
Can Electronegative LDL Act as a Multienzymatic Complex?

 , ,
, , 
Abstract
:1. LDL Modification and Atherosclerosis
2. Association of LDL(−) with Disease
3. Atherogenic Properties of LDL(−)
4. Some Potentially Protective Properties of LDL(−)
5. Structural Alterations in LDL(−)
6. Enzymatic Activities Associated with LDL(−)
7. Origin of PLC-Like and CDase-Like Activities Associated with LDL(−)
8. Could the Enzymatic Activities Associated with LDL(−) Act Cooperatively as a sort of Enzymatic Complex?
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ross, R. Atherosclerosis as an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Lipoprotein receptors and the control of plasma LDL cholesterol levels. Eur. Heart J. 1992, 13, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Kruth, H.S.; Jones, N.L.; Huang, W.; Zhao, B.; Ishii, I.; Chang, J.; Combs, C.A.; Malide, D.; Zhang, W.-Y. Macropinocytosis Is the Endocytic Pathway That Mediates Macrophage Foam Cell Formation with Native Low Density Lipoprotein. J. Biol. Chem. 2005, 280, 2352–2360. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I. 2016 Russell Ross Memorial Lecture in Vascular Biology: Molecular-Cellular Mechanisms in the Progression of Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 37, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N. Modified and Dysfunctional Lipoproteins in Atherosclerosis: Effectors or Biomarkers? Curr. Med. Chem. 2019, 26, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Quesada, J.L.; Rivas-Urbina, A.; Benitez, S.; Perez, A. Modified low-density lipoproteins as biomarkers in diabetes and metabolic syndrome. Front. Biosci. 2018, 23, 1220–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.-F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [Green Version]
- Ke, L.-Y.; Law, S.H.; Mishra, V.K.; Parveen, F.; Chan, H.-C.; Lu, Y.-H.; Chu, C.-S. Molecular and Cellular Mechanisms of Electronegative Lipoproteins in Cardiovascular Diseases. Biomedicines 2020, 8, 550. [Google Scholar] [CrossRef]
- Sanchez-Quesada, J.L.; Benítez, S.; Ordonez-Llanos, J. Electronegative low-density lipoprotein. Curr. Opin. Infect. Dis. 2004, 15, 329–335. [Google Scholar] [CrossRef]
- Mello, A.P.Q.; da Silva, I.T.; Abdalla, D.S.P.; Damasceno, N.R.T. Electronegative low-density lipoprotein: Origin and impact on health and disease. Atherosclerosis 2011, 215, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Quesada, J.L.; Benítez, S.; Otal, C.; Franco, M.; Blanco-Vaca, F.; Ordóñez-Llanos, J. Density distribution of electronegative LDL in normolipemic and hyperlipemic subjects. J. Lipid Res. 2002, 43, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Bittolo-Bon, G.; Cazzolato, G. Analytical capillary isotachophoresis of total plasma lipoproteins: A new tool to identify atherogenic low density lipoproteins. J. Lipid Res. 1999, 40, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Cazzolato, G.; Avogaro, P.; Bittolo-Bon, G. Characterization of a more electronegatively charged LDL subfraction by ion exchange HPLC. Free. Radic. Biol. Med. 1991, 11, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Akyol, S.; Lu, J.; Akyol, O.; Akcay, F.; Armutcu, F.; Ke, L.-Y.; Chen, C.-H. The role of electronegative low-density lipoprotein in cardiovascular diseases and its therapeutic implications. Trends Cardiovasc. Med. 2017, 27, 239–246. [Google Scholar] [CrossRef]
- Sánchez-Quesada, J.L.; Estruch, M.; Benítez, S.; Ordóñez-Llanos, J. Electronegative LDL: A useful biomarker of cardiovascular risk? Clin. Lipidol. 2012, 7, 345–359. [Google Scholar] [CrossRef]
- Rivas-Urbina, A.; Rull, A.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Electronegative LDL: An Active Player in Atherogenesis or a By- Product of Atherosclerosis? Curr. Med. Chem. 2019, 26, 1665–1679. [Google Scholar] [CrossRef]
- Vural, H.; Armutcu, F.; Akyol, O.; Weiskirchen, R. The potential pathophysiological role of altered lipid metabolism and electronegative low-density lipoprotein (LDL) in non-alcoholic fatty liver disease and cardiovascular diseases. Clin. Chim. Acta 2021, 523, 374–379. [Google Scholar] [CrossRef]
- Akyol, O.; Chowdhury, I.; Akyol, H.R.; Tessier, K.; Vural, H.; Akyol, S. Why are cardiovascular diseases more common among patients with severe mental illness? The potential involvement of electronegative low-density lipoprotein (LDL) L5. Med. Hypotheses 2020, 142, 109821. [Google Scholar] [CrossRef]
- Chen, C.; Ke, L.; Chan, H.; Chu, C.; Lee, A.-S.; Lin, K.-D.; Lee, M.; Hsiao, P.; Chen, C.; Shin, S. Electronegative low--density lipoprotein of patients with metabolic syndrome induces pathogenesis of aorta through disruption of the stimulated by retinoic acid 6 cascade. J. Diabetes Investig. 2019, 11, 535–544. [Google Scholar] [CrossRef]
- Chu, C.-S.; Law, S.H.; Lenzen, D.; Tan, Y.-H.; Weng, S.-F.; Ito, E.; Wu, J.-C.; Chen, C.-H.; Chan, H.-C.; Ke, L.-Y. Clinical Significance of Electronegative Low-Density Lipoprotein Cholesterol in Atherothrombosis. Biomedicines 2020, 8, 254. [Google Scholar] [CrossRef]
- Avogaro, P.; Cazzolato, G.; Bittolo-Bon, G. Some questions concerning a small, more electronegative LDL circulating in human plasma. Atherosclerosis 1991, 91, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.-C.; Ke, L.-Y.; Chu, C.-S.; Lee, A.-S.; Shen, M.-Y.; Cruz, M.A.; Hsu, J.-F.; Cheng, K.-H.; Chan, H.-C.B.; Lu, J.; et al. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.-Y.; Chen, F.-Y.; Hsu, J.-F.; Fu, R.-H.; Chang, C.-M.; Chang, C.-T.; Liu, C.-H.; Wu, J.-R.; Lee, A.-S.; Chan, H.-C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Gambino, R.; Uberti, B.; Alemanno, N.; Pisu, E.; Pagano, G.; Cassader, M. In vivo oxidizability of LDL in type 2 diabetic patients in good and poor glycemic control. Atherosclerosis 2004, 173, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Matsunaga, A.; Rainwater, D.L.; Miura, S.-I.; Noda, K.; Nishikawa, H.; Uehara, Y.; Shirai, K.; Ogawa, M.; Saku, K. Effects of rosuvastatin on electronegative LDL as characterized by capillary isotachophoresis: The ROSARY Study. J. Lipid Res. 2009, 50, 1832–1841. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, P.; Bon, G.B.; Cazzolato, G. Presence of a modified low density lipoprotein in humans. Arter. Off. J. Am. Hearth Assoc. Inc. 1988, 8, 79–87. [Google Scholar] [CrossRef] [Green Version]
- De Castellarnau, C.; Sánchez-Quesada, J.L.; Benítez, S.; Rosa, R.; Caveda, L.; Vila, L.; Ordóñez-Llanos, J. Electronegative LDL From Normolipemic Subjects Induces IL-8 and Monocyte Chemotactic Protein Secretion by Human Endothelial Cells. Arter. Thromb. Vasc. Biol. 2000, 20, 2281–2287. [Google Scholar] [CrossRef] [Green Version]
- de Castellarnau, C.; Bancells, C.; Benítez, S.; Reina, M.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Atherogenic and inflammatory profile of human arterial endothelial cells (HUAEC) in response to LDL subfractions. Clin. Chim. Acta 2007, 376, 233–236. [Google Scholar] [CrossRef]
- Pech-Amsellem, M.A.; Myara, I.; Storogenko, M.; DeMuth, K.; Proust, A.; Moatti, N. Enhanced modifications of low-density lipoproteins (LDL) by endothelial cells from smokers: A possible mechanism of smoking-related atherosclerosis. Cardiovasc. Res. 1996, 31, 975–983. [Google Scholar] [CrossRef]
- Stancel, N.; Chen, C.-C.; Ke, L.-Y.; Chu, C.-S.; Lu, J.; Sawamura, T.; Chen, C.-H. Interplay between CRP, Atherogenic LDL, and LOX-1 and Its Potential Role in the Pathogenesis of Atherosclerosis. Clin. Chem. 2016, 62, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-S.; Wang, Y.-C.; Lu, L.-S.; Walton, B.; Yilmaz, H.R.; Huang, R.Y.; Sawamura, T.; Dixon, R.A.F.; Lai, W.-T.; Chen, C.-H.; et al. Electronegative Low-Density Lipoprotein Increases C-Reactive Protein Expression in Vascular Endothelial Cells through the LOX-1 Receptor. PLoS ONE 2013, 8, e70533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estruch, M.; Bancells, C.; Beloki, L.; Sanchez-Quesada, J.L.; Ordóñez-Llanos, J.; Benitez, S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis 2013, 229, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Estruch, M.; Miñambres, I.; Sanchez-Quesada, J.L.; Soler, M.; Pérez, A.; Ordoñez-Llanos, J.; Benitez, S. Increased inflammatory effect of electronegative LDL and decreased protection by HDL in type 2 diabetic patients. Atherosclerosis 2017, 265, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Estruch, M.; Rajamäki, K.; Sanchez-Quesada, J.; Kovanen, P.; Öörni, K.; Benitez, S.; Ordoñez-Llanos, J. Electronegative LDL induces priming and inflammasome activation leading to IL-1β release in human monocytes and macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Estruch, M.; Sánchez-Quesada, J.L.; Ordóñez-Llanos, J.; Benítez, S. Ceramide-enriched LDL induces cytokine release through TLR4 and CD14 in monocytes. Similarities with electronegative LDL. Clin. Investig. Arter. Publ. Of. Soc. Espanola Arter. 2014, 26, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, J.-H.; Burns, A.R.; Chen, H.-H.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.-Y.; Sawamura, T.; Chen, C.-H. Mediation of Electronegative Low-Density Lipoprotein Signaling by LOX-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef]
- Tang, D.; Lu, J.; Walterscheid, J.P.; Chen, H.-H.; Engler, D.A.; Sawamura, T.; Chang, P.-Y.; Safi, H.J.; Yang, C.-Y.; Chen, C.-H. Electronegative LDL circulating in smokers impairs endothelial progenitor cell differentiation by inhibiting Akt phosphorylation via LOX-1. J. Lipid Res. 2008, 49, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Lee, A.-S.; Lu, L.-S.; Ke, L.-Y.; Chen, W.-Y.; Dong, J.-W.; Lu, J.; Chen, Z.; Chu, C.-S.; Chan, H.-C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef] [Green Version]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordoñez-Llanos, J.; Benitez, S. Inflammatory intracellular pathways activated by electronegative LDL in monocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 963–969. [Google Scholar] [CrossRef]
- Estruch, M.; Sanchez-Quesada, J.L.; Beloki, L.; Ordoñez-Llanos, J.; Benitez, S. The Induction of Cytokine Release in Monocytes by Electronegative Low-Density Lipoprotein (LDL) Is Related to Its Higher Ceramide Content than Native LDL. Int. J. Mol. Sci. 2013, 14, 2601–2616. [Google Scholar] [CrossRef] [Green Version]
- Benítez, S.; Camacho, M.; Arcelus, R.; Vila, L.; Bancells, C.; Ordonez-Llanos, J.; Sánchez-Quesada, J.L. Increased lysophosphatidylcholine and non-esterified fatty acid content in LDL induces chemokine release in endothelial cells: Relationship with electronegative LDL. Atherosclerosis 2004, 177, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Sevanian, A.; Bittolo-Bon, G.; Cazzolato, G.; Hodis, H.; Hwang, J.; Zamburlini, A.; Maiorino, M.; Ursini, F. LDL- is a lipid hydroperoxide-enriched circulating lipoprotein. J. Lipid Res. 1997, 38, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Abdalla, D.S.; Sevanian, A. Characterization of Cholesterol Oxidation Products Formed by Oxidative Modification of Low Density Lipoprotein. Free. Radic. Biol. Med. 1997, 23, 202–214. [Google Scholar] [CrossRef]
- Chang, S.-F.; Chang, P.-Y.; Chou, Y.-C.; Lu, S.-C. Electronegative LDL Induces M1 Polarization of Human Macrophages Through a LOX-1-Dependent Pathway. Inflammation 2020, 43, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-Y.; Chang, S.-F.; Chang, T.-Y.; Su, H.-M.; Lu, S.-C. Synergistic effects of electronegative-LDL- and palmitic-acid-triggered IL-1β production in macrophages via LOX-1- and voltage-gated-potassium-channel-dependent pathways. J. Nutr. Biochem. 2021, 97, 108767. [Google Scholar] [CrossRef]
- Demuth, K.; Myara, I.; Chappey, B.; Vedie, B.; Pech-Amsellem, M.A.; Haberland, M.E.; Moatti, N. A Cytotoxic Electronegative LDL Subfraction Is Present in Human Plasma. Arter. Thromb. Vasc. Biol. 1996, 16, 773–783. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Chen, H.-H.; Huang, M.T.; Raya, J.L.; Yang, J.-H.; Chen, C.-H.; Gaubatz, J.W.; Pownall, H.J.; Taylor, A.A.; Ballantyne, C.M.; et al. Pro-apoptotic low-density lipoprotein subfractions in type II diabetes. Atherosclerosis 2007, 193, 283–291. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Chen, Y.-F.; Chan, H.-C.; Chung, C.-H.; Peng, H.-Y.; Ho, Y.-C.; Chen, C.-H.; Chang, K.-C.; Tang, C.-H.; Lee, A.-S. Role of apolipoprotein E in electronegative low-density lipoprotein-induced mitochondrial dysfunction in cardiomyocytes. Metabolism 2020, 107, 154227. [Google Scholar] [CrossRef]
- Revuelta-López, E.; Cal, R.; Julve, J.; Rull, A.; Martínez-Bujidos, M.; Perez-Cuellar, M.; Ordoñez-Llanos, J.; Badimon, L.; Sanchez-Quesada, J.L.; Llorente-Cortés, V. Hypoxia worsens the impact of intracellular triglyceride accumulation promoted by electronegative low-density lipoprotein in cardiomyocytes by impairing perilipin 5 upregulation. Int. J. Biochem. Cell Biol. 2015, 65, 257–267. [Google Scholar] [CrossRef]
- Puig, N.; Montolio, L.; Camps-Renom, P.; Navarra, L.; Jiménez-Altayó, F.; Jiménez-Xarrié, E.; Sánchez-Quesada, J.L.; Benitez, S. Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages. Cells 2020, 9, 583. [Google Scholar] [CrossRef] [Green Version]
- Ligi, D.; Benitez, S.; Croce, L.; Rivas-Urbina, A.; Puig, N.; Ordóñez-Llanos, J.; Mannello, F.; Sanchez-Quesada, J.L. Electronegative LDL induces MMP-9 and TIMP-1 release in monocytes through CD14 activation: Inhibitory effect of glycosaminoglycan sulodexide. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Ziouzenkova, O.; Asatryan, L.; Sahady, D.; Orasanu, G.; Perrey, S.; Cutak, B.; Hassell, T.; Akiyama, T.E.; Berger, J.P.; Sevanian, A.; et al. Dual Roles for Lipolysis and Oxidation in Peroxisome Proliferation-Activator Receptor Responses to Electronegative Low Density Lipoprotein. J. Biol. Chem. 2003, 278, 39874–39881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benítez, S.; Bancells, C.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Pro-inflammatory action of LDL(−) on mononuclear cells is counteracted by increased IL10 production. Biochim. et Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 613–622. [Google Scholar] [CrossRef]
- Benítez, S.; Sanchez-Quesada, J.L.; Ribas, V.; Jorba, O.; Blanco-Vaca, F.; González-Sastre, F.; Ordóñez-Llanos, J. Platelet-Activating Factor Acetylhydrolase Is Mainly Associated with Electronegative Low-Density Lipoprotein Subfraction. Circulation 2003, 108, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancells, C.; Canals, F.; Benítez, S.; Colomé, N.; Julve, J.; Ordonez-Llanos, J.; Sánchez-Quesada, J.L. Proteomic analysis of electronegative low-density lipoprotein. J. Lipid Res. 2010, 51, 3508–3515. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Bujidos, M.; Rull, A.; González-Cura, B.; Pérez-Cuéllar, M.; Montoliu-Gaya, L.; Villegas, S.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2014, 29, 1688–1700. [Google Scholar] [CrossRef]
- Hevonoja, T.; Pentikäinen, M.O.; Hyvönen, M.T.; Kovanen, P.T.; Ala-Korpela, M. Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim. et Biophys. Acta Mol. Cell Biol. Lipids 2000, 1488, 189–210. [Google Scholar] [CrossRef]
- Sánchez-Quesada, J.L.; Villegas, S.; Ordóñez-Llanos, J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr. Opin. Infect. Dis. 2012, 23, 479–486. [Google Scholar] [CrossRef]
- Parasassi, T.; De Spirito, M.; Mei, G.; Brunelli, R.; Greco, G.; Lenzi, L.; Maulucci, G.; Nicolai, E.; Papi, M.; Arcovito, G.; et al. Low density lipoprotein misfolding and amyloidogenesis. FASEB J. 2008, 22, 2350–2356. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, R.; Spirito, M.; Mei, G.; Papi, M.; Perrone, G.; Stefanutti, C.; Parasassi, T. Misfolding of Apoprotein B-100, LDL Aggregation and 17-β -estradiol in Atherogenesis. Curr. Med. Chem. 2014, 21, 2276–2283. [Google Scholar] [CrossRef]
- Bancells, C.; Benítez, S.; Jauhiainen, M.; Ordonez-Llanos, J.; Kovanen, P.T.; Villegas, S.; Sanchez-Quesada, J.L.; Öörni, K. High binding affinity of electronegative LDL to human aortic proteoglycans depends on its aggregation level. J. Lipid Res. 2009, 50, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancells, C.; Benítez, S.; Ordóñez-Llanos, J.; Öörni, K.; Kovanen, P.T.; Milne, R.W.; Sánchez-Quesada, J.L. Immunochemical Analysis of the Electronegative LDL Subfraction Shows That Abnormal N-terminal Apolipoprotein B Conformation Is Involved in Increased Binding to Proteoglycans. J. Biol. Chem. 2011, 286, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.; Villegas, S.; Benítez, S.; Bancells, C.; Diercks, T.; Ordonez-Llanos, J.; Sánchez-Quesada, J.L. 2D-NMR reveals different populations of exposed lysine residues in the apoB-100 protein of electronegative and electropositive fractions of LDL particles. J. Lipid Res. 2010, 51, 1560–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, L.-Y.; Engler, D.A.; Lu, J.; Matsunami, R.K.; Chan, H.-C.; Wang, G.-J.; Yang, C.-Y.; Chang, J.-G.; Chen, C.-H. Chemical composition-oriented receptor selectivity of L5, a naturally occurring atherogenic low-density lipoprotein. Pure Appl. Chem. 2011, 83, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Scraba, D.G.; Ryan, R.O. Prevention of phospholipase-C induced aggregation of low density lipoprotein by amphipathic apolipoproteins. FEBS Lett. 1993, 316, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Khoo, J.C.; Miller, E.; McLoughlin, P.; Steinberg, D. Prevention of low density lipoprotein aggregation by high density lipoprotein or apolipoprotein A-I. J. Lipid Res. 1990, 31, 645–652. [Google Scholar] [CrossRef]
- Brunelli, R.; Balogh, G.; Costa, G.; De Spirito, M.; Greco, G.; Mei, G.; Nicolai, E.; Vigh, L.; Ursini, F.; Parasassi, T. Estradiol Binding Prevents ApoB-100 Misfolding in Electronegative LDL(−). Biochemistry 2010, 49, 7297–7302. [Google Scholar] [CrossRef]
- Benítez, S.; Villegas, V.; Bancells, C.; Jorba, O.; González-Sastre, F.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Impaired Binding Affinity of Electronegative Low-Density Lipoprotein (LDL) to the LDL Receptor Is Related to Nonesterified Fatty Acids and Lysophosphatidylcholine Content. Biochemistry 2004, 43, 15863–15872. [Google Scholar] [CrossRef]
- Bancells, C.; Villegas, S.; Blanco, F.; Benítez, S.; Gállego, I.; Beloki, L.; Pérez-Cuellar, M.; Ordonez-Llanos, J.; Sánchez-Quesada, J.L. Aggregated Electronegative Low Density Lipoprotein in Human Plasma Shows a High Tendency toward Phospholipolysis and Particle Fusion. J. Biol. Chem. 2010, 285, 32425–32435. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Davies, K.J.; Maiorino, M.; Parasassi, T.; Sevanian, A. Atherosclerosis: Another protein misfolding disease? Trends Mol. Med. 2002, 8, 370–374. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Gursky, O. Effects of phospholipase A2 and its products on structural stability of human LDL: Relevance to formation of LDL-derived lipid droplets. J. Lipid Res. 2011, 52, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaubatz, J.W.; Gillard, B.K.; Massey, J.B.; Hoogeveen, R.C.; Huang, M.; Lloyd, E.E.; Raya, J.L.; Yang, C.-Y.; Pownall, H.J. Dynamics of dense electronegative low density lipoproteins and their preferential association with lipoprotein phospholipase A2. J. Lipid Res. 2007, 48, 348–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tselepis, A.D.; Dentan, C.; Karabina, S.-A.P.; Chapman, M.J.; Ninio, E. PAF-Degrading Acetylhydrolase Is Preferentially Associated with Dense LDL and VHDL-1 in Human Plasma. Catalytic characteristics and relation to the monocyte-derived enzyme. Arter. Thromb. Vasc. Biol. 1995, 15, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Stafforini, D.M. Plasma PAF-AH (PLA2G7). Biochemical Properties, Association with LDLs and HDLs, and Regulation of Expression. Enzymes 2015, 38, 71–93. [Google Scholar] [CrossRef]
- Chen, C.-H. Platelet-activating factor acetylhydrolase: Is it good or bad for you? Curr. Opin. Infect. Dis. 2004, 15, 337–341. [Google Scholar] [CrossRef]
- Stafforini, D.M.; Zimmerman, G.A. Unraveling the PAF-AH/Lp-PLA2 controversy. J. Lipid Res. 2014, 55, 1811–1814. [Google Scholar] [CrossRef] [Green Version]
- MacPhee, C.H.; Moores, K.E.; Boyd, H.F.; Dhanak, D.; Ife, R.J.; Leach, C.A.; Leake, D.S.; Milliner, K.J.; Patterson, R.A.; Suckling, K.E.; et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: Use of a novel inhibitor. Biochem. J. 1999, 338, 479–487. [Google Scholar] [CrossRef]
- Bancells, C.; Sánchez-Quesada, J.L.; Birkelund, R.; Ordóñez-Llanos, J.; Benítez, S. HDL and electronegative LDL exchange anti- and pro-inflammatory properties. J. Lipid Res. 2010, 51, 2947–2956. [Google Scholar] [CrossRef] [Green Version]
- Bancells, C.; Benítez, S.; Villegas, S.; Jorba, O.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Novel Phospholipolytic Activities Associated with Electronegative Low-Density Lipoprotein Are Involved in Increased Self-Aggregation. Biochemistry 2008, 47, 8186–8194. [Google Scholar] [CrossRef]
- Rivas-Urbina, A.; Rull, A.; Montoliu-Gaya, L.; Pérez-Cuellar, M.; Ordóñez-Llanos, J.; Villegas, S.; Sánchez-Quesada, J.L. Low-density lipoprotein aggregation is inhibited by apolipoprotein J-derived mimetic peptide D-[113–122]apoJ. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158541. [Google Scholar] [CrossRef]
- Puig, N.; Rives, J.; Estruch, M.; Aguilera-Simon, A.; Rotllan, N.; Camacho, M.; Colomé, N.; Canals, F.; Sánchez-Quesada, J.L.; Benitez, S. Presence of Ceramidase Activity in Electronegative LDL. Int. J. Mol. Sci. 2022, 24, 165. [Google Scholar] [CrossRef]
- Lu, M.; Gursky, O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol. Concepts 2013, 4, 501–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnunen, P.K.J. Sphingomyelinase Activity of LDL A Link between Atherosclerosis, Ceramide, and Apoptosis? Trends Cardiovasc. Med. 2002, 12, 37–42. [Google Scholar] [CrossRef]
- Puig, N.; Estruch, M.; Jin, L.; Sanchez-Quesada, J.L.; Benitez, S. The Role of Distinctive Sphingolipids in the Inflammatory and Apoptotic Effects of Electronegative LDL on Monocytes. Biomolecules 2019, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Holopainen, J.M.; Medina, O.P.; Metso, A.J.; Kinnunen, P.K. Sphingomyelinase Activity Associated with Human Plasma Low Density Lipoprotein. J. Biol. Chem. 2000, 275, 16484–16489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, L.-Y.; Chan, H.-C.; Chen, C.-C.; Lu, J.; Marathe, G.K.; Chu, C.-S.; Chan, H.-C.; Wang, C.-Y.; Tung, Y.-C.; McIntyre, T.M.; et al. Enhanced Sphingomyelinase Activity Contributes to the Apoptotic Capacity of Electronegative Low-Density Lipoprotein. J. Med. Chem. 2016, 59, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Zawadzki, Z.; Milne, R.W.; Marcel, Y.L. Cu2(+)-mediated oxidation of dialyzed plasma: Effects on low and high density lipoproteins and cholesteryl ester transfer protein. J. Lipid Res. 1991, 32, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wilensky, R.L.; Macphee, C.H. Lipoprotein-associated phospholipase A2 and atherosclerosis. Curr. Opin. Infect. Dis. 2009, 20, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Stafforini, D.M.; Tjoelker, L.W.; McCormick, S.P.A.; Vaitkus, D.; McIntyre, T.M.; Gray, P.W.; Young, S.G.; Prescott, S.M. Molecular Basis of the Interaction between Plasma Platelet-activating Factor Acetylhydrolase and Low Density Lipoprotein. J. Biol. Chem. 1999, 274, 7018–7024. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Atkinson, D. Immuno-electron cryo-microscopy imaging reveals a looped topology of apoB at the surface of human LDL. J. Lipid Res. 2011, 52, 1111–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]


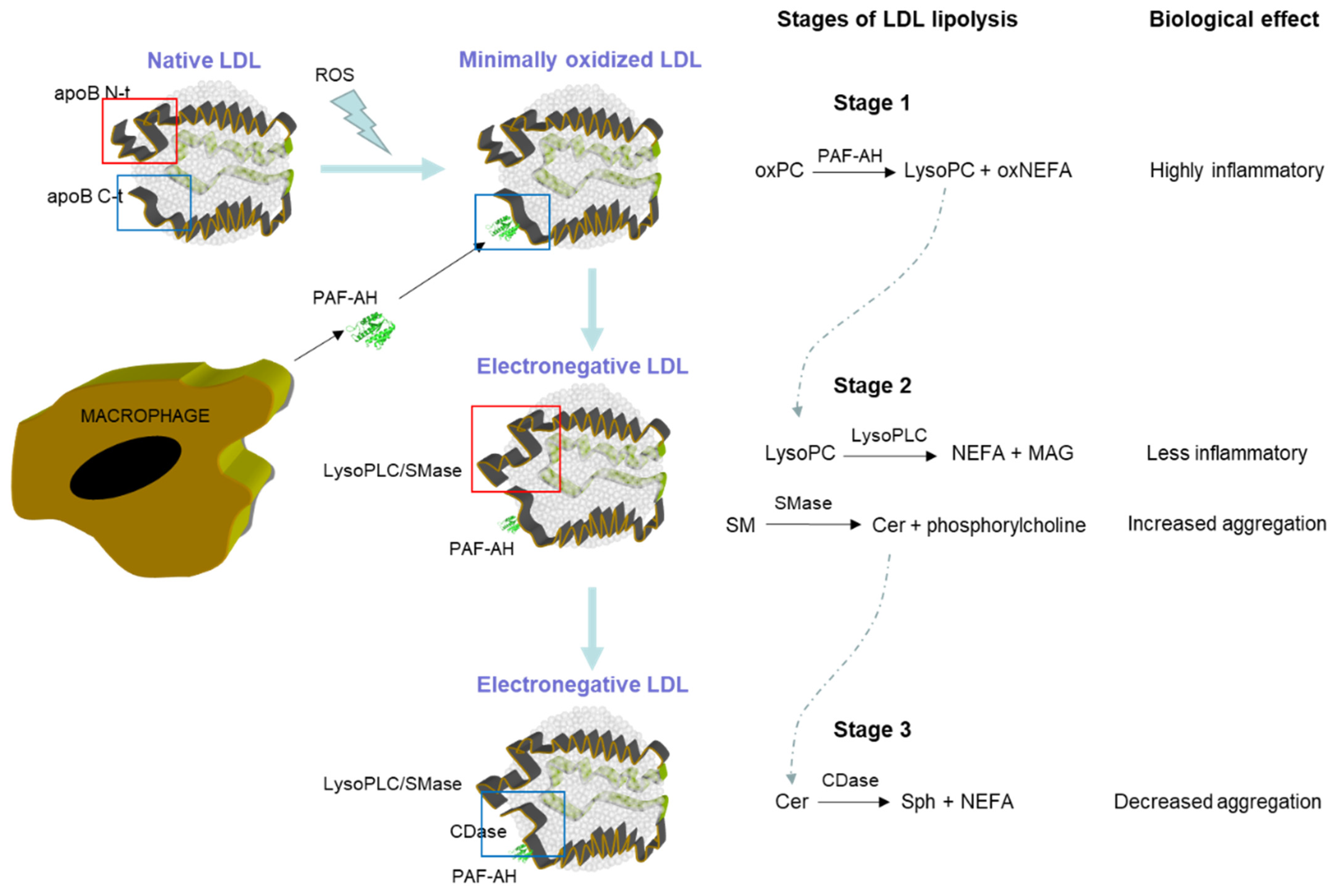{kind=link}
| Nonpathological | Chronic Diseases | Acute Phase Diseases |
|---|---|---|
|
|
|
| Component | Alteration |
|---|---|
| Proteins |
|
| Lipids |
|
| Enzymatic Activity | Substrate | Product |
|---|---|---|
| PAF-AH activity | Oxidized PC | LysoPC Oxidized short-chain NEFA |
| PLC-like activity | LysoPC | Monoacylglycerol Phosphorylcholine |
| SMase-like activity | SM | Ceramide Phosphorylcholine |
| CDase-like activity | Ceramide | Sphingosine NEFA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benitez, S.; Puig, N.; Rives, J.; Solé, A.; Sánchez-Quesada, J.L. Can Electronegative LDL Act as a Multienzymatic Complex? Int. J. Mol. Sci. 2023, 24, 7074. https://doi.org/10.3390/ijms24087074
Benitez S, Puig N, Rives J, Solé A, Sánchez-Quesada JL. Can Electronegative LDL Act as a Multienzymatic Complex? International Journal of Molecular Sciences. 2023; 24(8):7074. https://doi.org/10.3390/ijms24087074
Chicago/Turabian StyleBenitez, Sonia, Núria Puig, José Rives, Arnau Solé, and José Luis Sánchez-Quesada. 2023. "Can Electronegative LDL Act as a Multienzymatic Complex?" International Journal of Molecular Sciences 24, no. 8: 7074. https://doi.org/10.3390/ijms24087074
APA StyleBenitez, S., Puig, N., Rives, J., Solé, A., & Sánchez-Quesada, J. L. (2023). Can Electronegative LDL Act as a Multienzymatic Complex? International Journal of Molecular Sciences, 24(8), 7074. https://doi.org/10.3390/ijms24087074