
Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review


Abstract
:1. Introduction
2. Methods
2.1. The Literature Search Strategy
2.2. Selection Criteria
2.2.1. Inclusion Criteria
- Proteomic studies
- Genome-wide transcriptomic studies
- Information on differentially expressed proteins/genes/pathways related to control conditions
- Samples employed either from human patients or cell lines of human origin
- Non-review articles.
2.2.2. Exclusion Criteria
- Studies conducted on nonhuman tissue or cell lines
- Interventional studies
- Literature reviews.
2.3. Data Extraction and Management
3. Results
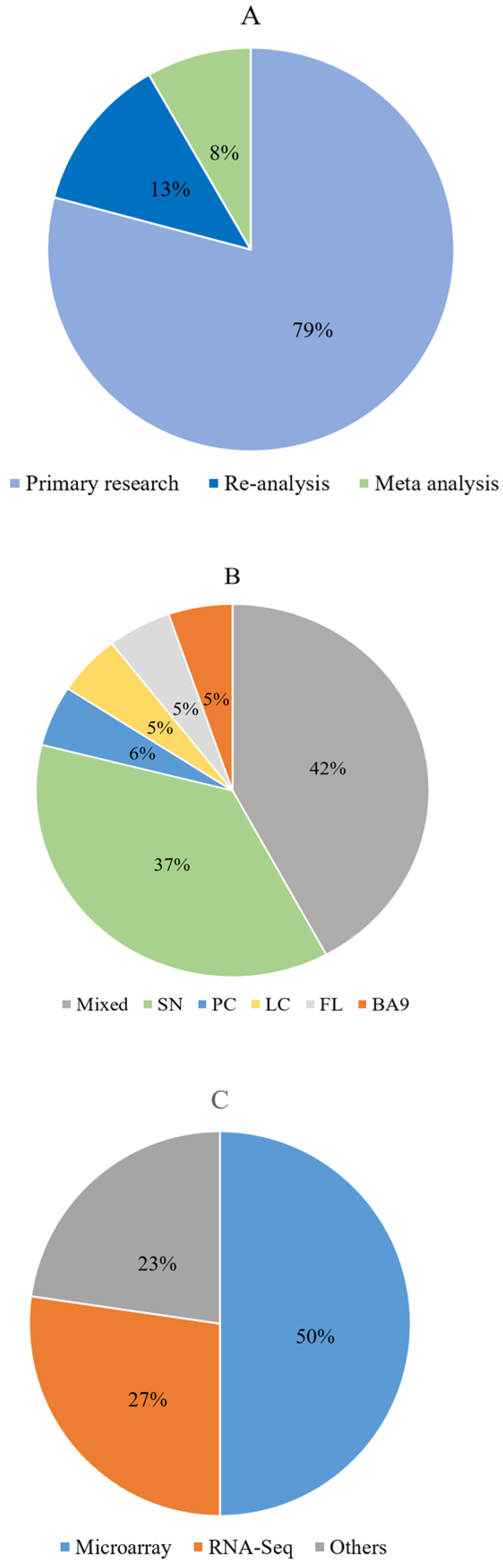
3.1. Literature Search Results
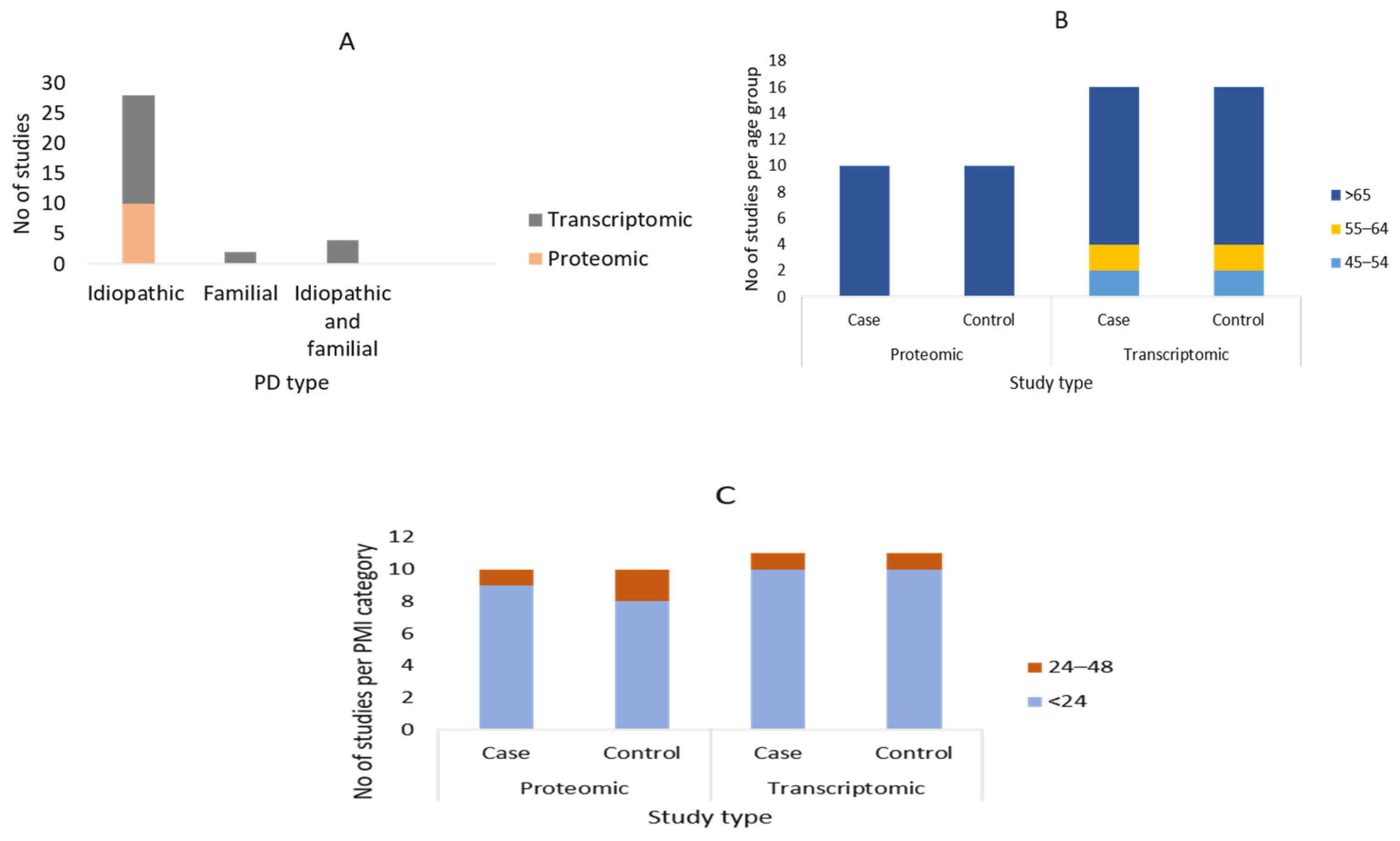
3.2. Summary of the Demographic Characteristics of the Study Participants
3.3. Description of the Included Articles
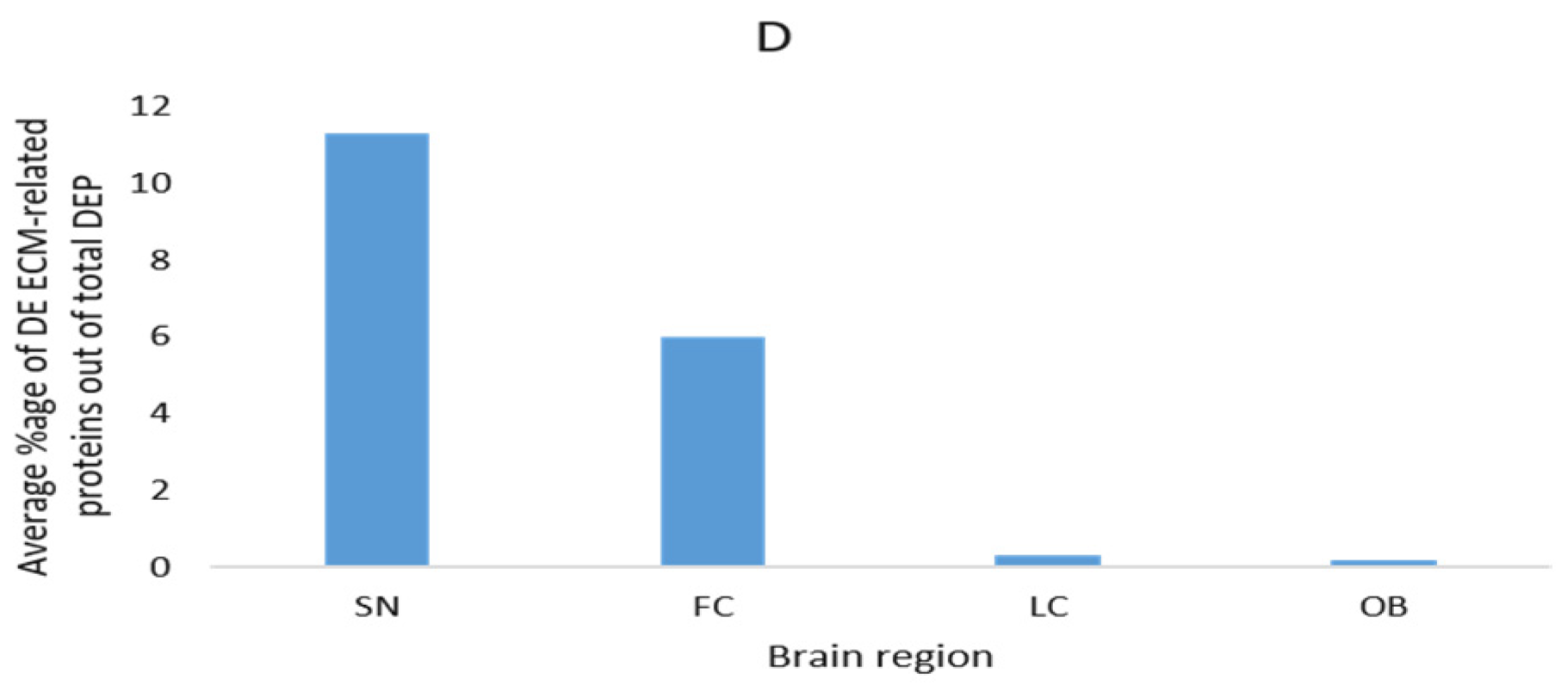
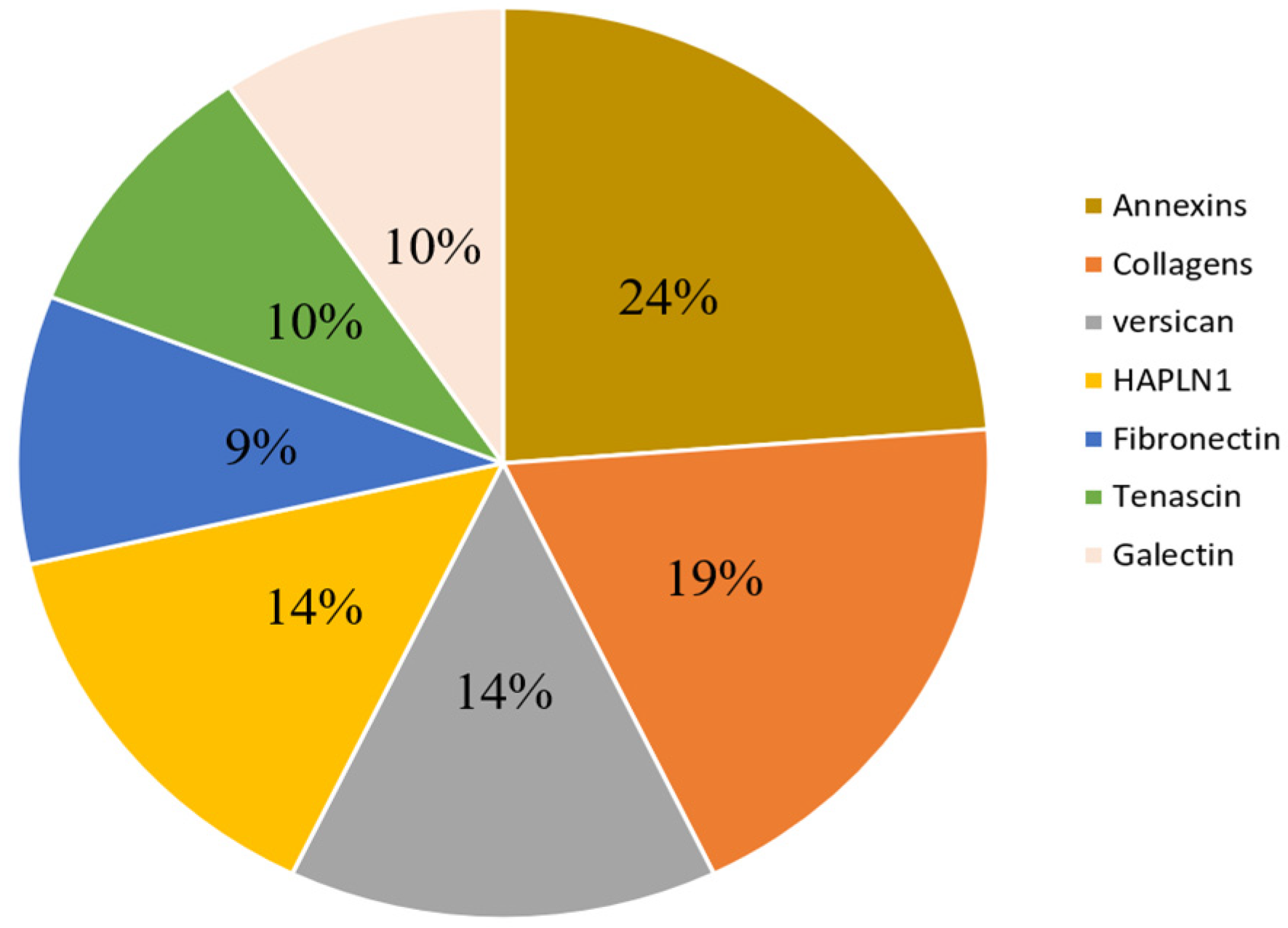
3.4. Description of Commonly Reported Differentially Expressed ECM and ECM-Related Proteins
3.5. Description of Commonly Reported Differentially Expressed ECM and ECM-Related Gene Groups/Pathways/Processes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lang, A.E.; Obeso, J.A. Time to move beyond nigrostriatal dopamine deficiency in Parkinson’s disease. Ann. Neurol. 2004, 55, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E. A critical appraisal of the premotor symptoms of Parkinson’s disease: Potential usefulness in early diagnosis and design of neuroprotective trials. Mov. Disord. 2011, 26, 775–783. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S.J.N. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Quik, M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci. 2004, 27, 561–568. [Google Scholar] [CrossRef]
- Dulski, J.; Uitti, R.J.; Ross, O.A.; Wszolek, M.D.; Zbigniew, K. Genetic architecture of Parkinson’s disease subtypes—Review of the literature. Front. Aging Neurosci. 2022, 14, 1243. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2023, 68, 125–130. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, G.; Zhou, Q.; Xie, Y.; Wang, Z.; Fang, Z.; Lu, B.; Qin, L.; Zhao, Y.; Zhang, R.; et al. Gene4PD: A Comprehensive Genetic Database of Parkinson’s Disease. Front. Neurosci. 2021, 15, 679568. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Parkinson’s Disease. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 2021–2035. [Google Scholar]
- Tewari, B.P.; Chaunsali, L.; Prim, C.E.; Sontheimer, H. A glial perspective on the extracellular matrix and perineuronal net remodeling in the central nervous system. Front. Aging Neurosci. 2022, 16, 1022754. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Michell, A.W.; Barker, R.A. Parkinson’s disease—The continuing search for biomarkers. Clin. Chem. Lab. Med. 2011, 49, 393–401. [Google Scholar] [CrossRef]
- Cragg, B. Brain extracellular space fixed for electron microscopy. Neurosci. Lett. 1979, 15, 301–306. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016. 97, 4–27. [CrossRef]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef]
- Novak, U.; Kaye, A.H. Extracellular matrix and the brain: Components and function. J. Clin. Neurosci. 2000, 7, 280–290. [Google Scholar] [CrossRef]
- Nicholson, C.; Syková, E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998, 21, 207–215. [Google Scholar] [CrossRef]
- Yue, B. Biology of the Extracellular Matrix: An Overview. J. Glaucoma 2014, 23, S20. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Wong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef]
- Krishnaswamy, V.R.; Benbenishty, A.; Blinder, P.; Sagi, I. Demystifying the extracellular matrix and its proteolytic remodeling in the brain: Structural and functional insights. Cell. Mol. Life Sci. 2019, 76, 3229–3248. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000, 80, 1267–1290. [Google Scholar] [CrossRef]
- Pintér, P.; Alpár, A. The Role of Extracellular Matrix in Human Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 11085. [Google Scholar] [CrossRef]
- Dankovich, T.M.; Rizzoli, S.O. The Synaptic Extracellular Matrix: Long-Lived, Stable, and Still Remarkably Dynamic. Front. Synaptic. Neurosci. 2022, 14, 854956. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M. Extracellular matrix molecules and synaptic plasticity. Nat. Rev. Neurosci. 2003, 4, 456–468. [Google Scholar] [CrossRef]
- Frischknecht, R.; Gundelfinger, E.D. The Brain’s Extracellular Matrix and Its Role in Synaptic Plasticity. In Synaptic Plasticity: Dynamics, Development and Disease; Kreutz, M.R., Sala, C., Eds.; Springer: Vienna, Austria, 2012; pp. 153–171. [Google Scholar]
- Dityatev, A.; Wehrle-Haller, B.; Pitkänen, A. Brain Extracellular Matrix in Health and Disease/Edited by Alexander Dityatev, Bernhard Wehrle-Haller, Asla Pitkänen, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Ruoslahti, E. Brain extracellular matrix. Glycobiology 1996, 6, 489–492. [Google Scholar] [CrossRef]
- Yang, S.; Kwok, J.C.; Fawcett, J.W. Neural ECM in regeneration and rehabilitation. Prog. Brain Res. 2014, 214, 179–192. [Google Scholar] [PubMed]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef]
- Sonbol, H.S. Extracellular Matrix Remodeling in Human Disease. J. Microsc. Ultrastruct. 2018, 6, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kurshan, P.T.; Phan, A.Q.; Wang, G.J.; Crane, M.M.; Lu, H.; Shen, K. Regulation of synaptic extracellular matrix composition is critical for proper synapse morphology. J. Neurosci. 2014, 34, 12678–12689. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R. The basement membrane/basal lamina of skeletal muscle. J. Biol. Chem. 2003, 278, 12601–12604. [Google Scholar] [CrossRef] [PubMed]
- Hockfield, S.; Kalb, R.G.; Zaremba, S.; Fryer, H. Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harb. Symp. Quant. Biol. 1990, 55, 505–514. [Google Scholar] [CrossRef]
- Kochlamazashvili, G.; Henneberger, C.; Bukalo, O.; Dvoretskova, E.; Senkov, O.; Lievens, P.M.; Westenbroek, R.; Engel, A.K.; Catterall, W.A.; Rusakov, D.A.; et al. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca(2+) channels. Neuron 2010, 67, 116–128. [Google Scholar] [CrossRef]
- Dansie, L.E.; Ethell, I.M. Casting a net on dendritic spines: The extracellular matrix and its receptors. Dev. Neurobiol. 2011, 71, 956–981. [Google Scholar] [CrossRef]
- Howell, M.D.; Gottschall, P.E. Lectican proteoglycans, their cleaving metalloproteinases, and plasticity in the central nervous system extracellular microenvironment. Neuroscience 2012, 217, 6–18. [Google Scholar] [CrossRef]
- Vargová, L.; Syková, E. Astrocytes and extracellular matrix in extrasynaptic volume transmission. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130608. [Google Scholar] [CrossRef]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Cargill, R.; Kohama, S.G.; Struve, J.; Su, W.; Banine, F.; Witkowski, E.; Back, S.A.; Sherman, L.S. Astrocytes in aged nonhuman primate brain gray matter synthesize excess hyaluronan. Neurobiol. Aging 2012, 33, 830.e13–830.e24. [Google Scholar] [CrossRef] [PubMed]
- Strackeljan, L.; Baczynska, E.; Cangalaya, C.; Baidoe-Ansah, D.; Wlodarczyk, J.; Kaushik, R.; Dityatev, A. Microglia Depletion-Induced Remodeling of Extracellular Matrix and Excitatory Synapses in the Hippocampus of Adult Mice. Cells 2021, 10, 1862. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403.e15. [Google Scholar] [CrossRef]
- Song, I.; Dityatev, A. Crosstalk between glia, extracellular matrix and neurons. Brain Res. Bull. 2018, 136, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Tansley, S.; Gu, N.; Guzmán, A.U.; Cai, W.; Wong, C.; Lister, K.C.; Muñoz-Pino, E.; Yousefpour, N.; Roome, R.B.; Heal, J.; et al. Microglia-mediated degradation of perineuronal nets promotes pain. Science 2022, 377, 80–86. [Google Scholar] [CrossRef]
- Soria, F.N.; Paviolo, C.; Doudnikoff, E.; Arotcarena, M.-L.; Lee, A.; Danné, N.; Mandal, A.K.; Gosset, P.; Dehay, B.; Groc, L.; et al. Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan remodeling. Nat. Commun. 2020, 11, 3440. [Google Scholar] [CrossRef]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Orlando, C.; Ster, J.; Gerber, U.; Fawcett, J.W.; Raineteau, O. Perisynaptic chondroitin sulfate proteoglycans restrict structural plasticity in an integrin-dependent manner. J. Neurosci. 2012, 32, 18009–18017. [Google Scholar] [CrossRef]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef]
- Hasegawa, S.; Yamaguchi, M.; Nagao, H.; Mishina, M.; Mori, K. Enhanced cell-to-cell contacts between activated microglia and pyramidal cell dendrites following kainic acid-induced neurotoxicity in the hippocampus. J. Neuroimmunol. 2007, 186, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood–Brain Barrier Pathology in Alzheimer’s and Parkinson’s Disease: Implications for Drug Therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Heindryckx, F.; Li, J.-P. Role of proteoglycans in neuro-inflammation and central nervous system fibrosis. Matrix Biol. 2018, 68–69, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from α-Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J. Cell Commun. Signal 2009, 3, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Mirza, B.; Hadberg, H.; Thomsen, P.; Moos, T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 1999, 95, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Prager, O.; Kamintsky, L.; Hasam-Henderson, L.A.; Schoknecht, K.; Wuntke, V.; Papageorgiou, I.; Swolinsky, J.; Muoio, V.; Bar-Klein, G.; Vazana, U.; et al. Seizure-induced microvascular injury is associated with impaired neurovascular coupling and blood-brain barrier dysfunction. Epilepsia 2019, 60, 322–336. [Google Scholar] [CrossRef]
- Itoh, Y.; Toriumi, H.; Yamada, S.; Hoshino, H.; Suzuki, N. Astrocytes and pericytes cooperatively maintain a capillary-like structure composed of endothelial cells on gel matrix. Brain Res. 2011, 1406, 74–83. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Nisa Awan, M.U.; Bai, J. The Mechanism and Function of Glia in Parkinson’s Disease. Front. Cell. Neurosci. 2022, 16, 903469. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Iwase, M.; Sasaki, B.; Suzuki, N. The molecular regulation of oligodendrocyte development and CNS myelination by ECM proteins. Front. Cell Dev. Biol. 2022, 10, 952135. [Google Scholar] [CrossRef] [PubMed]
- Wilems, T.; Vardhan, S.; Wu, S.; Sakiyama-Elbert, S. The influence of microenvironment and extracellular matrix molecules in driving neural stem cell fate within biomaterials. Brain Res. Bull. 2019, 148, 25–33. [Google Scholar] [CrossRef]
- Stern, S.; Lau, S.; Manole, A.; Rosh, I.; Percia, M.M.; Ben Ezer, R.; Shokhirev, M.N.; Qiu, F.; Schafer, S.; Mansour, A.A.; et al. Reduced synaptic activity and dysregulated extracellular matrix pathways in midbrain neurons from Parkinson’s disease patients. NPJ Park. Dis. 2022, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, U.; Rosh, I.; Ezer, R.B.; Nayak, R.; Choudhary, A.; Djamus, J.; Manole, A.; Haulden, H.; Gage, F.H.; Stern, S. Upregulated extracellular matrix-related genes and impaired synaptic activity in dopaminergic and hippocampal neurons derived from Parkinson’s disease patients with PINK1 and PARK2 mutations. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Wessely, F.; Connor-Robson, N.; Rinaldi, F.; Vowles, J.; Browne, C.; Evetts, S.G.; Hu, M.T.; Cowley, S.A.; Webber, C.; et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. Neurobiol. Dis. 2019, 129, 56–66. [Google Scholar] [CrossRef]
- Steinberg, D.J.; Aqeilan, R.I. WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives. Cells 2021, 10, 3082. [Google Scholar] [CrossRef]
- Brant, B.; Stern, T.; Shekhidem, H.A.; Mizrahi, L.; Rosh, I.; Stern, Y.; Ofer, P.; Asleh, A.; Umanah, G.K.E.; Jada, R.; et al. IQSEC2 mutation associated with epilepsy, intellectual disability, and autism results in hyperexcitability of patient-derived neurons and deficient synaptic transmission. Mol. Psychiatr. 2021, 26, 7498–7508. [Google Scholar] [CrossRef]
- Choudhary, A.; Peles, D.; Nayak, R.; Mizrahi, L.; Stern, S. Current progress in understanding schizophrenia using genomics and pluripotent stem cells: A meta-analytical overview. Schizophr. Res. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Marotta, N.; Kim, S.; Krainc, D. Organoid and pluripotent stem cells in Parkinson’s disease modeling: An expert view on their value to drug discovery. Expert Opin. Drug Discov. 2020, 15, 427–441. [Google Scholar] [CrossRef]
- Smits, L.M.; Schwamborn, J.C. Midbrain Organoids: A New Tool to Investigate Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 359. [Google Scholar] [CrossRef]
- Repudi, S.; Kustanovich, I.; Abu-Swai, S.; Stern, S.; Aqeilan, R.I. Neonatal neuronal WWOX gene therapy rescues Wwox null phenotypes. EMBO Mol. Med. 2021, 13, e14599. [Google Scholar] [CrossRef] [PubMed]
- Nayak, R.; Rosh, I.; Kustanovich, I.; Stern, S. Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems. Int. J. Mol. Sci. 2021, 22, 9315. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Petretto, A.; Angelini, G.; Pfeffer, U. Chapter 4—Advancements in Omics Sciences. In Translational Medicine; Shahzad, A., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 67–108. [Google Scholar]
- Li, X.; Wang, W.; Chen, J. Recent progress in mass spectrometry proteomics for biomedical research. Sci. China Life Sci. 2017, 60, 1093–1113. [Google Scholar] [CrossRef]
- Dixit, A.; Srivastava, G.; Verma, D.; Mishra, M.; Singh, P.K.; Prakash, O.; Singh, M.P. Minocycline, levodopa and MnTMPyP induced changes in the mitochondrial proteome profile of MPTP and maneb and paraquat mice models of Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1227–1240. [Google Scholar] [CrossRef]
- Licker, V.; Burkhard, P.R. Proteomics as a new paradigm to tackle Parkinson’s disease research challenges. Transl. Proteom. 2014, 4, 1–17. [Google Scholar] [CrossRef]
- Kasap, M.; Akpinar, G.; Kanli, A. Proteomic studies associated with Parkinson’s disease. Expert Rev. Proteom. 2017, 14, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef]
- Cicchetti, F.; Lapointe, N.; Roberge-Tremblay, A.; Saint-Pierre, M.; Jimenez, L.; Ficke, B.W.; Gross, R.E. Systemic exposure to paraquat and maneb models early Parkinson’s disease in young adult rats. Neurobiol. Dis. 2005, 20, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.P.; Patel, S.; Yadav, S.; Singh, A.K.; Singh, S.; Singh, M.P. Involvement of nitric oxide in maneb- and paraquat-induced Parkinson’s disease phenotype in mouse: Is there any link with lipid peroxidation? Neurochem. Res. 2010, 35, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Licker, V.; Kövari, E.; Hochstrasser, D.F.; Burkhard, P.R. Proteomics in human Parkinson’s disease research. J. Proteom. 2009, 73, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Berretta, S.; Heckers, S.; Benes, F.M. Searching human brain for mechanisms of psychiatric disorders. Implications for studies on schizophrenia. Schizophr. Res. 2015, 167, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Sorg, B.A.; Berretta, S.; Blacktop, J.M.; Fawcett, J.W.; Kitagawa, H.; Kwok, J.C.; Miquel, M. Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J. Neurosci. 2016, 36, 11459–11468. [Google Scholar] [CrossRef]
- Oohashi, T.; Edamatsu, M.; Bekku, Y.; Carulli, D. The hyaluronan and proteoglycan link proteins: Organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp. Neurol. 2015, 274, 134–144. [Google Scholar] [CrossRef]
- Mizumoto, S.; Yamada, S.; Sugahara, K. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 2015, 34, 35–42. [Google Scholar] [CrossRef]
- Levy, A.D.; Omar, M.H.; Koleske, A.J. Extracellular matrix control of dendritic spine and synapse structure and plasticity in adulthood. Front. Neuroanat. 2014, 8, 116. [Google Scholar] [CrossRef]
- Sethi, M.K.; Zaia, J. Extracellular matrix proteomics in schizophrenia and Alzheimer’s disease. Anal. Bioanal. Chem. 2017, 409, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804. [Google Scholar] [CrossRef] [PubMed]
- Downs, M.; Sethi, M.K.; Raghunathan, R.; Layne, M.D.; Zaia, J. Matrisome changes in Parkinson’s disease. Anal. Bioanal. Chem. 2022, 414, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol. 2019, 2, 43. [Google Scholar] [CrossRef]
- Avenoso, A.; Bruschetta, G.; D’Ascola, A.; Scuruchi, M.; Mandraffino, G.; Gullace, R.; Saitta, A.; Campo, S.; Campo, G.M. Hyaluronan fragments produced during tissue injury: A signal amplifying the inflammatory response. Arch. Biochem. Biophys. 2019, 663, 228–238. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Popovich, P.G. Extracellular matrix regulation of inflammation in the healthy and injured spinal cord. Exp. Neurol. 2014, 258, 24–34. [Google Scholar] [CrossRef]
- Hill, R.C.; Calle, E.A.; Dzieciatkowska, M.; Niklason, L.E.; Hansen, K.C. Quantification of Extracellular Matrix Proteins from a Rat Lung Scaffold to Provide a Molecular Readout for Tissue Engineering. Mol. Cell. Proteom. 2015, 14, 961–973. [Google Scholar] [CrossRef]
- Naba, A. 10 years of extracellular matrix proteomics: Accomplishments, challenges, and future perspectives. Mol. Cell. Proteom. 2023, 22, 100528. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Hu, M.; Ling, Z.; Ren, X. Extracellular matrix dynamics: Tracking in biological systems and their implications. J. Biol. Eng. 2022, 16, 13. [Google Scholar] [CrossRef]
- Fathi, A.; Bakshy, K.; Zieghami, L.; Fiene, R.; Bradley, R.; Dickerson, S.; Carlson, C.; Schachtele, S.; Liu, J. Diverging Parkinson’s Disease Pathology between patient-derived GBAN370S, LRRK2G2019S and engineered SNCAA53T iPSC-derived Dopaminergic Neurons. bioRxiv, 2023; preprint. [Google Scholar] [CrossRef]
- Rahman, A.A.; Soto-Avellaneda, A.; Yong Jin, H.; Stojkovska, I.; Lai, N.K.; Albright, J.E.; Webb, A.R.; Oe, E.; Valarde, J.P.; Oxford, A.E.; et al. Enhanced Hyaluronan Signaling and Autophagy Dysfunction by VPS35 D620N. Neuroscience 2020, 441, 33–45. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, K.D.; Berendse, H.W.; Drukarch, B.; Fratantoni, S.A.; Pham, T.V.; Piersma, S.R.; Huisman, E.; Breve, J.J.; Groenewegen, H.J.; Jimenez, C.R.; et al. The proteome of the locus ceruleus in Parkinson’s disease: Relevance to pathogenesis. Brain Pathol. 2012, 22, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Licker, V.; Cote, M.; Lobrinus, J.A.; Rodrigo, N.; Kovari, E.; Hochstrasser, D.F.; Turck, N.; Sanchez, J.C.; Burkhard, P.R. Proteomic profiling of the substantia nigra demonstrates CNDP2 overexpression in Parkinson’s disease. J. Proteom. 2012, 75, 4656–4667. [Google Scholar] [CrossRef]
- Licker, V.; Turck, N.; Kovari, E.; Burkhardt, K.; Cote, M.; Surini-Demiri, M.; Lobrinus, J.A.; Sanchez, J.C.; Burkhard, P.R. Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 2014, 14, 784–794. [Google Scholar] [CrossRef]
- Shi, M.J.J.; Wang, Y.; Beyer, R.P.; Kitsou, E.; Albin, R.L.; Gearing, M.; Pan, C.; Zhang, J. Mortalin: A Protein Associated With Progression of Parkinson Disease? J. Neuropathol. Exp. Neurol. 2008, 67, 117–124. [Google Scholar] [CrossRef]
- Werner, C.J.; Heyny-von Haussen, R.; Mall, G.; Wolf, S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008, 6, 8. [Google Scholar] [CrossRef]
- Lachen-Montes, M.; Gonzalez-Morales, A.; Iloro, I.; Elortza, F.; Ferrer, I.; Gveric, D.; Fernandez-Irigoyen, J.; Santamaria, E. Unveiling the olfactory proteostatic disarrangement in Parkinson’s disease by proteome-wide profiling. Neurobiol. Aging 2019, 73, 123–134. [Google Scholar] [CrossRef]
- Dumitriu, A.; Golji, J.; Labadorf, A.T.; Gao, B.; Beach, T.G.; Myers, R.H.; Longo, K.A.; Latourelle, J.C. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med. Genom. 2016, 9, 5. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Q.; Tang, M.; Fu, N.; Shao, W.; Zhang, S.; Yin, Y.; Zeng, R.; Wang, X.; Hu, G.; et al. Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol. Aging 2015, 36, 1686–1691. [Google Scholar] [CrossRef]
- Bossers, K.; Meerhoff, G.; Balesar, R.; van Dongen, J.W.; Kruse, C.G.; Swaab, D.F.; Verhaagen, J. Analysis of gene expression in Parkinson’s disease: Possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol. 2009, 19, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z. An Integrated Network Analysis of mRNA and Gene Expression Profiles in Parkinson’s Disease. Med. Sci. Monit. 2020, 26, e920846. [Google Scholar] [PubMed]
- Edwards, Y.J.; Beecham, G.W.; Scott, W.K.; Khuri, S.; Bademci, G.; Tekin, D.; Martin, E.R.; Jiang, Z.; Mash, D.C.; Ffrench-Mullen, J.; et al. Identifying consensus disease pathways in Parkinson’s disease using an integrative systems biology approach. PLoS ONE 2011, 6, e16917. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Bonchev, D. A network view on Parkinson’s disease. Comput. Struct. Biotechnol. J. 2013, 7, e201304004. [Google Scholar] [CrossRef] [PubMed]
- Botta-Orfila, T.; Sanchez-Pla, A.; Fernandez, M.; Carmona, F.; Ezquerra, M.; Tolosa, E. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson’s disease. Brain Res. 2012, 1466, 152–157. [Google Scholar] [CrossRef]
- Grunblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural. Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef]
- Durrenberger, P.F.; Grunblatt, E.; Fernando, F.S.; Monoranu, C.M.; Evans, J.; Riederer, P.; Reynolds, R.; Dexter, D.T. Inflammatory Pathways in Parkinson’s Disease; A BNE Microarray Study. Park. Dis. 2012, 2012, 214714. [Google Scholar] [CrossRef]
- Riley, B.E.; Gardai, S.J.; Emig-Agius, D.; Bessarabova, M.; Ivliev, A.E.; Schule, B.; Alexander, J.; Wallace, W.; Halliday, G.M.; Langston, J.W.; et al. Systems-based analyses of brain regions functionally impacted in Parkinson’s disease reveals underlying causal mechanisms. PLoS ONE 2014, 9, e102909. [Google Scholar] [CrossRef]
- Zhang, B.; Xia, C.; Lin, Q.; Huang, J. Identification of key pathways and transcription factors related to Parkinson disease in genome wide. Mol. Biol. Rep. 2012, 39, 10881–10887. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, X. Systematic analysis of microarray datasets to identify Parkinson’s disease-associated pathways and genes. Mol. Med. Rep. 2017, 15, 1252–1262. [Google Scholar] [CrossRef]
- Dijkstra, A.A.; Ingrassia, A.; de Menezes, R.X.; van Kesteren, R.E.; Rozemuller, A.J.; Heutink, P.; van de Berg, W.D. Evidence for Immune Response, Axonal Dysfunction and Reduced Endocytosis in the Substantia Nigra in Early Stage Parkinson’s Disease. PLoS ONE 2015, 10, e0128651. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.M.; Xu, H.; Schmid, S.; Alexandrova, R.; Kaur, G.; Thiruvahindrapuram, B.; Pereira, S.L.; Jog, M.; Hebb, M.O. Expanding the search for genetic biomarkers of Parkinson’s disease into the living brain. Neurobiol. Dis. 2020, 140, 104872. [Google Scholar] [CrossRef] [PubMed]
- Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L.T.; Krichevsky, A.M.; Andersen, S.L.; Stephens, R.M.; Benes, F.M.; Sonntag, K.C. Gene expression profiling of substantia nigra dopamine neurons: Further insights into Parkinson’s disease pathology. Brain 2009, 132, 1795–1809. [Google Scholar] [CrossRef]
- Stamper, C.; Siegel, A.; Liang, W.S.; Pearson, J.V.; Stephan, D.A.; Shill, H.; Connor, D.; Caviness, J.N.; Sabbagh, M.; Beach, T.G.; et al. Neuronal gene expression correlates of Parkinson’s disease with dementia. Mov. Disord. 2008, 23, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Cullen, D.K.; Lessing, M.C.; LaPlaca, M.C. Collagen-Dependent Neurite Outgrowth and Response to Dynamic Deformation in Three-Dimensional Neuronal Cultures. Ann. Biomed. Eng. 2007, 35, 835–846. [Google Scholar] [CrossRef]
- Paiva, I.; Jain, G.; Lázaro, D.F.; Jerčić, K.G.; Hentrich, T.; Kerimoglu, C.; Pinho, R.; Szegő, È.M.; Burkhardt, S.; Capece, V.; et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol. Dis. 2018, 119, 121–135. [Google Scholar] [CrossRef]
- Sievers, J.; Pehlemann, F.W.; Gude, S.; Berry, M. Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J. Neurocytol. 1994, 23, 135–149. [Google Scholar] [CrossRef]
- Cescon, M.; Chen, P.; Castagnaro, S.; Gregorio, I.; Bonaldo, P. Lack of collagen VI promotes neurodegeneration by impairing autophagy and inducing apoptosis during aging. Aging 2016, 8, 1083–1101. [Google Scholar] [CrossRef]
- Cheng, J.S.; Dubal, D.B.; Kim, D.H.; Legleiter, J.; Cheng, I.H.; Yu, G.-Q.; Tesseur, I.; Wyss-Coray, T.; Bonaldo, P.; Mucke, L. Collagen VI protects neurons against Aβ toxicity. Nat. Neurosci. 2009, 12, 119–121. [Google Scholar] [CrossRef]
- Jochim, A.; Zech, M.; Gora-Stahlberg, G.; Winkelmann, J.; Haslinger, B. The clinical phenotype of early-onset isolated dystonia caused by recessive COL6A3 mutations (DYT27). Mov. Disord. 2016, 31, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.-Y.; Zheng, R.; Lin, Z.-H.; Xue, N.-J.; Chen, Y.; Gao, T.; Yan, Y.-Q.; Fang, Y.; Yan, Y.-P.; Yin, X.-Z.; et al. Study of the collagen type VI alpha 3 (COL6A3) gene in Parkinson’s disease. BMC Neurol. 2021, 21, 187. [Google Scholar] [CrossRef]
- Cheng, I.H.; Lin, Y.C.; Hwang, E.; Huang, H.T.; Chang, W.H.; Liu, Y.L.; Chao, C.Y. Collagen VI protects against neuronal apoptosis elicited by ultraviolet irradiation via an Akt/Phosphatidylinositol 3-kinase signaling pathway. Neuroscience 2011, 183, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Végh, M.J.; Heldring, C.M.; Kamphuis, W.; Hijazi, S.; Timmerman, A.J.; Li, K.W.; van Nierop, P.; Mansvelder, H.D.; Hol, E.M.; Smit, A.B.; et al. Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 76. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kang, I.; Merrilees, M.J. Versican and the control of inflammation. Matrix Biol. 2014, 35, 152–161. [Google Scholar] [CrossRef]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the regulation of cell phenotype in disease. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2441–2451. [Google Scholar] [CrossRef]
- Arroyo, A.G.; Iruela-Arispe, M.L. Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc. Res. 2010, 86, 226–235. [Google Scholar] [CrossRef]
- Adair-Kirk, T.L.; Senior, R.M. Fragments of extracellular matrix as mediators of inflammation. Int. J. Biochem. Cell Biol. 2008, 40, 1101–1110. [Google Scholar] [CrossRef]
- van Horssen, J.; Dijkstra, C.D.; de Vries, H.E. The extracellular matrix in multiple sclerosis pathology. J. Neurochem. 2007, 103, 1293–1301. [Google Scholar] [CrossRef]
- Gutowski, N.J.; Newcombe, J.; Cuzner, M.L. Tenascin-R and C in multiple sclerosis lesions: Relevance to extracellular matrix remodelling. Neuropathol. Appl. Neurobiol. 1999, 25, 207–214. [Google Scholar] [CrossRef]
- Siddiqui, S.; Kamal, A.; Khan, F.; Jamali, K.S.; Saify, Z.S. Gallic and vanillic acid suppress inflammation and promote myelination in an in vitro mouse model of neurodegeneration. Mol. Biol. Rep. 2019, 46, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Liu, Y.; Hao, W.; Walter, S.; Penke, B.; Hartmann, T.; Schachner, M.; Fassbender, K. Tenascin-C deficiency ameliorates Alzheimer’s disease-related pathology in mice. Neurobiol. Aging 2013, 34, 2389–2398. [Google Scholar] [CrossRef]
- Sottile, J.; Hocking, D.C. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell 2002, 13, 3546–3559. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, R.; Kojima, K.; Utsumi, H.; Ogawa, H.; Matsumoto, I. Glycosaminoglycan binding properties of annexin IV, V, and VI. J. Biol. Chem. 1998, 273, 9935–9941. [Google Scholar] [CrossRef]
- Gauthier-Kemper, A.; Suárez Alonso, M.; Sündermann, F.; Niewidok, B.; Fernandez, M.P.; Bakota, L.; Heinisch, J.J.; Brandt, R. Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau’s axonal localization. J. Biol. Chem. 2018, 293, 8065–8076. [Google Scholar] [CrossRef]
- Ries, M.; Watts, H.; Mota, B.C.; Lopez, M.Y.; Donat, C.K.; Baxan, N.; Pickering, J.A.; Chau, T.W.; Semmler, A.; Gurung, B.; et al. Annexin A1 restores cerebrovascular integrity concomitant with reduced amyloid-β and tau pathology. Brain J. Neurol. 2021, 144, 1526–1541. [Google Scholar] [CrossRef]
- Bartolome, F.; Krzyzanowska, A.; de la Cueva, M.; Pascual, C.; Antequera, D.; Spuch, C.; Villarejo-Galende, A.; Rabano, A.; Fortea, J.; Alcolea, D.; et al. Annexin A5 prevents amyloid-β-induced toxicity in choroid plexus: Implication for Alzheimer’s disease. Sci. Rep. 2020, 10, 9391. [Google Scholar] [CrossRef]
- Burridge, K. Focal adhesions: A personal perspective on a half century of progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef]
- Wu, C. Focal adhesion: A focal point in current cell biology and molecular medicine. Cell Adhes. Migr. 2007, 1, 13–18. [Google Scholar] [CrossRef]
- Berezin, V.; Walmod, P.S.; Filippov, M.; Dityatev, A. Chapter 15—Targeting of ECM molecules and their metabolizing enzymes and receptors for the treatment of CNS diseases. Prog. Brain Res. 2014, 214, 353–388. [Google Scholar] [PubMed]
- Pokutta, S.; Weis, W.I. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu. Rev. Cell Dev. Biol. 2007, 23, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Motallebnejad, P.; Azarin, S.M. Chemically defined human vascular laminins for biologically relevant culture of hiPSC-derived brain microvascular endothelial cells. Fluids Barriers CNS 2020, 17, 54. [Google Scholar] [CrossRef]
- Mierke, C.T. The Pertinent Role of Cell and Matrix Mechanics in Cell Adhesion and Migration. Front. Cell Dev. Biol. 2021, 9, 720494. [Google Scholar] [CrossRef]
- Millard, M.; Odde, S.; Neamati, N. Integrin targeted therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef]
- Kim, C.; Ye, F.; Ginsberg, M.H. Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 2011, 27, 321–345. [Google Scholar] [CrossRef]
- Blair, J.A.; Wang, C.; Hernandez, D.; Siedlak, S.L.; Rodgers, M.S.; Achar, R.K.; Fahmy, L.M.; Torres, S.L.; Petersen, R.B.; Zhu, X.; et al. Individual Case Analysis of Postmortem Interval Time on Brain Tissue Preservation. PLoS ONE 2016, 11, e0151615. [Google Scholar] [CrossRef]
- Hynd, M.R.; Lewohl, J.M.; Scott, H.L.; Dodd, P.R. Biochemical and molecular studies using human autopsy brain tissue. J. Neurochem. 2003, 85, 543–562. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miao, L.; Wang, L. Inflammation amplification by Versican: The first mediator. Int. J. Mol. Sci. 2012, 13, 6873–6882. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Loiola, E.C.; da Costa, R.M.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef]
- Kim, J.; Ye, F.; Ginsberg, M.H. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Steinberg, D.J.; Repudi, S.; Saleem, A.; Kustanovich, I.; Viukov, S.; Abudiab, B.; Banne, E.; Mahajnah, M.; Hanna, J.H.; Stern, S.; et al. Modeling genetic epileptic encephalopathies using brain organoids. EMBO Mol. Med. 2021, 13, e13610. [Google Scholar] [CrossRef]
- Wang, H. Modeling Neurological Diseases With Human Brain Organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yang, J.; Xiang, Y. Modeling human neurodevelopmental diseases with brain organoids. Cell Regen 2022, 11, 1. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Kim, K.; Kang, H. Matrigel uses in cell biology and for the identification of thymosin β4, a mediator of tissue regeneration. Appl. Biol. Chem. 2018, 61, 703–708. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef]
- Heo, J.H.; Kang, D.; Seo, S.J.; Jin, Y. Engineering the Extracellular Matrix for Organoid Culture. Int. J. Stem. Cells 2022, 15, 60–69. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Study Participants | Average Age | Sex | PMI (h) | Remark | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Case | Control | Case | Control | Male | Female | Case | Control | ||
| 6 | 6 | 77.8 | 77.8 | 0 | 12 | 5.75 | 7.15 | Matched * | [107] |
| 3 | 3 | 81.7 | 83.3 | 6 | 0 | 19.7 | 20 | [108] | |
| 3 | 3 | 79 | 72.7 | 3 | 3 | 18.7 | 24 | [109] | |
| 20 | 5 | - | - | - | - | <12 h | <12 h | Matched * | [110] |
| 28 | 37 | 77.6 | 68 | 28 | 37 | 10.25 | 14.1 | Matched */cohort | [96] |
| 28 | 36 | 77.6 | 68 | 28 | 36 | 10.25 | 14.1 | Matched */cohort | [97] |
| 5 | 5 | 84.2 | 77.4 | 6 | 4 | 35.6 | 30.2 | Matched *. PMI controlled | [111] |
| 21 | 8 | 79.9 | 77.8 | 17 | 12 | 10.6 | 17.4 | [112] | |
| 12 | 12 | 76.8 | 79.5 | 24 | 0 | 4.16 | 5.66 | ** controlled | [113] |
| 3 | 3 | 4 | 2 | Matched * | [114] | ||||
| Brain Region | Method | DEP | DEP (ECM-Related) | DEP (ECM-Related) % | Ref. |
|---|---|---|---|---|---|
| Locus ceruleus | LC-MS | 87 | 2 | 1.1 | [107] |
| Substantia nigra | MS/MS | 23 | 2 | 8.7 | [108] |
| Substantia nigra | LC-MS/MS | 204 | 12 | 5.9 | [109] |
| Frontal cortex (middle frontal gyrus) | LC-MS/MS | 200 | 2 | 1 | [110] |
| Frontal cortex Brodmann area 9 | Q-Extractive HF MS | 89 | 14 | 15.7 | [96] |
| Frontal cortex Brodmann area 9 | Q-Extractive HF MS | 112 * | 8 | 7.1 | [97] |
| Substantia nigra | LC-MS/MS | 11 | 2 | 18.2 | [114] |
| Substantia nigra | 2D-GE, MS/MS | 16 | 2 | 12.5 | [111] |
| Olfactory bulbs | LC-MS/MS | 168 | 1 | 0.6 | [112] |
| Frontal cortex Brodmann area 9 | LC-MS/MS | 283 | 1 | 0.2 | [113] |
| Components | Protein Names | Gene Names | Up/Down | Ref |
|---|---|---|---|---|
| Versican family | Versican core protein | VCAN, CSPG2 | up | [96,97,107] |
| Collagen family | Collagen alpha-1(I) chain | COL1A1 | up | [96,97] |
| Collagen alpha-2(I) chain | COL1A2 | up | [96,97] | |
| Collagen alpha-1(IV) chain | COL4A1 | up | [96] | |
| Collagen alpha-2(IV) chain | COL4A2 | up | [96,97,112] | |
| Collagen alpha-3(VI) chain | COL6A3 | up | [96,97] | |
| Annexin family | Annexin A1 | ANXA1 | up | [109,114] |
| Annexin A2 | ANXA2 | up | [97,109] | |
| Annexin A5 | ANXA5, ANX5 | up | [97,111] | |
| Annexin A6 | ANXA6, ANX6 | down | [97,108] | |
| Hyaluronan and proteoglycan link protein family | Hyaluronan and proteoglycan link protein 1 | HAPLN1 (CRTL1) | up | [96] |
| Hyaluronan and proteoglycan link protein 2 | HAPLN2 | Down and up | [96,109,114] | |
| Hyaluronan and proteoglycan link protein 4 | HAPLN4 | down | [109] | |
| Fibronectin family | Fibronectin (FN) | FN1 (FN) | Up and down | [97,109] |
| Tenascin family | Tenascin, TN | TNC (HXB) | up | [96] |
| Tenascin-R (TN-R) | TNR | up | [110] | |
| Galectin family | Galectin-1, Gal-1 | LGALS1 | up | [111] |
| Galectin-3, Gal-3 | LGALS3, MAC2 | Down | [107] | |
| Galectin-3-binding protein | LGALS3BP M2BP | up | [109] |
| Genes/Gene Groups/Pathways/Biological Processes | Sample | Up/Down | References |
|---|---|---|---|
| ECM–receptor interaction | Brain tissue, post-mitotic catecholaminergic neuron-like cells, iPSC-derived DA neurons | up | [68,106,115] |
| Focal adhesion | Brain tissue and iPSC-derived DA neurons | up | [68,115,116,117,118] |
| iPSC-derived DA neurons | down | [67] | |
| Cell adhesion molecules | Brain tissue and iPSC-derived DA neurons | up | [67,115,117,119] |
| Cell adhesion | Brain tissue | up | [113,120,121] |
| iPSC-derived DA neurons | Down | [69] | |
| Cell–matrix adhesion | Brain tissue | up | [115] |
| Brain tissue | down | [122] | |
| Glycosaminoglycan degradation | Brain tissue | up | [123,124] |
| Collagen and related processes | iPSC-derived DA neurons | down | [67,69] |
| Integrin signaling | Brain tissue | up | [113] |
| ITGA1, ITGA 3, ITGA 4, ITGA 5, ITGA 7, ITGA 11, ITGAM, ITGB3BP | Brain tissue and iPSC-derived DA neurons | up | [67,68,120] |
| COL1A2, COL4A1, COL4A2, COL6A3, COL12A1 | iPSC-derived DA neurons | down | [67,125] |
| COL1A2, COL4A1, COLA2, COL18A1 | Brain tissue and iPSC-derived DA neurons | up | [68,120] |
| LAMA1, LAMA2, LAMB1, LAMB2 | Brain tissue and iPSC-derived DA neurons | up | [67,120,126,127,128] |
| LAMA3 | Brain tissue | Down | [126] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rike, W.A.; Stern, S. Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 7435. https://doi.org/10.3390/ijms24087435
Rike WA, Stern S. Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(8):7435. https://doi.org/10.3390/ijms24087435
Chicago/Turabian StyleRike, Wote Amelo, and Shani Stern. 2023. "Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review" International Journal of Molecular Sciences 24, no. 8: 7435. https://doi.org/10.3390/ijms24087435
APA StyleRike, W. A., & Stern, S. (2023). Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review. International Journal of Molecular Sciences, 24(8), 7435. https://doi.org/10.3390/ijms24087435