Protein Kinase C Isoforms and Secondary Messengers in Astrocytes. J. Biol. Chem. 2005, 280, 27662–27669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, M.J.; Papasergi-Scott, M.M.; He, F.; Seven, A.B.; Meyerowitz, J.G.; Panova, O.; Peroto, M.C.; Che, T.; Skiniotis, G. Structure Determination of Inactive-State GPCRs with a Universal Nanobody. Nat. Struct. Mol. Biol. 2022, 29, 1188–1195. [Google Scholar] [CrossRef]
- Wang, H.; Hetzer, F.; Huang, W.; Qu, Q.; Meyerowitz, J.; Kaindl, J.; Hübner, H.; Skiniotis, G.; Kobilka, B.K.; Gmeiner, P. Structure-Based Evolution of G Protein-Biased Μ-Opioid Receptor Agonists. Angew. Chem. Int. Ed. 2022, 61, e202200269. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular Recognition of Morphine and Fentanyl by the Human μ-Opioid Receptor. Cell 2022, 185, 4361–4375.e19. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural Insights into Μ-Opioid Receptor Activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the Μ-Opioid Receptor–Gi Protein Complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Qu, Q.; Huang, W.; Aydin, D.; Paggi, J.M.; Seven, A.B.; Wang, H.; Chakraborty, S.; Che, T.; DiBerto, J.F.; Robertson, M.J.; et al. Insights into Distinct Signaling Profiles of the ΜOR Activated by Diverse Agonists. Nat. Chem. Biol. 2022, 19, 423–430. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis: UCSF ChimeraX Visualization System. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Maestro; Maestro, Schrödinger, LLC: New York, NY, USA, 2021.
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakkiah, S.; Guo, W.; Pan, B.; Ji, Z.; Yavas, G.; Azevedo, M.; Hawes, J.; Patterson, T.A.; Hong, H. Elucidating Interactions Between SARS-CoV-2 Trimeric Spike Protein and ACE2 Using Homology Modeling and Molecular Dynamics Simulations. Front. Chem. 2021, 8, 622632. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Kusko, R.; Pan, B.; Guo, W.; Ge, W.; Tong, W.; Hong, H. Structural Changes Due to Antagonist Binding in Ligand Binding Pocket of Androgen Receptor Elucidated Through Molecular Dynamics Simulations. Front. Pharmacol. 2018, 9, 492. [Google Scholar] [CrossRef]
- Selvaraj, C.; Sakkiah, S.; Tong, W.; Hong, H. Molecular Dynamics Simulations and Applications in Computational Toxicology and Nanotoxicology. Food Chem. Toxicol. 2018, 112, 495–506. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, W.; Fang, H.; Perkins, R.; Tong, W.; Hong, H. Homology Modeling, Molecular Docking, and Molecular Dynamics Simulations Elucidated α-Fetoprotein Binding Modes. BMC Bioinform. 2013, 14, S6. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.; Coop, A.; MacKerell, A.D. Molecular Details of the Activation of the μ Opioid Receptor. J. Phys. Chem. B 2013, 117, 7907–7917. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Vogel, H.; Filipek, S. The Role of Water and Sodium Ions in the Activation of the μ-Opioid Receptor. Angew. Chem. Int. Ed. 2013, 52, 10112–10115. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Activation and Allosteric Modulation of Human μ Opioid Receptor in Molecular Dynamics. J. Chem. Inf. Model. 2015, 55, 2421–2434. [Google Scholar] [CrossRef]
- Yuan, S.; Palczewski, K.; Peng, Q.; Kolinski, M.; Vogel, H.; Filipek, S. The Mechanism of Ligand-Induced Activation or Inhibition of μ- and κ-Opioid Receptors. Angew. Chem. Int. Ed. 2015, 54, 7560–7563. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Cheng, T.; Li, W.; Liu, G.; Zhu, W.; Tang, Y. Computational Insights into the G-Protein-Biased Activation and Inactivation Mechanisms of the μ Opioid Receptor. Acta Pharm. Sin. 2018, 39, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, K.A.; Shang, Y.; Filizola, M. Insights into the Function of Opioid Receptors from Molecular Dynamics Simulations of Available Crystal Structures. Br. J. Pharmacol. 2018, 175, 2834–2845. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wang, Y.; Hunkele, A.; Provasi, D.; Pasternak, G.W.; Filizola, M. Kinetic and Thermodynamic Insights into Sodium Ion Translocation through the μ-Opioid Receptor from Molecular Dynamics and Machine Learning Analysis. PLoS Comput. Biol. 2019, 15, e1006689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipiński, P.F.J.; Jarończyk, M.; Dobrowolski, J.C.; Sadlej, J. Molecular Dynamics of Fentanyl Bound to μ-Opioid Receptor. J. Mol. Model. 2019, 25, 144. [Google Scholar] [CrossRef] [Green Version]
- Dumitrascuta, M.; Bermudez, M.; Ballet, S.; Wolber, G.; Spetea, M. Mechanistic Understanding of Peptide Analogues, DALDA, [Dmt1]DALDA, and KGOP01, Binding to the Mu Opioid Receptor. Molecules 2020, 25, 2087. [Google Scholar] [CrossRef]
- Hu, X.; Provasi, D.; Ramsey, S.; Filizola, M. Mechanism of μ-Opioid Receptor-Magnesium Interaction and Positive Allosteric Modulation. Biophys. J. 2020, 118, 909–921. [Google Scholar] [CrossRef]
- Mondal, D.; Kolev, V.; Warshel, A. Exploring the Activation Pathway and G i -Coupling Specificity of the μ-Opioid Receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 26218–26225. [Google Scholar] [CrossRef]
- Zhao, Z.; Huang, T.; Li, J. Molecular Dynamics Simulations to Investigate How PZM21 Affects the Conformational State of the μ-Opioid Receptor Upon Activation. IJMS 2020, 21, 4699. [Google Scholar] [CrossRef]
- Podlewska, S.; Bugno, R.; Kudla, L.; Bojarski, A.J.; Przewlocki, R. Molecular Modeling of µ Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl—Simulated Interaction Patterns Confronted with Experimental Data. Molecules 2020, 25, 4636. [Google Scholar] [CrossRef]
- Liao, S.; Tan, K.; Floyd, C.; Bong, D.; Pino, M.J.; Wu, C. Probing Biased Activation of Mu-Opioid Receptor by the Biased Agonist PZM21 Using All Atom Molecular Dynamics Simulation. Life Sci. 2021, 269, 119026. [Google Scholar] [CrossRef]
- Xie, B.; Le Rouzic, V.P.; Goldberg, A.; Tsai, M.-H.M.; Chen, L.; Zhang, T.; Sinha, A.; Pan, Y.-X.; Baumann, M.H.; Shi, L. Binding Preference at the μ-Opioid Receptor Underlies Distinct Pharmacology of Cyclopropyl versus Valeryl Analogs of Fentanyl. Neuropharmacology 2023, 227, 109442. [Google Scholar] [CrossRef]
- Freier, E.; Wolf, S.; Gerwert, K. Proton Transfer via a Transient Linear Water-Molecule Chain in a Membrane Protein. Proc. Natl. Acad. Sci. USA 2011, 108, 11435–11439. [Google Scholar] [CrossRef] [Green Version]
- Fried, S.D.E.; Hewage, K.S.K.; Eitel, A.R.; Struts, A.V.; Weerasinghe, N.; Perera, S.M.D.C.; Brown, M.F. Hydration-Mediated G-Protein–Coupled Receptor Activation. Proc. Natl. Acad. Sci. USA 2022, 119, e2117349119. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular Signatures of G-Protein-Coupled Receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Filipek, S.; Palczewski, K.; Vogel, H. Activation of G-Protein-Coupled Receptors Correlates with the Formation of a Continuous Internal Water Pathway. Nat. Commun. 2014, 5, 4733. [Google Scholar] [CrossRef] [Green Version]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Angel, T.E.; Chance, M.R.; Palczewski, K. Conserved Waters Mediate Structural and Functional Activation of Family A (Rhodopsin-like) G Protein-Coupled Receptors. Proc. Natl. Acad. Sci. USA 2009, 106, 8555–8560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raehal, K.M.; Walker, J.K.L.; Bohn, L.M. Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharm. Exp. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.-T.; Lefkowitz, R.J.; Caron, M.G. μ-Opioid Receptor Desensitization by β-Arrestin-2 Determines Morphine Tolerance but Not Dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.-T. Enhanced Morphine Analgesia in Mice Lacking β-Arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Kliewer, A.; Gillis, A.; Hill, R.; Schmiedel, F.; Bailey, C.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S. Morphine-induced Respiratory Depression Is Independent of Β-arrestin2 Signalling. Br. J. Pharm. 2020, 177, 2923–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, K.E.; Traynor, J.R. Allostery at Opioid Receptors: Modulation with Small Molecule Ligands. Br. J. Pharmacol. 2018, 175, 2846–2856. [Google Scholar] [CrossRef] [Green Version]
- Kathmann, M.; Flau, K.; Redmer, A.; Tränkle, C.; Schlicker, E. Cannabidiol Is an Allosteric Modulator at Mu- and Delta-Opioid Receptors. Naunyn. Schmied. Arch. Pharm. 2006, 372, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Rothman, R.B.; Murphy, D.L.; Xu, H.; Godin, J.A.; Dersch, C.M.; Partilla, J.S.; Tidgewell, K.; Schmidt, M.; Prisinzano, T.E. Salvinorin A: Allosteric Interactions at the μ-Opioid Receptor. J. Pharm. Exp. 2007, 320, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular Control of δ-Opioid Receptor Signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Miller-Gallacher, J.L.; Nehmé, R.; Warne, T.; Edwards, P.C.; Schertler, G.F.X.; Leslie, A.G.W.; Tate, C.G. The 2.1 Å Resolution Structure of Cyanopindolol-Bound Β1-Adrenoceptor Identifies an Intramembrane Na+ Ion That Stabilises the Ligand-Free Receptor. PLoS ONE 2014, 9, e92727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-Resolution Crystal Structure of Human Protease-Activated Receptor 1. Nature 2012, 492, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; LeRouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic Insights into the Allosteric Modulation of Opioid Receptors by Sodium Ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef] [Green Version]
- Vickery, O.N.; Carvalheda, C.A.; Zaidi, S.A.; Pisliakov, A.V.; Katritch, V.; Zachariae, U. Intracellular Transfer of Na+ in an Active-State G-Protein-Coupled Receptor. Structure 2018, 26, 171–180.e2. [Google Scholar] [CrossRef] [Green Version]
- Vickery, O.N.; Machtens, J.-P.; Tamburrino, G.; Seeliger, D.; Zachariae, U. Structural Mechanisms of Voltage Sensing in G Protein-Coupled Receptors. Structure 2016, 24, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomès, R.; García, A.E.; Ernst, O.P.; et al. Mechanistic Insights into Allosteric Regulation of the A2A Adenosine G Protein-Coupled Receptor by Physiological Cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef]
- Qiu, Y.; Wang, Y.; Law, P.-Y.; Chen, H.-Z.; Loh, H.H. Cholesterol Regulates μ-Opioid Receptor-Induced β-Arrestin 2 Translocation to Membrane Lipid Rafts. Mol. Pharm. 2011, 80, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; El-Fakahany, E.E. Allosteric Modulation of GPCRs of Class A by Cholesterol. IJMS 2021, 22, 1953. [Google Scholar] [CrossRef]
- Levitt, E.S.; Clark, M.J.; Jenkins, P.M.; Martens, J.R.; Traynor, J.R. Differential Effect of Membrane Cholesterol Removal on μ- and δ-Opioid Receptors. J. Biol. Chem. 2009, 284, 22108–22122. [Google Scholar] [CrossRef] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the Active δ-Opioid Receptor Crystal Structure with Peptide and Small-Molecule Agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [Green Version]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [Green Version]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A 2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.F.; Spiess, K.; Jude, K.M.; Tsutsumi, N.; Burg, J.S.; Ingram, J.R.; Waghray, D.; Hjorto, G.M.; Larsen, O.; Ploegh, H.L.; et al. Viral GPCR US28 Can Signal in Response to Chemokine Agonists of Nearly Unlimited Structural Degeneracy. eLife 2018, 7, e35850. [Google Scholar] [CrossRef]
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of Positive Allosteric Modulators and Silent Allosteric Modulators of the μ-Opioid Receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, K.E.; Traynor, J.R. Disruption of the Na + Ion Binding Site as a Mechanism for Positive Allosteric Modulation of the Mu-Opioid Receptor. Proc. Natl. Acad. Sci. USA 2014, 111, 18369–18374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Interplay between Two Allosteric Sites and Their Influence on Agonist Binding in Human μ Opioid Receptor. J. Chem. Inf. Model. 2016, 56, 563–570. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kędzierska, E.; Kaczor, A.A.; Schmidhammer, H.; Matosiuk, D. Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 8463. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
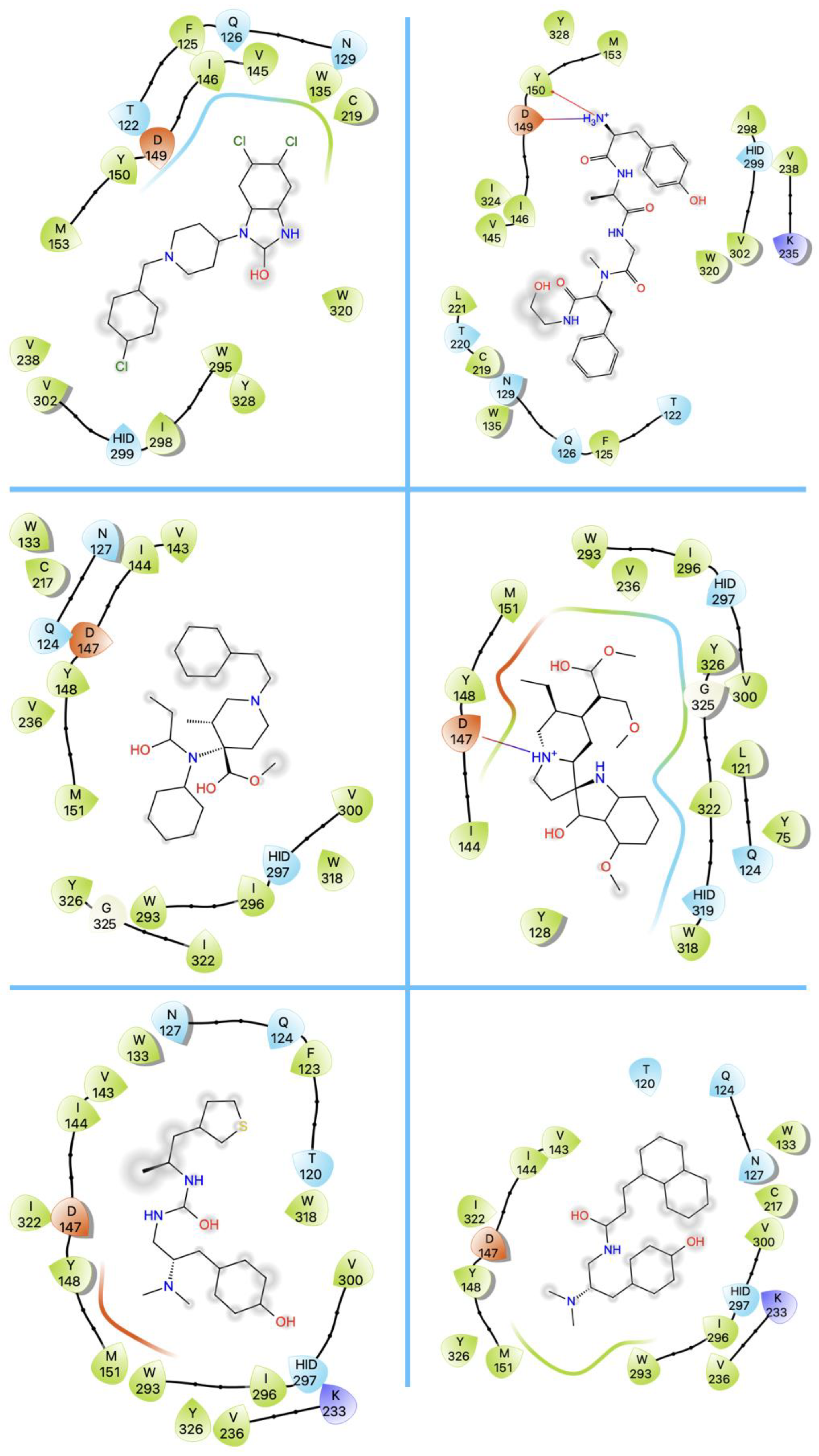{kind=link}
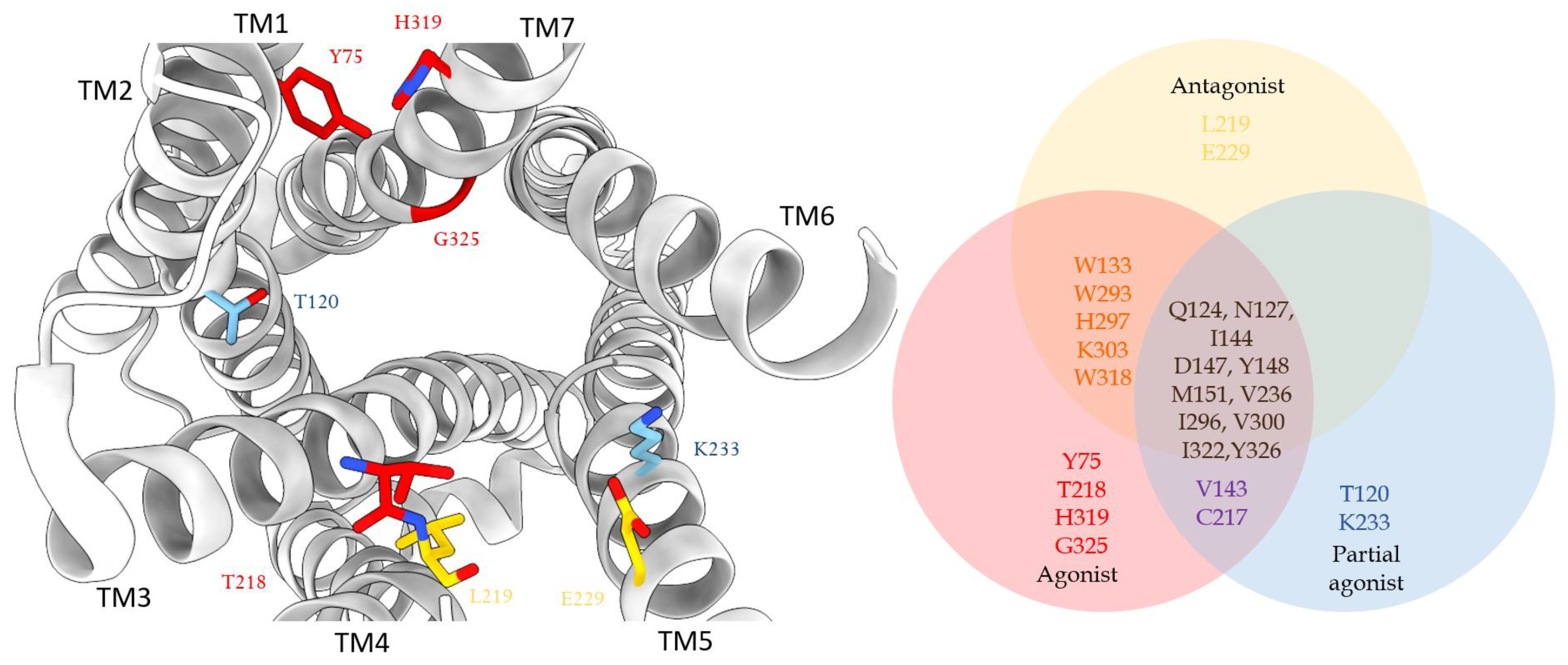{kind=link}
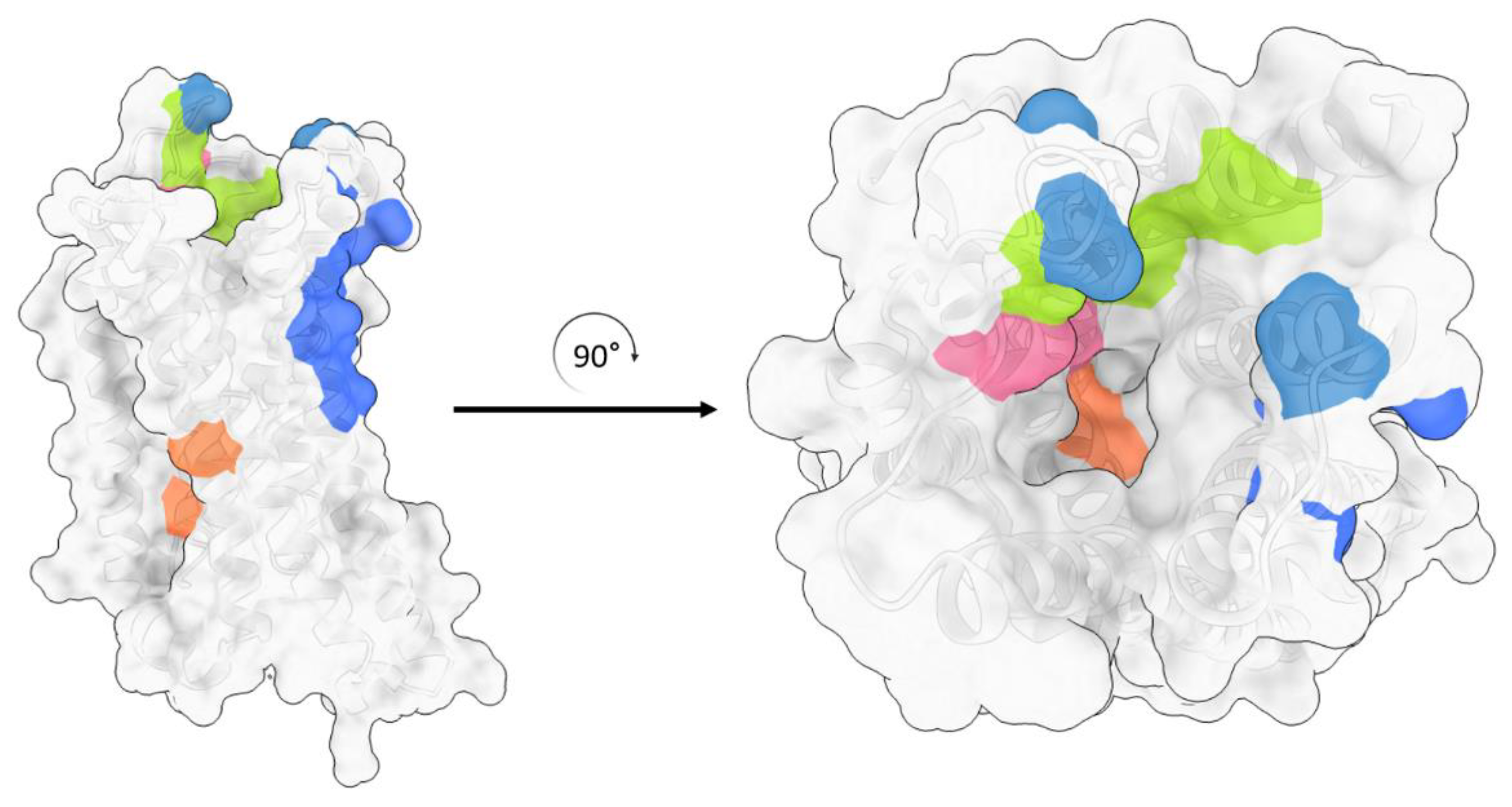{kind=link}
| Opioid Receptor Subtype | Endogenous Ligand |
|---|---|
| μ | β-endorphin, enkephalins, endomorphin-1, endomorphin-2 |
| δ | β-endorphin, enkephalins |
| κ | dynorphin A, dynorphin B, α-neoendorphin |
| NOR 1 | N/OFQ (nociceptin/orphanin FQ) |
| PDB ID | Ligand | Ligand Type | In Complex with | Structure Type | Resolution |
|---|---|---|---|---|---|
| 4DKL | β-FNA | Antagonist | Lysozyme chimera | X-RAY | 2.8 |
| 7UL4 | Alvimopan | Antagonist | Megabody 6 | Cryo-EM | 2.8 |
| 5C1M | BU72 | Full agonist | Nanobody 39 | X-RAY | 2.07 |
| 8EF5 | Fentanyl | Full agonist | Gαi-1, Gβ-1, Gγ-2 | Cryo-EM | 3.3 |
| 8EF6 | Morphine | Full agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.2 |
| 6DDE | DAMGO | Full agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.5 |
| 6DDF | DAMGO | Full agonist | Gαi-1, Gβ-1, Gγ-2 | Cryo-EM | 3.5 |
| 8EFQ | DAMGO | Full agonist | Gαi-1, Gβ-1, Gγ-2 | Cryo-EM | 3.3 |
| 7T2H | Lofentanil | Full agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.2 |
| 7T2G | Mitragynine pseudoindoxyl | Full agonist | Gαi-1, Gβ-1, Gγ-2 | Cryo-EM | 2.5 |
| 8EFB | Oliceridine (TRV130) | Partial agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.2 |
| 8EFL | SR17018 | Partial agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.2 |
| 8EFO | PZM21 | Partial agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 2.8 |
| 7SBF | PZM21 | Partial agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 2.9 |
| 7SCG | FH210 | Partial agonist | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.0 |
| 7U2L | NNPPP 1 | Bitopic ligand | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | 3.2 |
| Ligand Type | 2D Structure | |
|---|---|---|
| Agonist |  BU72 |  DAMGO |
 Morphine |  Fentanyl | |
 Lofentanil |  Mitragynine pseudoindoxyl | |
| Partial agonist |  PZM21 |  FH210 |
 Oliceridine (TRV130) |  SR17018 | |
| Antagonist |  β-FNA |  Alvimopan |
| PDB ID | Ligand | Charged Residues | Polar Residues | Hydrophobic Residues |
|---|---|---|---|---|
| 4DKL | β-FNA | 5 | 2 | 8 |
| 7UL4 | Alvimopan | 3 | 6 | 11 |
| 5C1M | BU72 | 4 | 4 | 10 |
| 8EF5 | Fentanyl | 1 | 3 | 13 |
| 8EF6 | Morphine | 2 | 1 | 9 |
| 6DDE | DAMGO | 4 | 5 | 10 |
| 6DDF6DDE | DAMGODAMGO | 44 | 55 | 1010 |
| 6DDF8EFQ | DAMGO | 24 | 55 | 1410 |
| 7T2H | Lofentanil | 2 | 4 | 12 |
| 7T2G | Mitragynine pseudoindoxyl | 3 | 5 | 10 |
| 8EFB | Oliceridine (TRV130) | 1 | 3 | 15 |
| 8EFL | SR17018 | 1 | 4 | 13 |
| 8EFO | PZM21 | 2 | 4 | 13 |
| 7SBF | PZM21 | 3 | 5 | 11 |
| 7SCG | FH210 | 3 | 5 | 9 |
| PDB ID | Ligand | SASA (Å2) | SAV (Å3) |
|---|---|---|---|
| 4DKL | β-FNA | 724 | 926 |
| 7UL4 | Alvimopan | 1016 | 1296 |
| 5C1M | BU72 | 1212 | 619 |
| 8EF5 | Fentanyl | 1062 | 1324 |
| 8EF6 | Morphine | 960 | 1097 |
| 6DDE | DAMGO | 716 | 682 |
| 6DDF | DAMGO | 631 | 588 |
| 8EFQ | DAMGO | 827 | 938 |
| 7T2H | Lofentanil | 821 | 1024 |
| 7T2G | Mitragynine pseudoindoxyl | 612 | 674 |
| 8EFB | Oliceridine (TRV130) | 1052 | 1067 |
| 8EFL | SR17018 | 1284 | 1875 |
| 8EFO | PZM21 | 949 | 989 |
| 7SBF | PZM21 | 1341 | 2126 |
| 7SCG | FH210 | 990 | 1330 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Liu, J.; Dong, F.; Chang, N.; Huang, R.; Xia, M.; Patterson, T.A.; Hong, H. Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor. Int. J. Mol. Sci. 2023, 24, 7042. https://doi.org/10.3390/ijms24087042
Li Z, Liu J, Dong F, Chang N, Huang R, Xia M, Patterson TA, Hong H. Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor. International Journal of Molecular Sciences. 2023; 24(8):7042. https://doi.org/10.3390/ijms24087042
Chicago/Turabian StyleLi, Zoe, Jie Liu, Fan Dong, Nancy Chang, Ruili Huang, Menghang Xia, Tucker A. Patterson, and Huixiao Hong. 2023. "Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor" International Journal of Molecular Sciences 24, no. 8: 7042. https://doi.org/10.3390/ijms24087042
APA StyleLi, Z., Liu, J., Dong, F., Chang, N., Huang, R., Xia, M., Patterson, T. A., & Hong, H. (2023). Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor. International Journal of Molecular Sciences, 24(8), 7042. https://doi.org/10.3390/ijms24087042