
Preclinical Evaluation of TB/FLU-04L—An Intranasal Influenza Vector-Based Boost Vaccine against Tuberculosis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
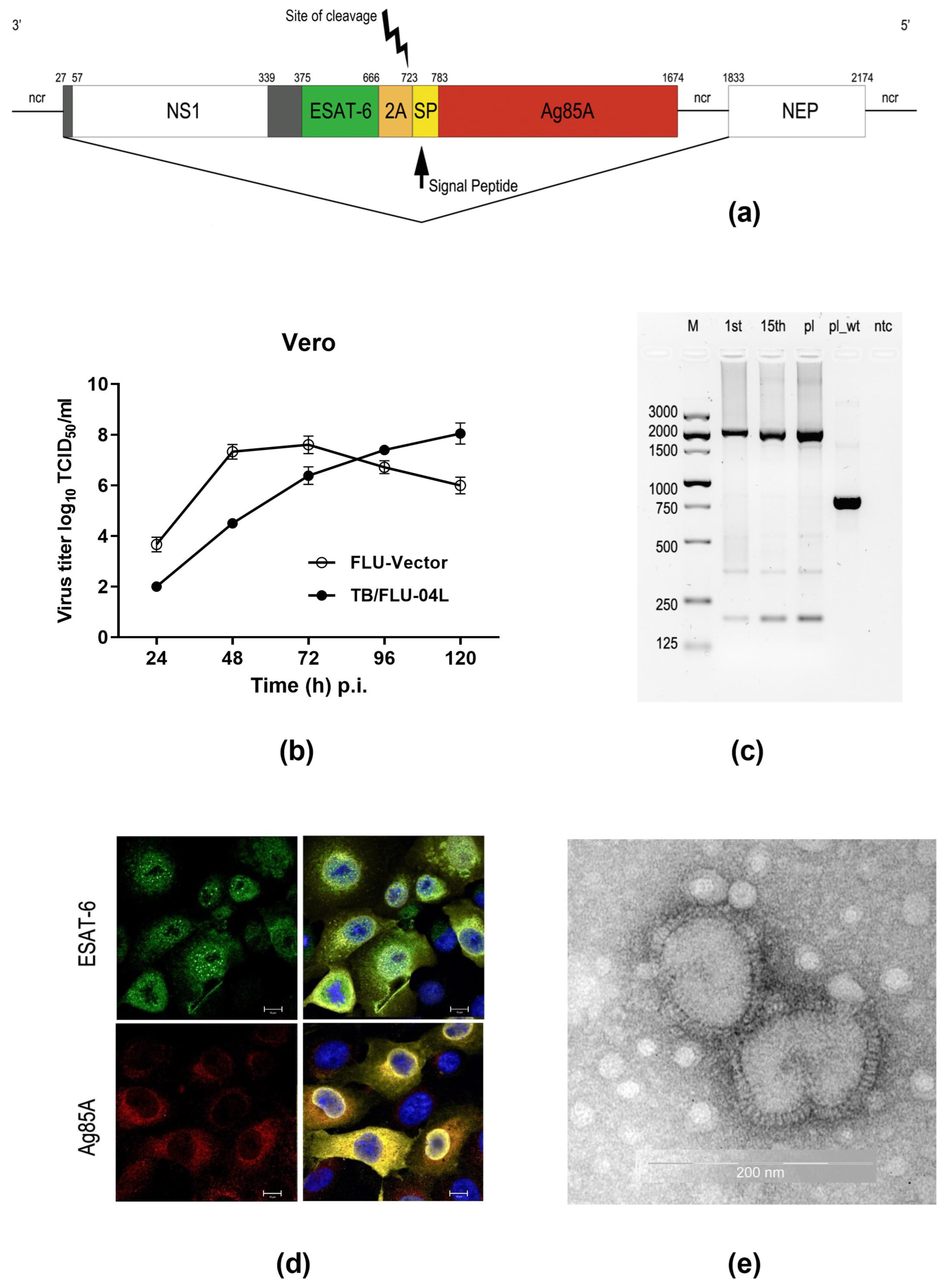
2.1. TB/FLU-04L Construction and Characterization
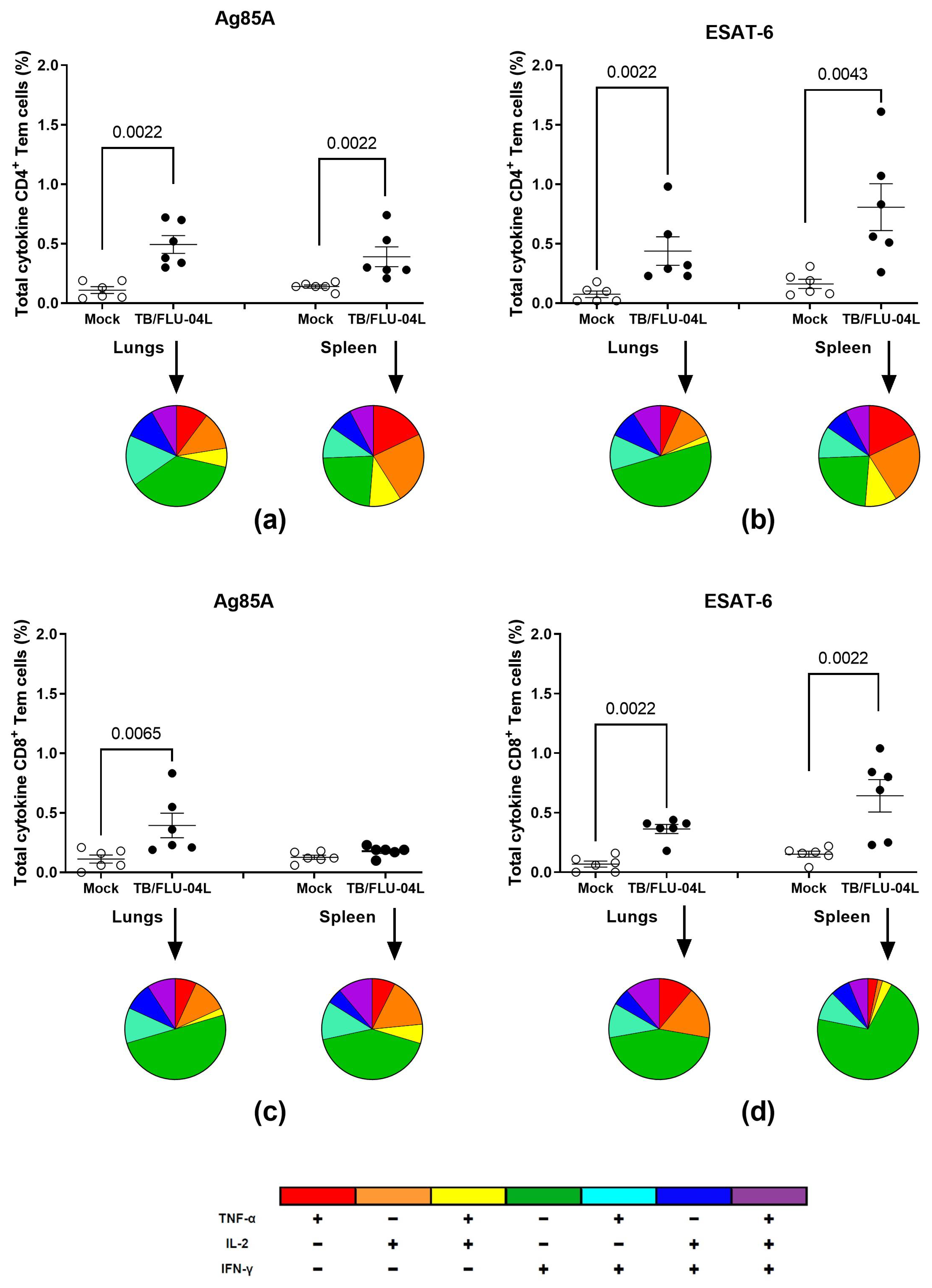
2.2. TB/FLU-04L Is Safe and Immunogenic in Mice
2.3. Safety and Immunogenicity of TB/FLU-04L in Macaca Fascicularis
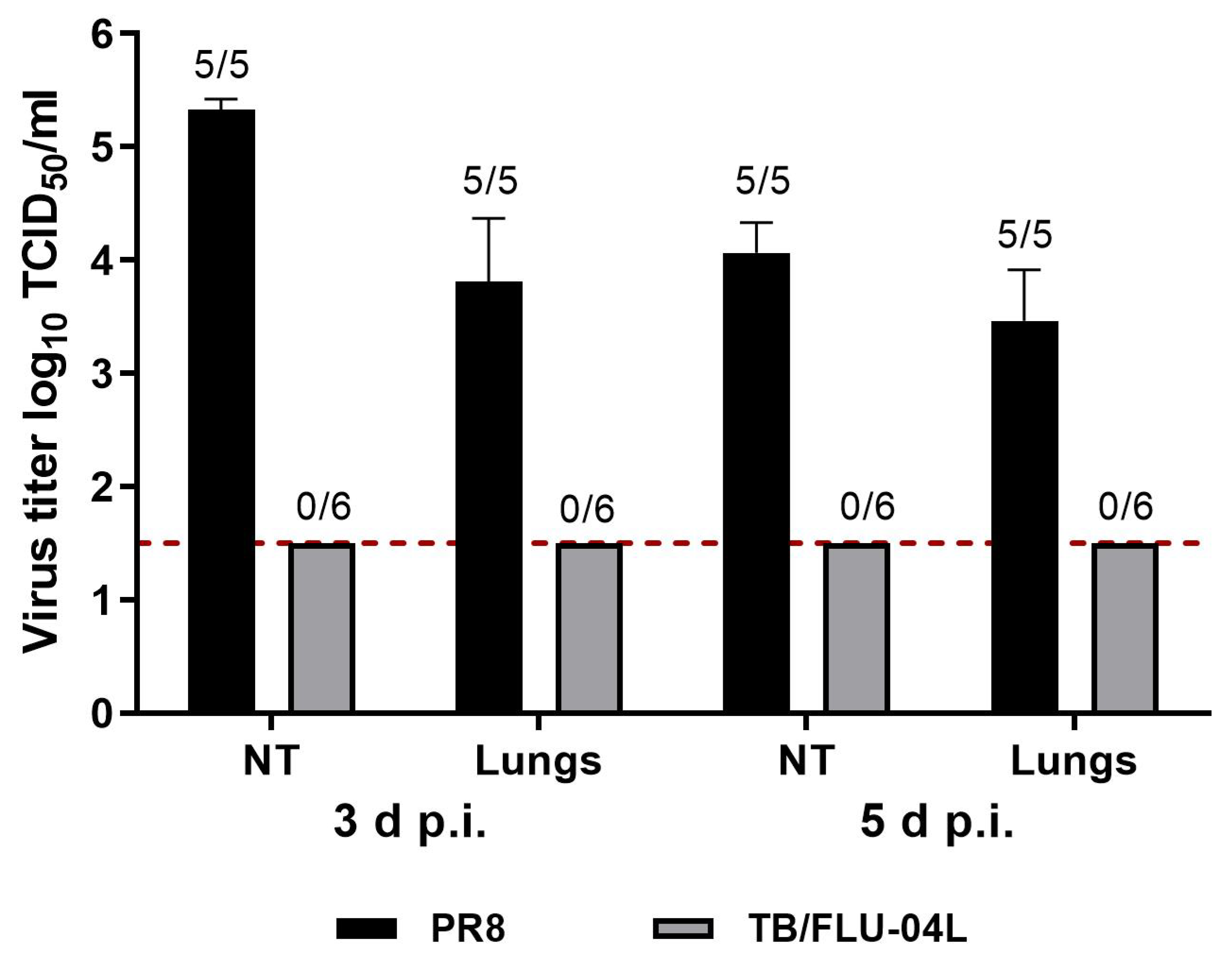
2.4. Two Intranasal Immunizations with the Influenza Vector Protect from Intravenous M. tuberculosis Infection at a Level Comparable with M. bovis BCG
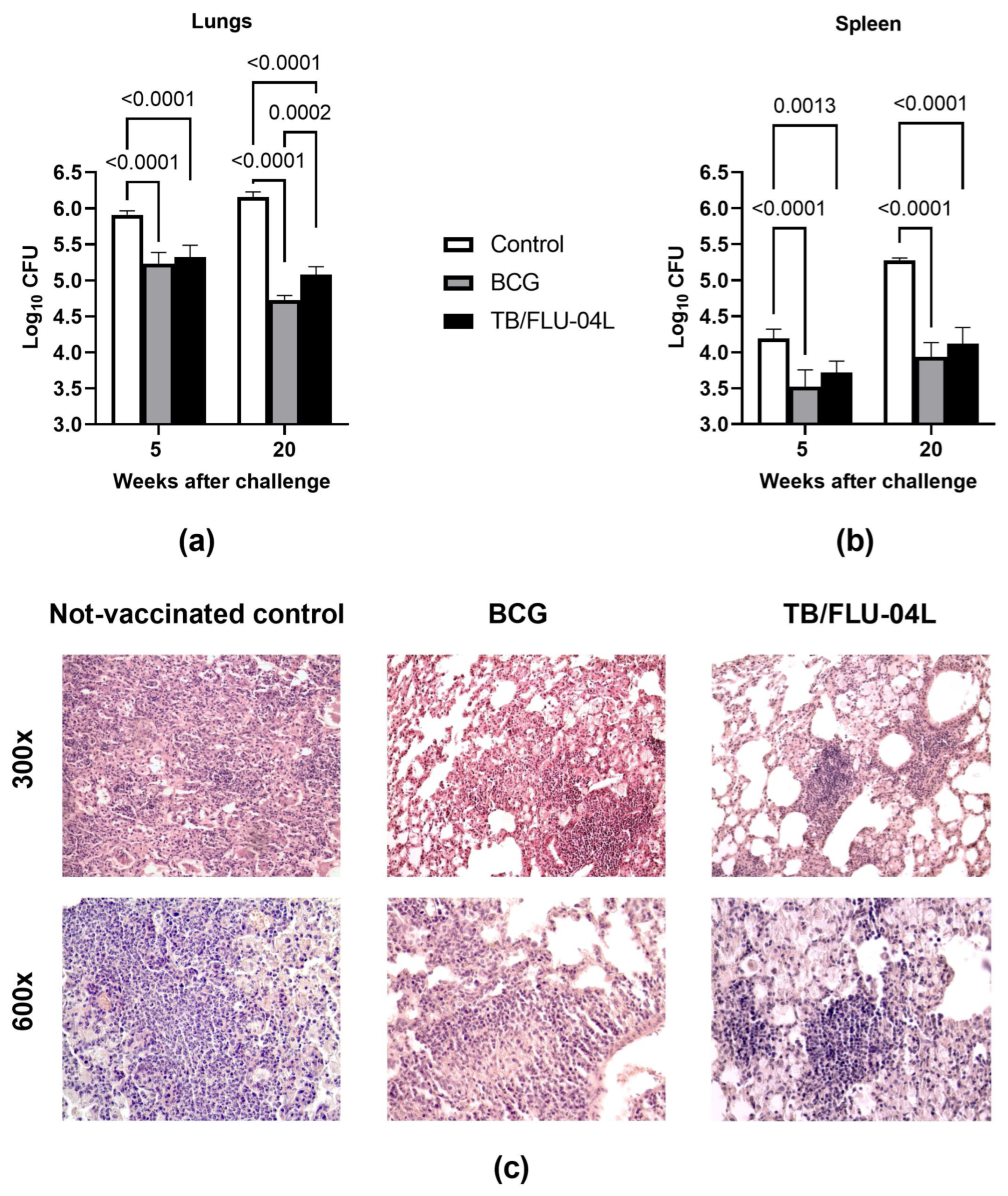
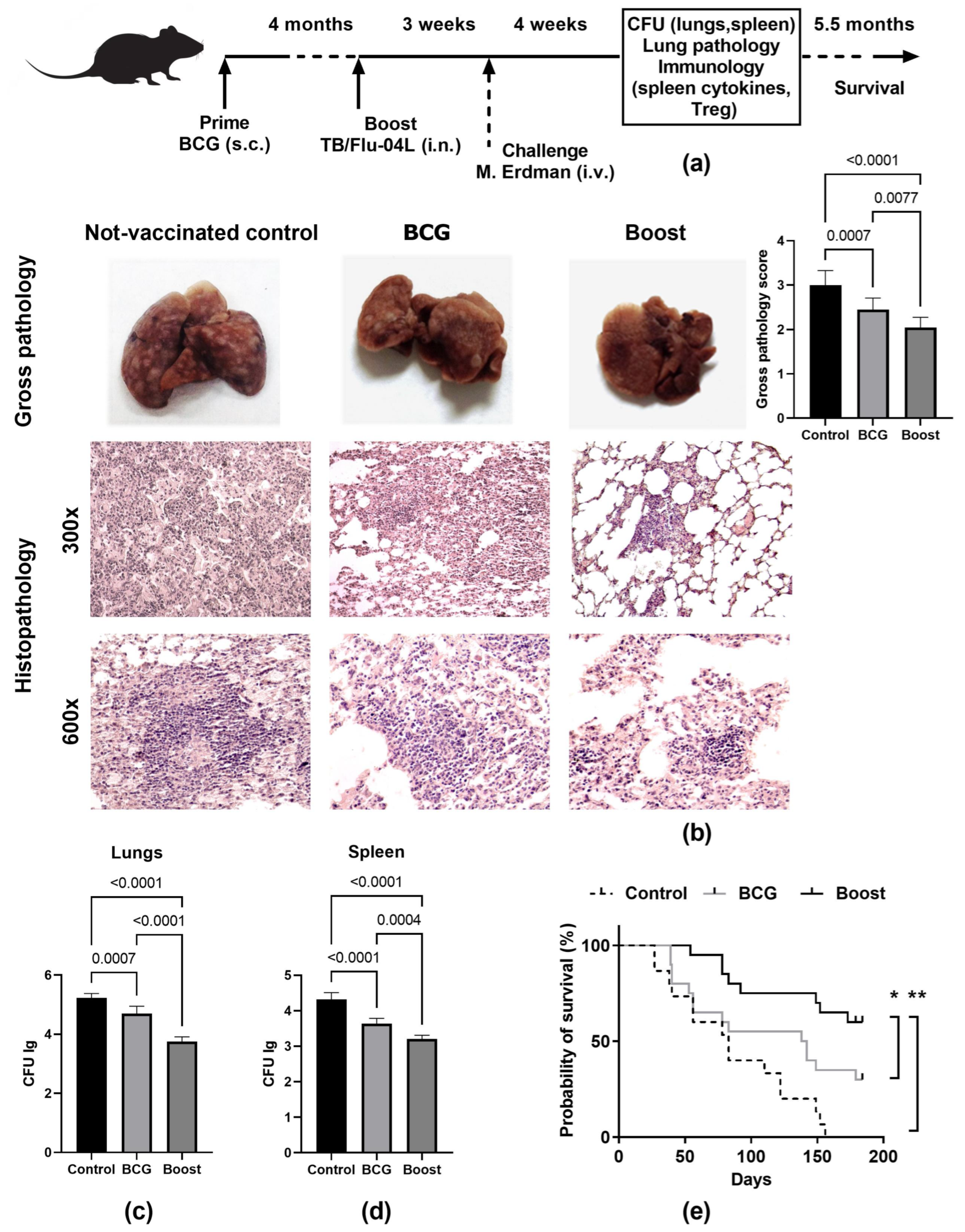
2.5. Single Intranasal Boost Immunization with the TB/FLU-04L Influenza Vector Enhances the Protective Efficacy of BCG against Intravenous M. tuberculosis Challenge
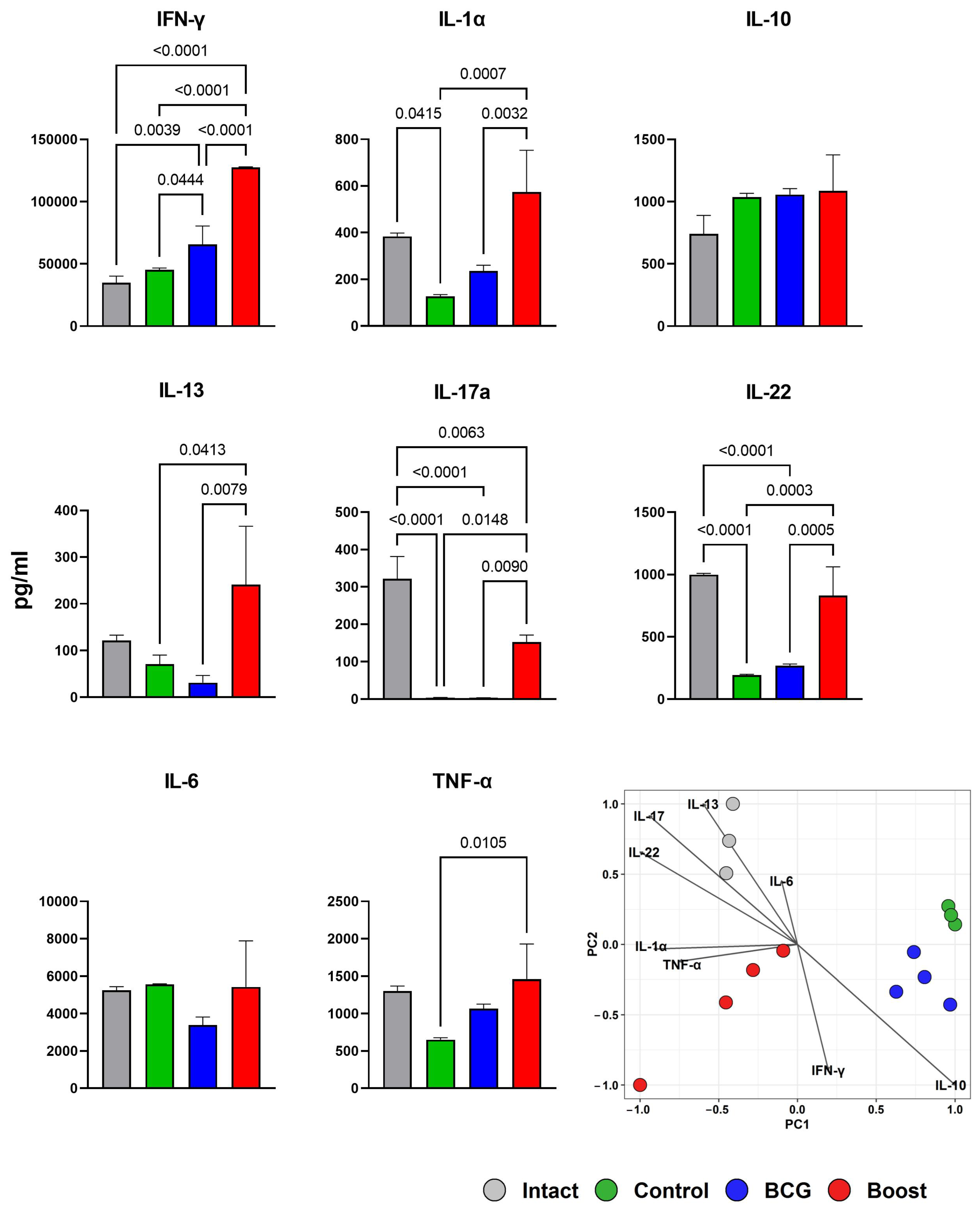
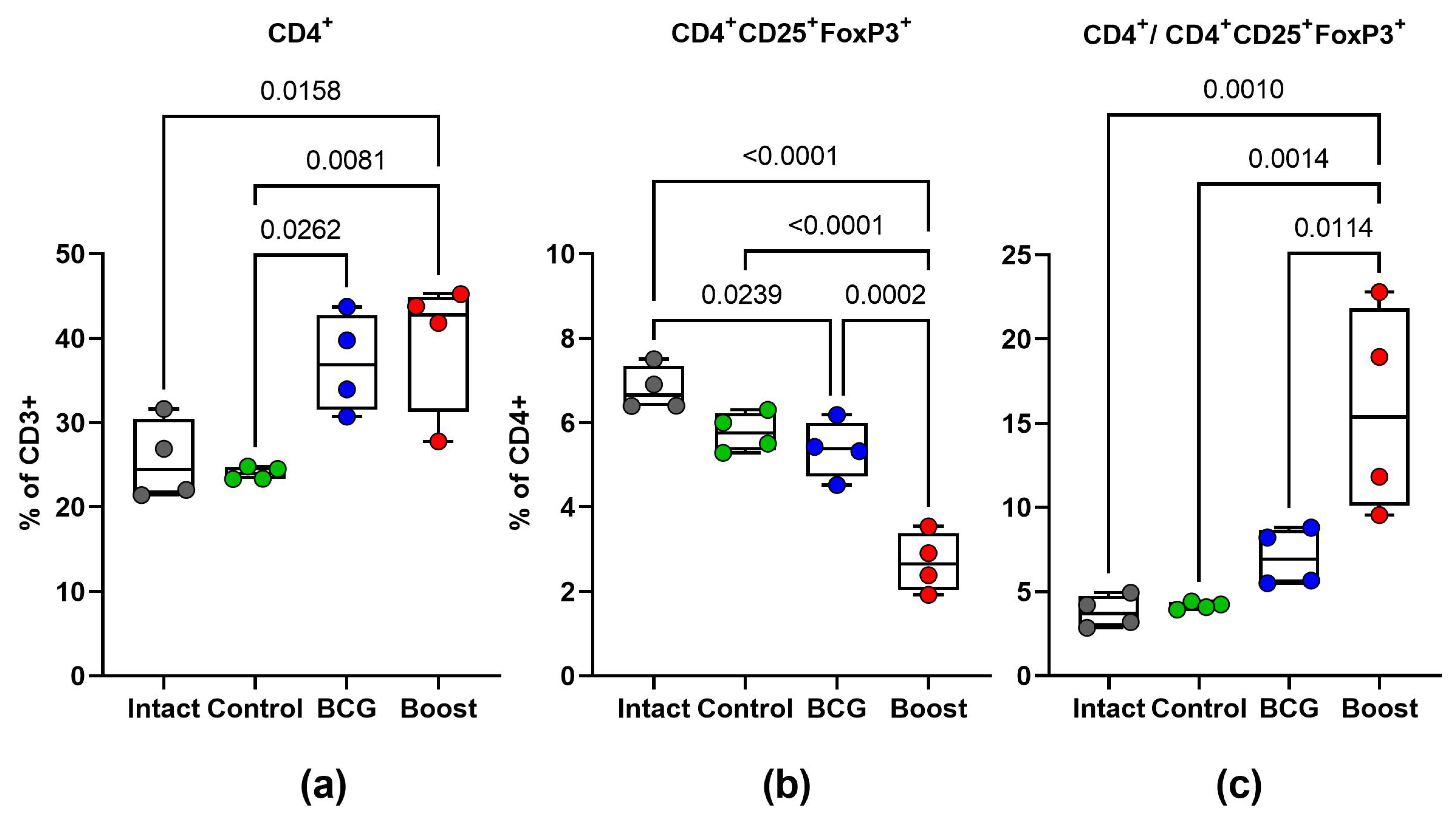
2.6. Type 1 and Type 17 Cytokine Responses and Treg-Cell Frequency during M. tuberculosis Challenge (Spleen)
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2022; IGO: Geneva, Switzerland, 2022; Volume 4, ISBN 9789240061729. [Google Scholar]
- Lipman, M.; McQuaid, C.F.; Abubakar, I.; Khan, M.; Kranzer, K.; McHugh, T.D.; Padmapriyadarsini, C.; Rangaka, M.X.; Stoker, N. The Impact of COVID-19 on Global Tuberculosis Control. Indian J. Med. Res. 2021, 153, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P. A Perspective on the Success and Failure of BCG. Front. Immunol. 2021, 12, 778028. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, E.C.; Ward, R.W. Mucosal Vaccines—Fortifying the Frontiers. Nat. Rev. Immunol. 2022, 22, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Ogongo, P.; Porterfield, J.Z.; Leslie, A. Lung Tissue Resident Memory T-Cells in the Immune Response to Mycobacterium Tuberculosis. Front. Immunol. 2019, 10, 992. [Google Scholar] [CrossRef] [PubMed]
- Shakya, A.K.; Chowdhury, M.Y.E.; Tao, W.; Gill, H.S. Mucosal Vaccine Delivery: Current State and a Pediatric Perspective. J. Control. Release 2016, 240, 394–413. [Google Scholar] [CrossRef]
- Abel, B.; Tameris, M.; Mansoor, N.; Gelderbloem, S.; Hughes, J.; Abrahams, D.; Makhethe, L.; Erasmus, M.; de Kock, M.; van der Merwe, L.; et al. The Novel Tuberculosis Vaccine, AERAS-402, Induces Robust and Polyfunctional CD4+ and CD8+ T Cells in Adults. Am. J. Respir. Crit. Care Med. 2010, 181, 1407–1417. [Google Scholar] [CrossRef]
- Wang, J.; Thorson, L.; Stokes, R.W.; Santosuosso, M.; Huygen, K.; Zganiacz, A.; Hitt, M.; Xing, Z. Single Mucosal, but Not Parenteral, Immunization with Recombinant Adenoviral-Based Vaccine Provides Potent Protection from Pulmonary Tuberculosis. J. Immunol. 2004, 173, 6357–6365. [Google Scholar] [CrossRef]
- Stylianou, E.; Griffiths, K.L.; Poyntz, H.C.; Harrington-Kandt, R.; Dicks, M.D.; Stockdale, L.; Betts, G.; McShane, H. Improvement of BCG Protective Efficacy with a Novel Chimpanzee Adenovirus and a Modified Vaccinia Ankara Virus Both Expressing Ag85A. Vaccine 2015, 33, 6800–6808. [Google Scholar] [CrossRef]
- Watanabe, K.; Matsubara, A.; Kawano, M.; Mizuno, S.; Okamura, T.; Tsujimura, Y.; Inada, H.; Nosaka, T.; Matsuo, K.; Yasutomi, Y. Recombinant Ag85B Vaccine by Taking Advantage of Characteristics of Human Parainfluenza Type 2 Virus Vector Showed Mycobacteria-Specific Immune Responses by Intranasal Immunization. Vaccine 2014, 32, 1727–1735. [Google Scholar] [CrossRef]
- Zhang, M.; Dong, C.; Xiong, S. Vesicular Stomatitis Virus-Vectored Multi-Antigen Tuberculosis Vaccine Limits Bacterial Proliferation in Mice Following a Single Intranasal Dose. Front. Cell. Infect. Microbiol. 2017, 7, 34. [Google Scholar] [CrossRef]
- Sereinig, S.; Stukova, M.; Zabolotnyh, N.; Ferko, B.; Kittel, C.; Romanova, J.; Vinogradova, T.; Katinger, H.; Kiselev, O.; Egorov, A. Influenza Virus NS Vectors Expressing the Mycobacterium Tuberculosis ESAT-6 Protein Induce CD4+ Th1 Immune Response and Protect Animals against Tuberculosis Challenge. Clin. Vaccine Immunol. 2006, 13, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Stukova, M.A.; Sereinig, S.; Zabolotnyh, N.V.; Ferko, B.; Kittel, C.; Romanova, J.; Vinogradova, T.I.; Katinger, H.; Kiselev, O.I.; Egorov, A. Vaccine Potential of Influenza Vectors Expressing Mycobacterium Tuberculosis ESAT-6 Protein. Tuberculosis 2006, 86, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.; Shurygina, A.-P.; Wolf, B.; Wolschek, M.; Enzmann, F.; Sansyzbay, A.; Khairullin, B.; Sandybayev, N.; Stukova, M.; Kiselev, O.; et al. Adaptive Mutation in Nuclear Export Protein Allows Stable Transgene Expression in a Chimaeric Influenza A Virus Vector. J. Gen. Virol. 2014, 95, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Hernandez, T.; Carnathan, D.G.; Moyle, P.M.; Toth, I.; West, N.P.; Young, P.R.; Silvestri, G.; Walker, M.J. The Contribution of Non-Human Primate Models to the Development of Human Vaccines. Discov. Med. 2014, 18, 313–322. [Google Scholar] [PubMed]
- Kaushal, D.; Mehra, S.; Didier, P.J.; Lackner, A.A. The Non-Human Primate Model of Tuberculosis. J. Med. Primatol. 2012, 41, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.J.; Hill, F.; Honeycutt, J.D.; Cottingham, M.G.; Bregu, M.; Rollier, C.S.; Furze, J.; Draper, S.J.; Søgaard, K.C.; Gilbert, S.C.; et al. Fusion of the Mycobacterium Tuberculosis Antigen 85A to an Oligomerization Domain Enhances Its Immunogenicity in Both Mice and Non-Human Primates. PLoS ONE 2012, 7, e33555. [Google Scholar] [CrossRef]
- Palin, A.C.; Alter, G.; Crotty, S.; Ellebedy, A.H.; Lane, M.C.; Lee, F.E.-H.; Locci, M.; Malaspina, A.; Mallia, C.; McElrath, M.J.; et al. The Persistence of Memory: Defining, Engineering, and Measuring Vaccine Durability. Nat. Immunol. 2022, 23, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Parkash, O.; Agrawal, S.; Madhan Kumar, M. T Regulatory Cells: Achilles’ Heel of Mycobacterium Tuberculosis Infection? Immunol. Res. 2015, 62, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, E.; Paul, M.J.; Reljic, R.; McShane, H. Mucosal Delivery of Tuberculosis Vaccines: A Review of Current Approaches and Challenges. Expert Rev. Vaccines 2019, 18, 1271–1284. [Google Scholar] [CrossRef]
- Pinschewer, D.D. Virally Vectored Vaccine Delivery: Medical Needs, Mechanisms, Advantages and Challenges. Swiss Med. Wkly. 2017, 147, w14465. [Google Scholar] [CrossRef]
- Vasilyev, K.A.; Yukhneva, M.A.; Shurygina, A.-P.S.; Stukova, M.A.; Egorov, A.Y. Enhancement of the Immunogenicity of Influenza A Virus by the Inhibition of Immunosuppressive Function of NS1 Protein. Microbiol. Indep. Res. J. 2018, 5, 36–47. [Google Scholar] [CrossRef]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef] [PubMed]
- Manjaly Thomas, Z.-R.; Satti, I.; Marshall, J.L.; Harris, S.A.; Lopez Ramon, R.; Hamidi, A.; Minhinnick, A.; Riste, M.; Stockdale, L.; Lawrie, A.M.; et al. Alternate Aerosol and Systemic Immunisation with a Recombinant Viral Vector for Tuberculosis, MVA85A: A Phase I Randomised Controlled Trial. PLoS Med. 2019, 16, e1002790. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Fritz, D.K.; Afkhami, S.; Aguirre, E.; Howie, K.J.; Zganiacz, A.; Dvorkin-Gheva, A.; Thompson, M.R.; Silver, R.F.; Cusack, R.P.; et al. Aerosol Delivery, but Not Intramuscular Injection, of Adenovirus-Vectored Tuberculosis Vaccine Induces Respiratory-Mucosal Immunity in Humans. JCI Insight 2022, 7, e155655. [Google Scholar] [CrossRef] [PubMed]
- Smorodincev, A.A. The Efficacy of Live Influenza Vaccines. Bull. World Health Organ. 1969, 41, 585–588. [Google Scholar]
- Marsh, G.A.; Watson, J.M.; White, W.E.; Tannock, G.A. An Evaluation of the Genetic Stability and Pathogenicity of the Russian Cold-Adapted Influenza A Donor Strains A/Leningrad/134/17/57 and A/Leningrad/134/47/57 in Ferrets. J. Virol. Methods 2003, 107, 63–69. [Google Scholar] [CrossRef]
- Carter, N.J.; Curran, M.P. Live Attenuated Influenza Vaccine (FluMist®; FluenzTM): A Review of Its Use in the Prevention of Seasonal Influenza in Children and Adults. Drugs 2011, 71, 1591–1622. [Google Scholar] [CrossRef]
- Wacheck, V.; Egorov, A.; Groiss, F.; Pfeiffer, A.; Fuereder, T.; Hoeflmayer, D.; Kundi, M.; Popow-Kraupp, T.; Redlberger-Fritz, M.; Mueller, C.A.; et al. A Novel Type of Influenza Vaccine: Safety and Immunogenicity of Replication-Deficient Influenza Virus Created by Deletion of the Interferon Antagonist NS1. J. Infect. Dis. 2010, 201, 354–362. [Google Scholar] [CrossRef]
- Mössler, C.; Groiss, F.; Wolzt, M.; Wolschek, M.; Seipelt, J.; Muster, T. Phase I/II Trial of a Replication-Deficient Trivalent Influenza Virus Vaccine Lacking NS1. Vaccine 2013, 31, 6194–6200. [Google Scholar] [CrossRef]
- Egorov, A.; Brandt, S.; Sereinig, S.; Romanova, J.; Ferko, B.; Katinger, D.; Grassauer, A.; Alexandrova, G.; Katinger, H.; Muster, T. Transfectant Influenza A Viruses with Long Deletions in the NS1 Protein Grow Efficiently in Vero Cells. J. Virol. 1998, 72, 6437–6441. [Google Scholar] [CrossRef]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A Virus Lacking the NS1 Gene Replicates in Interferon-Deficient Systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Desfarges, S.; Ciuffi, A. Viral Integration and Consequences on Host Gene Expression. In Viruses: Essential Agents of Life; Springer: Dordrecht, The Netherlands, 2012; pp. 147–175. [Google Scholar]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The Multifunctional NS1 Protein of Influenza A Viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Stasakova, J.; Ferko, B.; Kittel, C.; Sereinig, S.; Romanova, J.; Katinger, H.; Egorov, A. Influenza A Mutant Viruses with Altered NS1 Protein Function Provoke Caspase-1 Activation in Primary Human Macrophages, Resulting in Fast Apoptosis and Release of High Levels of Interleukins 1beta and 18. J. Gen. Virol. 2005, 86, 185–195. [Google Scholar] [CrossRef]
- Mu, J.; Jeyanathan, M.; Small, C.-L.; Zhang, X.; Roediger, E.; Feng, X.; Chong, D.; Gauldie, J.; Xing, Z. Immunization with a Bivalent Adenovirus-Vectored Tuberculosis Vaccine Provides Markedly Improved Protection over Its Monovalent Counterpart against Pulmonary Tuberculosis. Mol. Ther. 2009, 17, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, P.; Dierckx, J.P.; Frans, A.; Gillard, C.; Jovanović, D.; Stanescu, D. Live Influenza Vaccine in Patients with Chronic Bronchopulmonary Diseases. A Multicenter Study with Two Consecutive Vaccinal Strains. Dev. Biol. Stand. 1977, 39, 113–121. [Google Scholar] [PubMed]
- Buzitskaya, Z.; Stosman, K.; Khairullin, B.; Kassenov, M.; Nurpeisova, A.; Abylai Sansyzbay, A.; Shurygina, A.-P.; Aleksandrov, A.; Sivak, K.; Stukova, M. A New Intranasal Influenza Vector-Based Vaccine TB/FLU-04L Against Tuberculosis: Preclinical Safety Studies. Drug Res. 2022, 72, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.R.; Pathan, A.A.; Beveridge, N.E.R.; Poulton, I.; Minassian, A.; Alder, N.; Van Wijgerden, J.; Hill, A.V.S.; Gleeson, F.V.; Davies, R.J.O.; et al. Safety and Immunogenicity of a New Tuberculosis Vaccine, MVA85A, in Mycobacterium Tuberculosis-Infected Individuals. Am. J. Respir. Crit. Care Med. 2009, 179, 724–733. [Google Scholar] [CrossRef]
- Tena-Coki, N.G.; Scriba, T.J.; Peteni, N.; Eley, B.; Wilkinson, R.J.; Andersen, P.; Hanekom, W.A.; Kampmann, B. CD4 and CD8 T-Cell Responses to Mycobacterial Antigens in African Children. Am. J. Respir. Crit. Care Med. 2010, 182, 120–129. [Google Scholar] [CrossRef]
- Maphasa, R.E.; Meyer, M.; Dube, A. The Macrophage Response to Mycobacterium Tuberculosis and Opportunities for Autophagy Inducing Nanomedicines for Tuberculosis Therapy. Front. Cell. Infect. Microbiol. 2020, 10, 618414. [Google Scholar] [CrossRef]
- Chackerian, A.A.; Alt, J.M.; Perera, T.V.; Dascher, C.C.; Behar, S.M. Dissemination of Mycobacterium Tuberculosis Is Influenced by Host Factors and Precedes the Initiation of T-Cell Immunity. Infect. Immun. 2002, 70, 4501–4509. [Google Scholar] [CrossRef]
- Kaushal, D.; Foreman, T.W.; Gautam, U.S.; Alvarez, X.; Adekambi, T.; Rangel-Moreno, J.; Golden, N.A.; Johnson, A.-M.F.; Phillips, B.L.; Ahsan, M.H.; et al. Mucosal Vaccination with Attenuated Mycobacterium Tuberculosis Induces Strong Central Memory Responses and Protects against Tuberculosis. Nat. Commun. 2015, 6, 8533. [Google Scholar] [CrossRef] [PubMed]
- Copland, A.; Diogo, G.R.; Hart, P.; Harris, S.; Tran, A.C.; Paul, M.J.; Singh, M.; Cutting, S.M.; Reljic, R. Mucosal Delivery of Fusion Proteins with Bacillus Subtilis Spores Enhances Protection against Tuberculosis by Bacillus Calmette-Guérin. Front. Immunol. 2018, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T Cells in Nonlymphoid Tissue That Provide Enhanced Local Immunity during Infection with Herpes Simplex Virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T Cell Migration Program Provides Resident Memory within Intestinal Epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef]
- de Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium Tuberculosis: A Narrative Review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.L. CD8 T Cells and Mycobacterium Tuberculosis Infection. Semin. Immunopathol. 2015, 37, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Moliva, J.I.; Turner, J.; Torrelles, J.B. Immune Responses to Bacillus Calmette-Guérin Vaccination: Why Do They Fail to Protect against Mycobacterium Tuberculosis? Front. Immunol. 2017, 8, 407. [Google Scholar] [CrossRef]
- Hoffmann, E.; Neumann, G.; Kawaoka, Y.; Hobom, G.; Webster, R.G. A DNA Transfection System for Generation of Influenza A Virus from Eight Plasmids. Proc. Natl. Acad. Sci. USA 2000, 97, 6108–6113. [Google Scholar] [CrossRef]
- Hoffmann, E.; Webster, R.G. Unidirectional RNA Polymerase I-Polymerase II Transcription System for the Generation of Influenza A Virus from Eight Plasmids. J. Gen. Virol. 2000, 81, 2843–2847. [Google Scholar] [CrossRef]
- Percy, N.; Barclay, W.S.; García-Sastre, A.; Palese, P. Expression of a Foreign Protein by Influenza A Virus. J. Virol. 1994, 68, 4486–4492. [Google Scholar] [CrossRef]
- Wolschek, M.; Samm, E.; Seper, H.; Sturlan, S.; Kuznetsova, I.; Schwager, C.; Khassidov, A.; Kittel, C.; Muster, T.; Egorov, A.; et al. Establishment of a Chimeric, Replication-Deficient Influenza A Virus Vector by Modulation of Splicing Efficiency. J. Virol. 2011, 85, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A Simple Method of Estimating Fifty per Cent Endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shurygina, A.-P.; Zabolotnykh, N.; Vinogradova, T.; Khairullin, B.; Kassenov, M.; Nurpeisova, A.; Sarsenbayeva, G.; Sansyzbay, A.; Vasilyev, K.; Buzitskaya, J.; et al. Preclinical Evaluation of TB/FLU-04L—An Intranasal Influenza Vector-Based Boost Vaccine against Tuberculosis. Int. J. Mol. Sci. 2023, 24, 7439. https://doi.org/10.3390/ijms24087439
Shurygina A-P, Zabolotnykh N, Vinogradova T, Khairullin B, Kassenov M, Nurpeisova A, Sarsenbayeva G, Sansyzbay A, Vasilyev K, Buzitskaya J, et al. Preclinical Evaluation of TB/FLU-04L—An Intranasal Influenza Vector-Based Boost Vaccine against Tuberculosis. International Journal of Molecular Sciences. 2023; 24(8):7439. https://doi.org/10.3390/ijms24087439
Chicago/Turabian StyleShurygina, Anna-Polina, Natalia Zabolotnykh, Tatiana Vinogradova, Berik Khairullin, Markhabat Kassenov, Ainur Nurpeisova, Gulbanu Sarsenbayeva, Abylai Sansyzbay, Kirill Vasilyev, Janna Buzitskaya, and et al. 2023. "Preclinical Evaluation of TB/FLU-04L—An Intranasal Influenza Vector-Based Boost Vaccine against Tuberculosis" International Journal of Molecular Sciences 24, no. 8: 7439. https://doi.org/10.3390/ijms24087439
APA StyleShurygina, A. -P., Zabolotnykh, N., Vinogradova, T., Khairullin, B., Kassenov, M., Nurpeisova, A., Sarsenbayeva, G., Sansyzbay, A., Vasilyev, K., Buzitskaya, J., Egorov, A., & Stukova, M. (2023). Preclinical Evaluation of TB/FLU-04L—An Intranasal Influenza Vector-Based Boost Vaccine against Tuberculosis. International Journal of Molecular Sciences, 24(8), 7439. https://doi.org/10.3390/ijms24087439