
Immunopathological Alterations after Blast Injury and Hemorrhage in a Swine Model of Prolonged Damage Control Resuscitation

 , ,
, , 
Abstract
:1. Introduction
2. Results
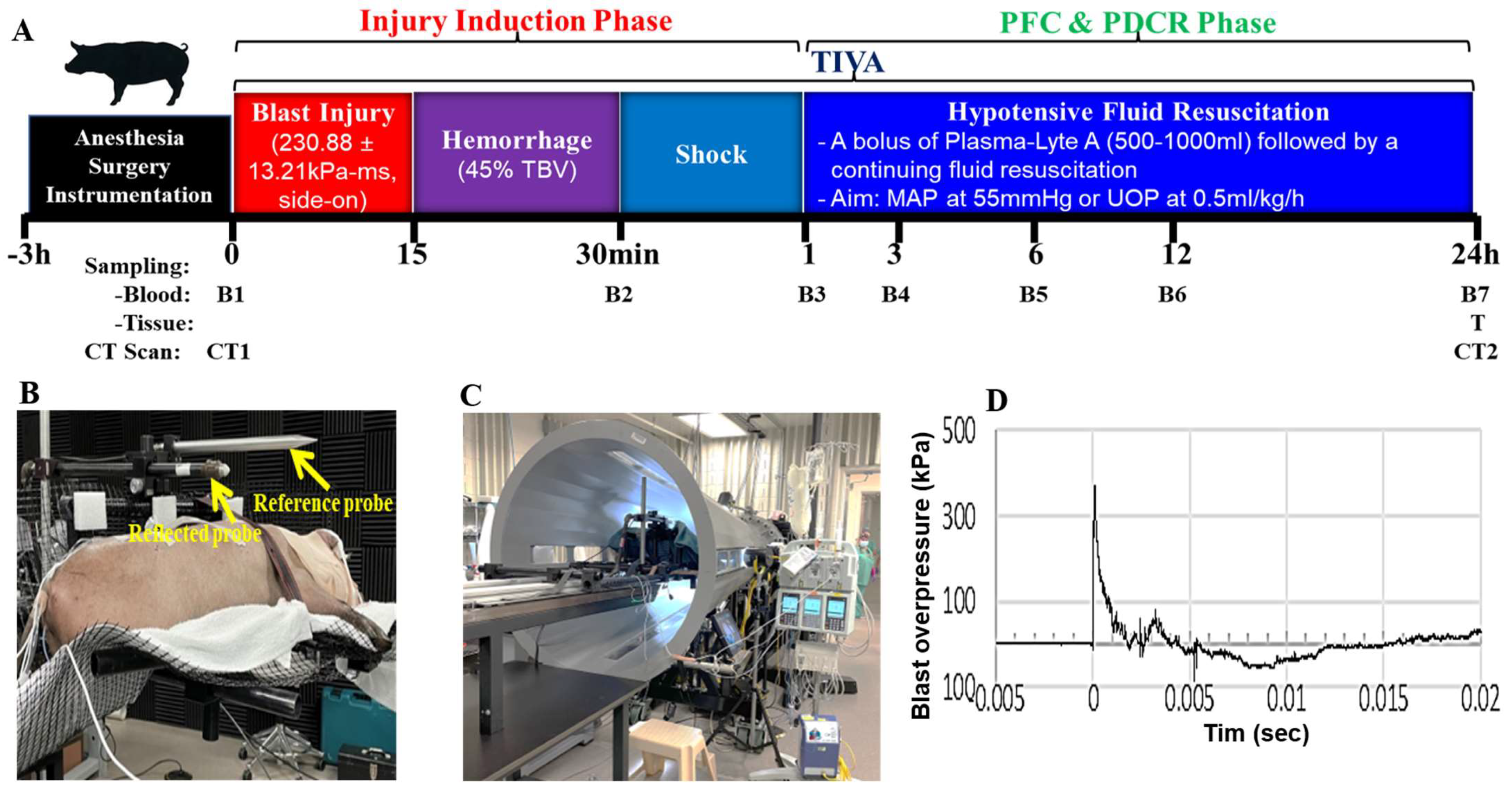
2.1. Blast Wave Parameters, Hemodynamic and Chemistry Changes after BI+HS
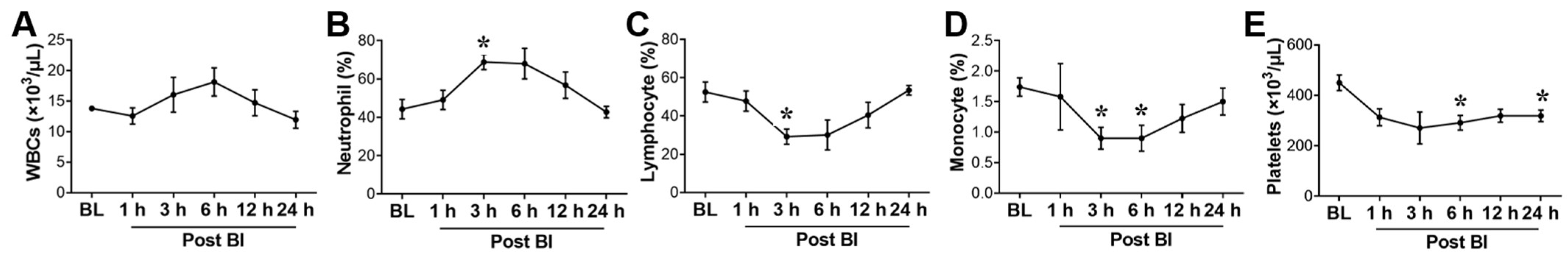
2.2. Effect of BI+HS on Circulating Complete Blood Count
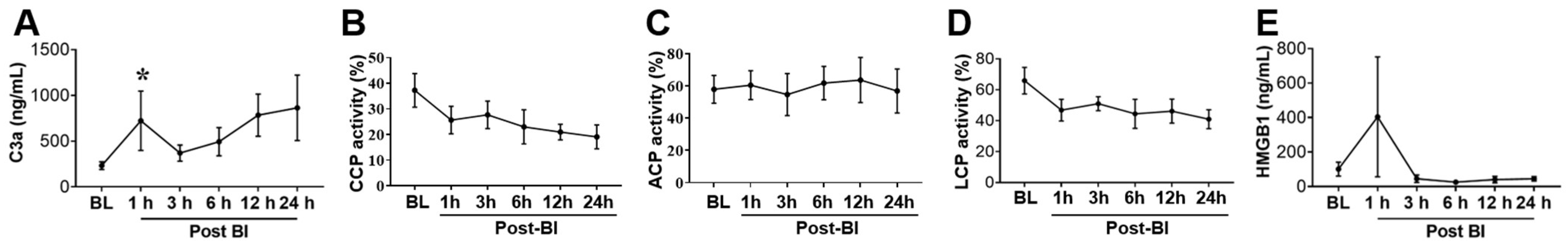
2.3. Effect of BI+HS on Circulating Complement Activation and HMGB1 Release
2.4. Effect of BI+HS on Circulating End Organ Damage Markers
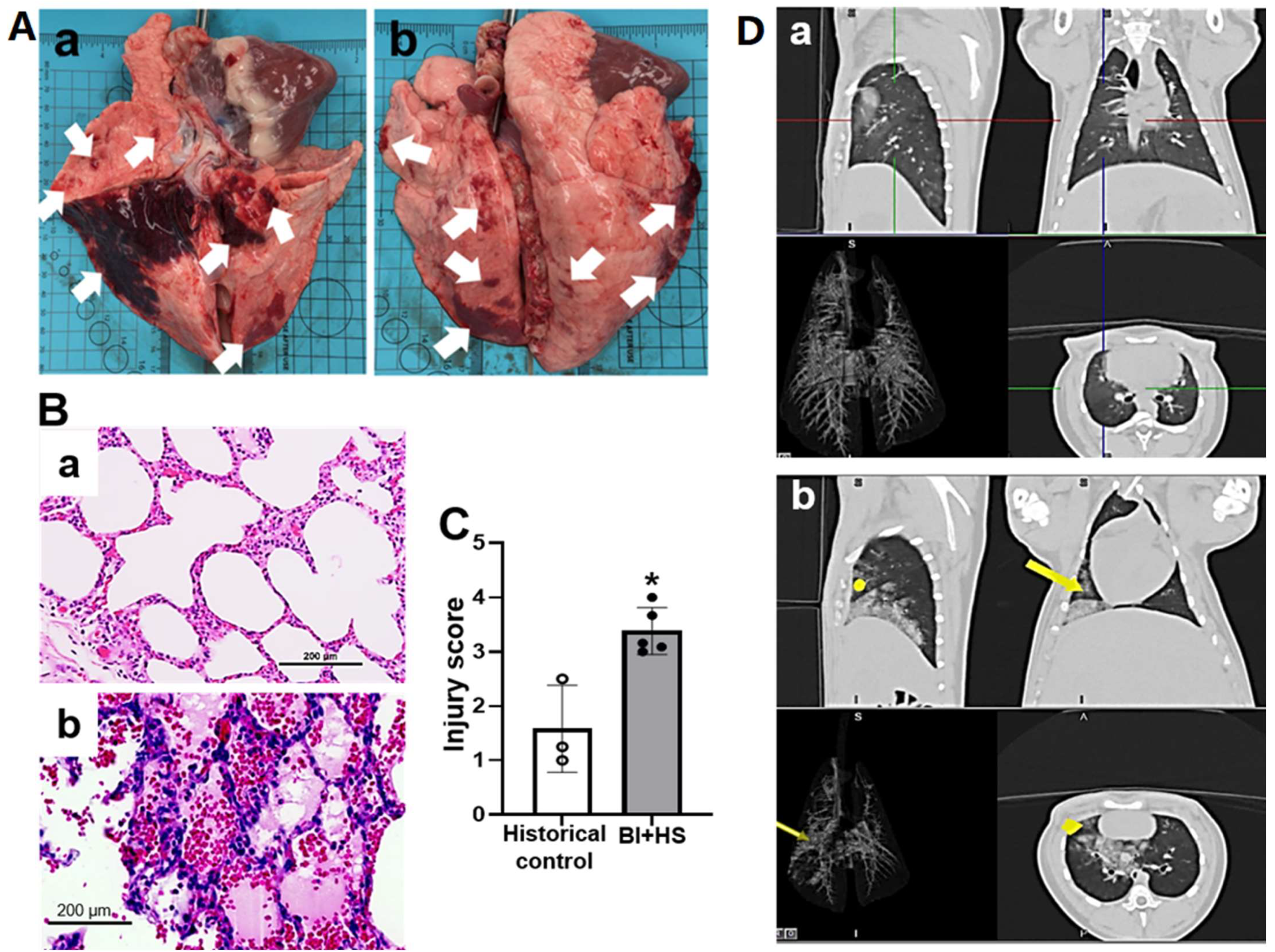
2.5. Effect of BI+HS on Acute Lung Injury
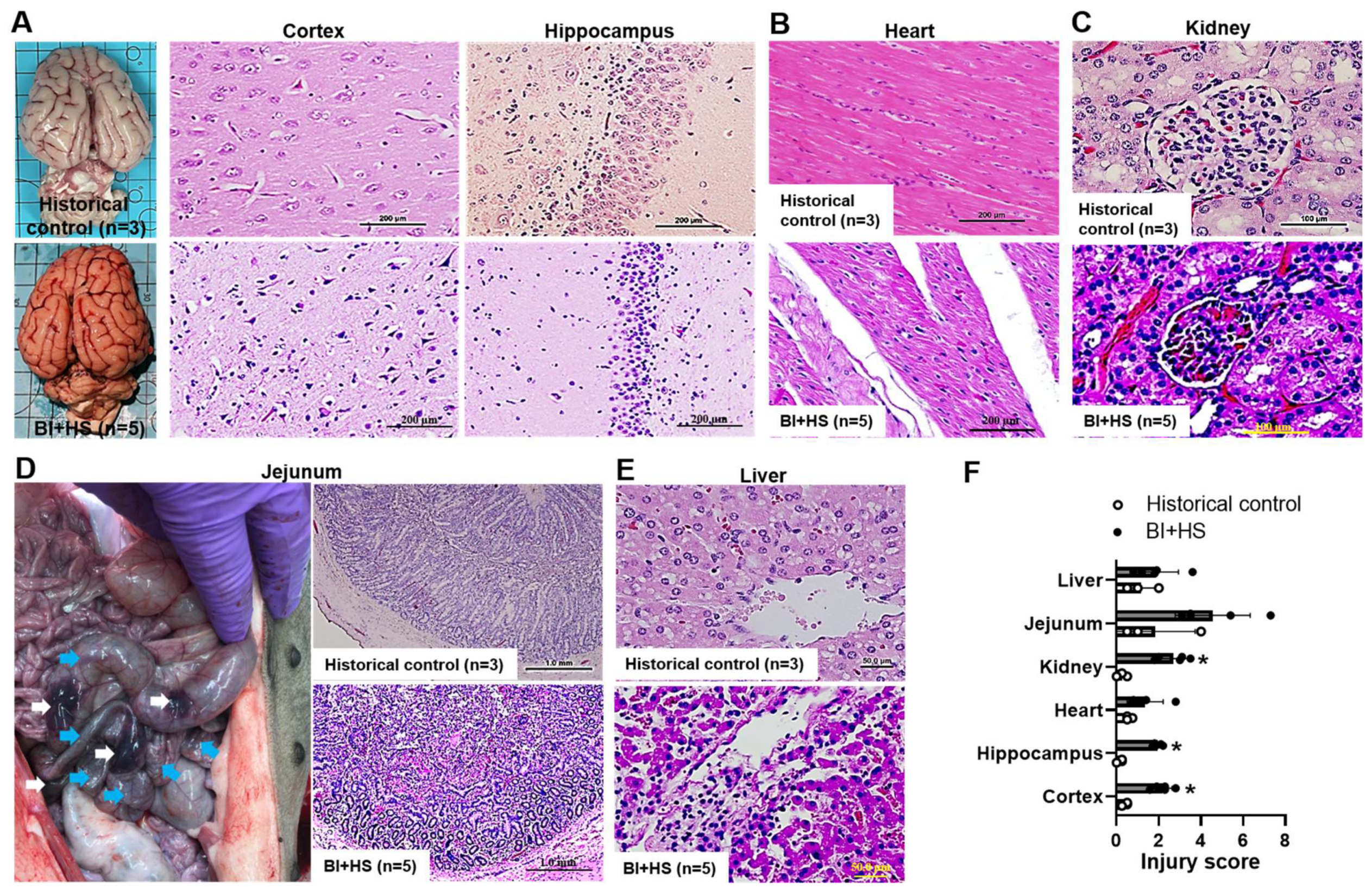
2.6. Effect of BI+HS on Other Organ Histopathological Alterations
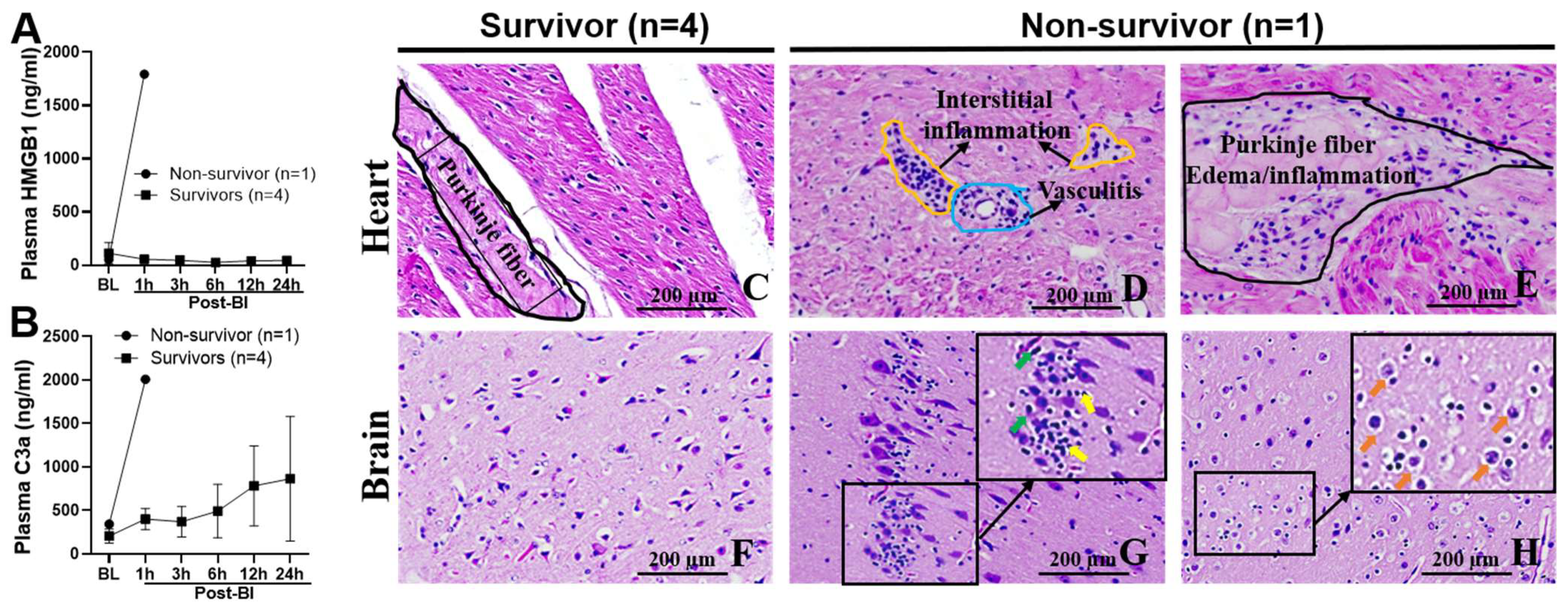
2.7. Blood Levels of C3a and HMGB1, and Myocarditis and Encephalitis in Non-Survival Animal
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.1.1. Animal Surgical, Injury, PDCR, and ICU Procedures
4.1.2. Biosampling
4.2. Assays
4.2.1. Analysis of Plasma C3a and HMGB1
4.2.2. Analysis of Complement Functional Activity
4.2.3. Complete Blood Count (CBC) and Coagulation Parameter Assessment
4.2.4. Assessment of End-Organ Damage Markers
4.2.5. Blood Gas and Chemistry Laboratory Assays
4.3. Histopathological Evaluation
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belmont, P.J.; McCriskin, B.J.; Hsiao, M.S.; Burks, R.; Nelson, K.J.; Schoenfeld, A.J. The Nature and Incidence of Musculoskeletal Combat Wounds in Iraq and Afghanistan (2005–2009). J. Orthop. Trauma 2013, 27, e107–e113. [Google Scholar] [CrossRef] [PubMed]
- Ritenour, A.E.; Blackbourne, L.H.; Kelly, J.F.; McLaughlin, D.F.; Pearse, L.A.; Holcomb, J.B.; Wade, C.E. Incidence of Primary Blast Injury in US Military Overseas Contingency Operations: A Retrospective Study. Ann. Surg. 2010, 251, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Cernak, I. The Importance of Systemic Response in the Pathobiology of Blast-Induced Neurotrauma. Front. Neurol. 2010, 1, 151. [Google Scholar] [CrossRef] [PubMed]
- Dubick, M.A. Current Concepts in Fluid Resuscitation for Prehospital Care of Combat Casualties. US Army Med. Dep. J. 2011, 18–24. [Google Scholar]
- Cannon, J.W. Hemorrhagic Shock. N. Engl. J. Med. 2018, 378, 370–379. [Google Scholar] [CrossRef]
- Lord, J.M.; Midwinter, M.J.; Chen, Y.-F.; Belli, A.; Brohi, K.; Kovacs, E.J.; Koenderman, L.; Kubes, P.; Lilford, R.J. The Systemic Immune Response to Trauma: An Overview of Pathophysiology and Treatment. Lancet 2014, 384, 1455–1465. [Google Scholar] [CrossRef]
- Duehrkop, C.; Rieben, R. Ischemia/Reperfusion Injury: Effect of Simultaneous Inhibition of Plasma Cascade Systems versus Specific Complement Inhibition. Biochem. Pharmacol. 2014, 88, 12–22. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate Immune Responses to Trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Huber-Lang, M.S.; Ignatius, A.; Köhl, J.; Mannes, M.; Braun, C.K. Complement in Trauma-Traumatised Complement? Br. J. Pharmacol. 2021, 178, 2863–2879. [Google Scholar] [CrossRef]
- Yang, Z.; Le, T.D.; Simovic, M.O.; Liu, B.; Fraker, T.L.; Cancio, T.S.; Cap, A.P.; Wade, C.E.; DalleLucca, J.J.; Li, Y. Traumatized Triad of Complementopathy, Endotheliopathy, and Coagulopathy-Impact on Clinical Outcomes in Severe Polytrauma Patients. Front. Immunol. 2022, 13, 991048. [Google Scholar] [CrossRef]
- Ganter, M.T.; Brohi, K.; Cohen, M.J.; Shaffer, L.A.; Walsh, M.C.; Stahl, G.L.; Pittet, J.-F. Role of the Alternative Pathway in the Early Complement Activation Following Major Trauma. Shock 2007, 28, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.-F. Early Release of High Mobility Group Box Nuclear Protein 1 after Severe Trauma in Humans: Role of Injury Severity and Tissue Hypoperfusion. Crit. Care 2009, 13, R174. [Google Scholar] [CrossRef] [PubMed]
- Peltz, E.D.; Moore, E.E.; Eckels, P.C.; Damle, S.S.; Tsuruta, Y.; Johnson, J.L.; Sauaia, A.; Silliman, C.C.; Banerjee, A.; Abraham, E. HMGB1 is Markedly Elevated within 6 Hours of Mechanical Trauma in Humans. Shock 2009, 32, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Burk, A.-M.; Martin, M.; Flierl, M.A.; Rittirsch, D.; Helm, M.; Lampl, L.; Bruckner, U.; Stahl, G.L.; Blom, A.M.; Perl, M.; et al. Early Complementopathy after Multiple Injuries in Humans. Shock 2012, 37, 348–354. [Google Scholar] [CrossRef]
- Coleman, L.G.; Maile, R.; Jones, S.W.; Cairns, B.A.; Crews, F.T. HMGB1/IL-1β Complexes in Plasma Microvesicles Modulate Immune Responses to Burn Injury. PLoS ONE 2018, 13, e0195335. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Q.; Liu, B.; Dixon, A.; Cancio, L.; Dubick, M.; Dalle Lucca, J. Early Complementopathy Predicts the Outcomes of Patients with Trauma. Trauma Surg. Acute Care Open 2019, 4, e000217. [Google Scholar] [CrossRef]
- Biancolella, M.; Testa, B.; Baghernajad Salehi, L.; D’Apice, M.R.; Novelli, G. Genetics and Genomics of Breast Cancer: Update and Translational Perspectives. Semin. Cancer Biol. 2021, 72, 27–35. [Google Scholar] [CrossRef]
- Yang, Z.; Simovic, M.O.; Edsall, P.R.; Liu, B.; Cancio, T.S.; Batchinsky, A.I.; Cancio, L.C.; Li, Y. HMGB1 Inhibition to Ameliorate Organ Failure and Increase Survival in Trauma. Biomolecules 2022, 12, 101. [Google Scholar] [CrossRef]
- Yang, Z.; Nunn, M.A.; Le, T.D.; Simovic, M.O.; Edsall, P.R.; Liu, B.; Barr, J.L.; Lund, B.J.; Hill-Pryor, C.D.; Pusateri, A.E.; et al. Immunopathology of Terminal Complement Activation and Complement C5 Blockade Creating a Pro-Survival and Organ-Protective Phenotype in Trauma. Br. J. Pharmacol. 2023, 180, 422–440. [Google Scholar] [CrossRef]
- Yang, Z.; Simovic, M.O.; Liu, B.; Burgess, M.B.; Cap, A.P.; DalleLucca, J.J.; Li, Y. Indices of Complement Activation and Coagulation Changes in Trauma Patients. Trauma Surg. Acute Care Open 2022, 7, e000927. [Google Scholar] [CrossRef]
- Dalle Lucca, J.J.; Chavko, M.; Dubick, M.A.; Adeeb, S.; Falabella, M.J.; Slack, J.L.; McCarron, R.; Li, Y. Blast-Induced Moderate Neurotrauma (BINT) Elicits Early Complement Activation and Tumor Necrosis Factor α (TNFα) Release in a Rat Brain. J. Neurol. Sci. 2012, 318, 146–154. [Google Scholar] [CrossRef]
- Li, Y.; Chavko, M.; Slack, J.L.; Liu, B.; McCarron, R.M.; Ross, J.D.; Dalle Lucca, J.J. Protective Effects of Decay-Accelerating Factor on Blast-Induced Neurotrauma in Rats. Acta Neuropathol. Commun. 2013, 1, 52. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Z.; Chavko, M.; Liu, B.; Aderemi, O.A.; Simovic, M.O.; Dubick, M.A.; Cancio, L.C. Complement Inhibition Ameliorates Blast-Induced Acute Lung Injury in Rats: Potential Role of Complement in Intracellular HMGB1-Mediated Inflammation. PLoS ONE 2018, 13, e0202594. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Aderemi, O.A.; Zhao, Q.; Edsall, P.R.; Simovic, M.O.; Lund, B.J.; Espinoza, M.D.; Woodson, A.M.; Li, Y.; Cancio, L.C. Early Complement and Fibrinolytic Activation in a Rat Model of Blast-Induced Multi-Organ Damage. Mil. Med. 2019, 184 (Suppl. 1), 282–290. [Google Scholar] [CrossRef]
- Szebeni, J.; Baranyi, L.; Savay, S.; Götze, O.; Alving, C.R.; Bünger, R.; Mongan, P.D. Complement Activation during Hemorrhagic Shock and Resuscitation in Swine. Shock 2003, 20, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Peckham, R.M.; Handrigan, M.T.; Bentley, T.B.; Falabella, M.J.; Chrovian, A.D.; Stahl, G.L.; Tsokos, G.C. C5-Blocking Antibody Reduces Fluid Requirements and Improves Responsiveness to Fluid Infusion in Hemorrhagic Shock Managed with Hypotensive Resuscitation. J. Appl. Physiol. 2007, 102, 673–680. [Google Scholar] [CrossRef]
- Ehrnthaller, C.; Amara, U.; Weckbach, S.; Kalbitz, M.; Huber-Lang, M.; Bahrami, S. Alteration of Complement Hemolytic Activity in Different Trauma and Sepsis Models. J. Inflamm. Res. 2012, 5, 59–66. [Google Scholar] [CrossRef]
- Simovic, M.O.; Falabella, M.J.; Le, T.D.; DalleLucca, J.J.; Li, Y. Decay-Accelerating Factor Creates an Organ-Protective Phenotype after Hemorrhage in Conscious Rats. Int. J. Mol. Sci. 2022, 23, 13563. [Google Scholar] [CrossRef] [PubMed]
- Dalle Lucca, J.J.; Li, Y.; Simovic, M.O.; Slack, J.L.; Cap, A.; Falabella, M.J.; Dubick, M.; Lebeda, F.; Tsokos, G.C. Decay-Accelerating Factor Limits Hemorrhage-Instigated Tissue Injury and Improves Resuscitation Clinical Parameters. J. Surg. Res. 2013, 179, 153–167. [Google Scholar] [CrossRef]
- Dalle Lucca, J.J.; Simovic, M.; Li, Y.; Moratz, C.; Falabella, M.; Tsokos, G.C. Decay-Accelerating Factor Mitigates Controlled Hemorrhage-Instigated Intestinal and Lung Tissue Damage and Hyperkalemia in Swine. J. Trauma 2011, 71 (Suppl. 1), S151–S160. [Google Scholar] [CrossRef]
- Campbell, J.C.; Li, Y.; van Amersfoort, E.; Relan, A.; Dubick, M.; Sheppard, F.; Pusateri, A.; Niemeyer, D.; Tsokos, G.C.; Dalle Lucca, J.J. C1 Inhibitor Limits Organ Injury and Prolongs Survival in Swine Subjected to Battlefield Simulated Injury. Shock 2016, 46 (3 Suppl. 1), 177–188. [Google Scholar] [CrossRef] [PubMed]
- Lupu, L.; Horst, K.; Greven, J.; Mert, Ü.; Ludviksen, J.A.K.; Pettersen, K.; Lau, C.; Li, Y.; Palmer, A.; Qin, K.; et al. Simultaneous C5 and CD14 Inhibition Limits Inflammation and Organ Dysfunction in Pig Polytrauma. Front. Immunol. 2022, 13, 952267. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.R.; Dodd, A.B.; Ling, J.M.; Stephenson, D.D.; Rannou-Latella, J.G.; Vermillion, M.S.; Mehos, C.J.; Johnson, V.E.; Gigliotti, A.P.; Dodd, R.J.; et al. Survival Rates and Biomarkers in a Large Animal Model of Traumatic Brain Injury Combined With Two Different Levels of Blood Loss. Shock 2021, 55, 554–562. [Google Scholar] [CrossRef]
- Butler, F.K.; Bennett, B.; Wedmore, C.I. Tactical Combat Casualty Care and Wilderness Medicine: Advancing Trauma Care in Austere Environments. Emerg. Med. Clin. N. Am. 2017, 35, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.; Montgomery, H.R.; Butler, F.K. Tactical Combat Casualty Care (TCCC) Guidelines for Medical Personnel: 05 November 2020. J. Spec. Oper. Med. 2020, 20, 144–151. [Google Scholar] [CrossRef]
- Watts, S.; Kirkman, E.; Bieler, D.; Bjarnason, S.; Franke, A.; Gupta, R.; Leggieri, M.J.; Orru, H.; Ouellet, S.; Philippens, M.; et al. Guidelines for Using Animal Models in Blast Injury Research. J. R. Army Med. Corps 2019, 165, 38–40. [Google Scholar] [CrossRef]
- Walters, E.M.; Prather, R.S. Advancing Swine Models for Human Health and Diseases. Mo. Med. 2013, 110, 212–215. [Google Scholar]
- Bassols, A.; Costa, C.; Eckersall, P.D.; Osada, J.; Sabrià, J.; Tibau, J. The Pig as an Animal Model for Human Pathologies: A Proteomics Perspective. Proteom. Clin. Appl. 2014, 8, 715–731. [Google Scholar] [CrossRef]
- Hawksworth, J.S.; Graybill, C.; Brown, T.S.; Gillern, S.M.; Wallace, S.M.; Davis, T.A.; Elster, E.A.; Tadaki, D.K. Lymphocyte Depletion in Experimental Hemorrhagic Shock in Swine. J. Inflamm. 2012, 9, 34. [Google Scholar] [CrossRef]
- Dalle Lucca, J.J.; Li, Y.; Simovic, M.; Pusateri, A.E.; Falabella, M.; Dubick, M.A.; Tsokos, G.C. Effects of C1 Inhibitor on Tissue Damage in a Porcine Model of Controlled Hemorrhage. Shock 2012, 38, 82–91. [Google Scholar] [CrossRef]
- Nunez, T.C.; Cotton, B.A. Transfusion Therapy in Hemorrhagic Shock. Curr. Opin. Crit. Care 2009, 15, 536–541. [Google Scholar] [CrossRef]
- Shafi, S.; Collinsworth, A.W.; Richter, K.M.; Alam, H.B.; Becker, L.B.; Bullock, M.R.; Ecklund, J.M.; Gallagher, J.; Gandhi, R.; Haut, E.R.; et al. Bundles of Care for Resuscitation from Hemorrhagic Shock and Severe Brain Injury in Trauma Patients-Translating Knowledge into Practice. J. Trauma Acute Care Surg. 2016, 81, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Hannon, J.P.; Wade, C.E.; Bossone, C.A.; Hunt, M.M.; Loveday, J.A. Oxygen Delivery and Demand in Conscious Pigs Subjected to Fixed-Volume Hemorrhage and Resuscitated with 7.5% NaCl in 6% Dextran. Circ. Shock 1989, 29, 205–217. [Google Scholar] [PubMed]
- Magder, S. Volume and Its Relationship to Cardiac Output and Venous Return. Crit. Care 2016, 20, 271. [Google Scholar] [CrossRef] [PubMed]
- Young, D.B. Control of Cardiac Output; Colloquium Series on Integrated Systems Physiology: From Molecule to Function to Disease; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Hauser, C.J. Preclinical Models of Traumatic, Hemorrhagic Shock. Shock 2005, 24 (Suppl. 1), 24–32. [Google Scholar] [CrossRef] [PubMed]
- Lomas-Niera, J.L.; Perl, M.; Chung, C.-S.; Ayala, A. Shock and Hemorrhage: An Overview of Animal Models. Shock 2005, 24 (Suppl. 1), 33–39. [Google Scholar] [CrossRef]
- Zahorec, R. Neutrophil-to-Lymphocyte Ratio, Past, Present and Future Perspectives. Bratisl. Lek. Listy 2021, 122, 474–488. [Google Scholar] [CrossRef]
- Hoffmann, T.; Böttger, E.C.; Baum, H.P.; Messner, M.; Hadding, U.; Bitter-Suermann, D. In Vivo Effects of C3a on Neutrophils and Its Contribution to Inflammatory Lung Processes in a Guinea-Pig Model. Clin. Exp. Immunol. 1988, 71, 486–492. [Google Scholar]
- Harboe, M.; Thorgersen, E.B.; Mollnes, T.E. Advances in Assay of Complement Function and Activation. Adv. Drug Deliv. Rev. 2011, 63, 976–987. [Google Scholar] [CrossRef]
- MacLeod, J.B.A.; Lynn, M.; McKenney, M.G.; Cohn, S.M.; Murtha, M. Early Coagulopathy Predicts Mortality in Trauma. J. Trauma 2003, 55, 39–44. [Google Scholar] [CrossRef]
- Woolley, T.; Gwyther, R.; Parmar, K.; Kirkman, E.; Watts, S.; Midwinter, M.; Lucca, J.D.; Hunt, B.J. A Prospective Observational Study of Acute Traumatic Coagulopathy in Traumatic Bleeding from the Battlefield. Transfusion 2020, 60 (Suppl. 3), S52–S61. [Google Scholar] [CrossRef] [PubMed]
- Bauman, R.A.; Ling, G.; Tong, L.; Januszkiewicz, A.; Agoston, D.; Delanerolle, N.; Kim, Y.; Ritzel, D.; Bell, R.; Ecklund, J.; et al. An Introductory Characterization of a Combat-Casualty-Care Relevant Swine Model of Closed Head Injury Resulting from Exposure to Explosive Blast. J. Neurotrauma 2009, 26, 841–860. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, E.; Watts, S. Haemodynamic Changes in Trauma. Br. J. Anaesth. 2014, 113, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, E.; Watts, S. Characterization of the Response to Primary Blast Injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 286–290. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.G.; Kenny, R.; Bennett, K.; Little, R.; Kirkman, E. Effect of Acute Traumatic Brain Injury on Baroreflex Function. Shock 2011, 35, 53–58. [Google Scholar] [CrossRef]
- Galvagno, S.M.; Fox, E.E.; Appana, S.N.; Baraniuk, S.; Bosarge, P.L.; Bulger, E.M.; Callcut, R.A.; Cotton, B.A.; Goodman, M.; Inaba, K.; et al. Outcomes after Concomitant Traumatic Brain Injury and Hemorrhagic Shock: A Secondary Analysis from the Pragmatic, Randomized Optimal Platelets and Plasma Ratios Trial. J. Trauma Acute Care Surg. 2017, 83, 668–674. [Google Scholar] [CrossRef]
- Liu, H.; Xiao, X.; Sun, C.; Sun, D.; Li, Y.; Yang, M. Systemic Inflammation and Multiple Organ Injury in Traumatic Hemorrhagic Shock. Front. Biosci. 2015, 20, 927–933. [Google Scholar] [CrossRef]
- Matsushita, Y.; Bramlett, H.M.; Kuluz, J.W.; Alonso, O.; Dietrich, W.D. Delayed Hemorrhagic Hypotension Exacerbates the Hemodynamic and Histopathologic Consequences of Traumatic Brain Injury in Rats. J. Cereb. Blood Flow Metab. 2001, 21, 847–856. [Google Scholar] [CrossRef]
- Giri, B.K.; Krishnappa, I.K.; Bryan, R.M.; Robertson, C.; Watson, J. Regional Cerebral Blood Flow after Cortical Impact Injury Complicated by a Secondary Insult in Rats. Stroke 2000, 31, 961–967. [Google Scholar] [CrossRef]
- Muller, C.R.; Courelli, V.; Lucas, A.; Williams, A.T.; Li, J.B.; Dos Santos, F.; Cuddington, C.T.; Moses, S.R.; Palmer, A.F.; Kistler, E.B.; et al. Resuscitation from Hemorrhagic Shock after Traumatic Brain Injury with Polymerized Hemoglobin. Sci. Rep. 2021, 11, 2509. [Google Scholar] [CrossRef]
- Brotfain, E.; Leibowitz, A.; Dar, D.E.; Krausz, M.M.; Shapira, Y.; Koyfman, L.; Klein, M.; Hess, S.; Zlotnik, A. Severe Traumatic Brain Injury and Controlled Hemorrhage in Rats: Quest for the Optimal Mean Arterial Blood Pressure after Whole Fresh Donor Blood Resuscitation. Shock 2012, 38, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Ramming, S.; Shackford, S.R.; Zhuang, J.; Schmoker, J.D. The Relationship of Fluid Balance and Sodium Administration to Cerebral Edema Formation and Intracranial Pressure in a Porcine Model of Brain Injury. J. Trauma 1994, 37, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, A.; Turóczi, Z.; Garbaisz, D.; Harsányi, L.; Szijártó, A. Experimental Models of Hemorrhagic Shock: A Review. Eur. Surg. Res. 2013, 50, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Q.; Wade, C.E. Influences of Traumatic Brain Injury on the Outcomes of Delayed and Repeated Hemorrhages. Circ. Shock 1991, 35, 231–236. [Google Scholar] [PubMed]
- Goodfellow, M.J.; Medina, J.A.; Proctor, J.L.; Xu, S.; Gullapalli, R.P.; Rangghran, P.; Miller, C.; Vesselinov, A.; Fiskum, G. Combined Traumatic Brain Injury and Hemorrhagic Shock in Ferrets Leads to Structural, Neurochemical, and Functional Impairments. J. Neurotrauma 2022, 39, 1442–1452. [Google Scholar] [CrossRef]
- Jin, G.; DeMoya, M.A.; Duggan, M.; Knightly, T.; Mejaddam, A.Y.; Hwabejire, J.; Lu, J.; Smith, W.M.; Kasotakis, G.; Velmahos, G.C.; et al. Traumatic Brain Injury and Hemorrhagic Shock: Evaluation of Different Resuscitation Strategies in a Large Animal Model of Combined Insults. Shock 2012, 38, 49–56. [Google Scholar] [CrossRef]
- Hicks, R.R.; Fertig, S.J.; Desrocher, R.E.; Koroshetz, W.J.; Pancrazio, J.J. Neurological Effects of Blast Injury. J. Trauma 2010, 68, 1257–1263. [Google Scholar] [CrossRef]
- Sziklavari, Z.; Molnar, T.F. Blast Injures to the Thorax. J. Thorac. Dis. 2019, 11 (Suppl. 2), S167–S171. [Google Scholar] [CrossRef]
- Wani, I.; Parray, F.Q.; Sheikh, T.; Wani, R.A.; Amin, A.; Gul, I.; Nazir, M. Spectrum of Abdominal Organ Injury in a Primary Blast Type. World J. Emerg. Surg. 2009, 4, 46. [Google Scholar] [CrossRef]
- Bass, C.R.; Panzer, M.B.; Rafaels, K.A.; Wood, G.; Shridharani, J.; Capehart, B. Brain Injuries from Blast. Ann. Biomed. Eng. 2012, 40, 185–202. [Google Scholar] [CrossRef]
- Mendez, M.F.; Owens, E.M.; Reza Berenji, G.; Peppers, D.C.; Liang, L.-J.; Licht, E.A. Mild Traumatic Brain Injury from Primary Blast vs. Blunt Forces: Post-Concussion Consequences and Functional Neuroimaging. NeuroRehabilitation 2013, 32, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.K.; Leonessa, F.; Ling, G.S.F. Blast TBI Models, Neuropathology, and Implications for Seizure Risk. Front. Neurol. 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Žurek, J.; Bartlová, L.; Fedora, M. Hyperphosphorylated Neurofilament NF-H as a Predictor of Mortality after Brain Injury in Children. Brain Inj. 2011, 25, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Okazaki, R.; Ishii, K.; Ueno, T.; Izawa, N.; Tanaka, Y.; Toyooka, S.; Matsuoka, N.; Morioka, K.; Ohori, Y.; et al. Phosphorylated Neurofilament Subunit NF-H as a Biomarker for Evaluating the Severity of Spinal Cord Injury Patients, a Pilot Study. Spinal Cord. 2012, 50, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Otani, N.; Morimoto, Y.; Kinoshita, M.; Ogata, T.; Mori, K.; Kobayashi, M.; Maeda, T.; Yoshino, A. Serial Changes in Serum Phosphorylated Neurofilament and Value for Prediction of Clinical Outcome after Traumatic Brain Injury. Surg. Neurol. Int. 2020, 11, 387. [Google Scholar] [CrossRef]
- Holmström, U.; Tsitsopoulos, P.P.; Holtz, A.; Salci, K.; Shaw, G.; Mondello, S.; Marklund, N. Cerebrospinal Fluid Levels of GFAP and PNF-H Are Elevated in Patients with Chronic Spinal Cord Injury and Neurological Deterioration. Acta Neurochir. 2020, 162, 2075–2086. [Google Scholar] [CrossRef]
- Ahadi, R.; Khodagholi, F.; Daneshi, A.; Vafaei, A.; Mafi, A.A.; Jorjani, M. Diagnostic Value of Serum Levels of GFAP, PNF-H, and NSE Compared With Clinical Findings in Severity Assessment of Human Traumatic Spinal Cord Injury. Spine 2015, 40, E823–E830. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Scheff, S.W.; Miller, K.M.; Roberts, K.N.; Gilmer, L.K.; Yang, C.; Shaw, G. The Phosphorylated Axonal Form of the Neurofilament Subunit NF-H (PNF-H) as a Blood Biomarker of Traumatic Brain Injury. J. Neurotrauma 2008, 25, 1079–1085. [Google Scholar] [CrossRef]
- Karesioglu, E.; Cikriklar, H.I.; Durak, V.A.; Aydin, B.; Ardic, A.; Armagan, E. Serum PNF-H Levels in the First Six Hours after Experimental Mild Traumatic Brain Injury in Rats. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 6928–6934. [Google Scholar] [CrossRef]
- Boylan, K.B.; Glass, J.D.; Crook, J.E.; Yang, C.; Thomas, C.S.; Desaro, P.; Johnston, A.; Overstreet, K.; Kelly, C.; Polak, M.; et al. Phosphorylated Neurofilament Heavy Subunit (PNF-H) in Peripheral Blood and CSF as a Potential Prognostic Biomarker in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 467–472. [Google Scholar] [CrossRef]
- Tsokos, M.; Paulsen, F.; Petri, S.; Madea, B.; Puschel, K.; Turk, E.E. Histologic, Immunohistochemical, and Ultrastructural Findings in Human Blast Lung Injury. Am. J. Respir. Crit. Care Med. 2003, 168, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Tovar, M.A.; Bell, R.S.; Neal, C.J. Epidemiology of Blast Neurotrauma: A Meta-Analysis of Blast Injury Patterns in the Military and Civilian Populations. World Neurosurg. 2021, 146, 308–314.e3. [Google Scholar] [CrossRef] [PubMed]
- Alstrup, A.K.O. Blood Lactate Concentrations in Göttingen Minipigs Compared with Domestic Pigs. J. Am. Assoc. Lab. Anim. Sci. 2016, 55, 18–20. [Google Scholar] [PubMed]
- Macku, D.; Hedvicak, P.; Quinn, J.M.; Bencko, V. Prehospital Medicine and the Future Will ECMO Ever Play a Role? J. Spec. Oper. Med. 2018, 18, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Butler, F.K.; Holcomb, J.B.; Schreiber, M.A.; Kotwal, R.S.; Jenkins, D.A.; Champion, H.R.; Bowling, F.; Cap, A.P.; DuBose, J.J.; Dorlac, W.C.; et al. Fluid Resuscitation for Hemorrhagic Shock in Tactical Combat Casualty Care: TCCC Guidelines Change 14-01–2 June 2014. J. Spec. Oper. Med. 2014, 14, 13–38. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dubick, M.A.; Yang, Z.; Barr, J.L.; Gremmer, B.J.; Lucas, M.L.; Necsoiu, C.; Jordan, B.S.; Batchinsky, A.I.; Cancio, L.C. Distal Organ Inflammation and Injury after Resuscitative Endovascular Balloon Occlusion of the Aorta in a Porcine Model of Severe Hemorrhagic Shock. PLoS ONE 2020, 15, e0242450. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overpressure | Reflected | |||||
|---|---|---|---|---|---|---|
| P0 (kPa) | t+ (ms) | I (kPa-ms) | P0 (kPa) | t+ (ms) | I (kPa-ms) | |
| BI+HS (n = 5) | 338.4 ± 22.55 | 2.09 ± 0.22 | 230.88 ± 29.55 | 616.92. ± 77.1 | 3.55 ± 1.19 | 472.58 ± 56.64 |
| Parameters | Baseline (BL) | 1 h Post-BI | 3 h Post-BI | 6 h Post-BI | 12 h Post-BI | 24 h Post-BI |
|---|---|---|---|---|---|---|
| MAP (mmHg) | 109.4 ± 13.30 | 55.20 ± 20.29 * | 73.25 ± 8.18 * | 79.50 ± 20.60 | 94.25 ± 11.15 | 87.50 ± 20.34 |
| PP (mmHg) | 37.8 ± 7.16 | 17.6 ± 9.74 * | 29.25 ± 4.79 | 13.75 ± 3.59 * | 24.25 ± 9.47 | 31.5 ± 13.63 |
| Shock Index | 1.09 ± 0.12 | 2.40 ± 0.54 * | 1.93 ± 0.34* | 1.76 ± 0.31 * | 1.43 ± 0.30 | 1.13 ± 0.22 |
| SpO2 (%) | 96.40 ± 2.70 | 86.00 ± 25.88 | 95.50 ± 2.09 | 96.75 ± 4.27 | 96.25 ± 2.50 | 98.50 ± 1.73 |
| PFR | 476.97 ± 82.18 | 366.36 ± 120.02 | 477.67 ± 64.00 | 478.76 ± 26.34 | 472.00 ± 48.77 | 507.33 ± 84.68 |
| pH | 7.37 ± 0.06 | 7.36 ± 0.03 | 7.45 ± 0.06 | 7.47 ± 0.06 | 7.49 ± 0.03 * | 7.45 ± 0.02 |
| BE/BD | 4.80 ± 1.48 | 3.0 ± 2.65 | 3.75 ± 3.59 | 5.25 ± 4.50 | 6.00 ± 2.45 | 4.75 ± 3.30 |
| Lactate (mmol/L) | 2.13 ± 0.70 | 4.47 ± 4.21 | 3.98 ± 3.00 | 2.35 ± 3.03 | 0.71 ± 0.19 * | 1.08 ± 0.51 |
| K+ (mmol/L) | 3.96 ± 0.15 | 4.78 ± 2.04 | 4.13 ± 0.45 | 4.03 ± 0.62 | 3.70 ± 0.82 | 3.65 ± 0.40 |
| Glucose (mg/dL) | 76.00 ± 14.34 | 83.40 ± 33.89 | 111.50 ± 17.37 * | 122.25 ± 46.65 * | 77.75 ± 18.23 | 63.25 ± 7.32 |
| Hct (%) | 26.20 ± 1.48 | 30.60 ± 1.52 * | 25.50 ± 4.20 | 24.25 ± 3.78 | 18.50 ± 4.04 * | 16.75 ± 2.22 * |
| Hgb (mmol/L) | 8.90 ± 0.49 | 10.40 ± 0.50 * | 8.65 ± 1.42 | 8.05 ± 1.62 | 7.50 ± 0.00 | 4.18 ± 2.25 * |
| Biomarkers | Baseline (BL) | 1 h Post-BI | 3 h Post-BI | 6 h Post-BI | 12 h Post-BI | 24 h Post-BI |
|---|---|---|---|---|---|---|
| Troponin I (pg/L) | 17.9 ± 13.59 | 40.28 ± 30.09 | 75.67 ± 38.25 | 374.48 ± 392.49 * | 2461 ± 2648.02 * | 1711.3 ± 2525.21 |
| Myoglobin (ng/mL) | 52.75 ± 9.03 | 90.00 ± 10.23 * | 62.50 ± 10.08 | 188.75 ± 204.98 * | 491.25 ± 333.75 * | 905.67 ± 964.93 |
| GFAP (ng/mL) | 0.20 ± 0.34 | 0.59 ± 0.56 | 0.86 ± 0.99 | 3.72 ± 5.34 | 0.86 ± 3.72 | 0.66 ± 0.74 |
| p-NF-H (ng/mL) | 14.54 ± 1.93 | 14.60 ± 1.56 | 15.26 ± 0.88 | 15.20 ± 0.16 | 15.56 ± 0.74 | 15.40 ± 0.90 |
| Total bilirubin (mg/dL) | 0.09 ± 0.04 | 0.14 ± 0.08 | 0.12 ± 0.03 | 0.10 ± 0.07 | 0.20 ± 0.08 | 0.60 ± 0.44 * |
| AST (U/L) | 27.20 ± 22.71 | 110.00 ± 164.91 | 35.50 ± 26.34 | 58.75 ± 6.90 * | 91.75 ± 21.69 * | 174.00 ± 206.41 |
| Creatinine (mg/dL) | 0.57 ± 0.29 | 0.72 ± 0.40 | 0.73 ± 0.06 | 0.79 ± 0.12 | 0.75 ± 0.15 | 0.58 ± 0.22 |
| PT (second) | 13.38 ± 0.35 | 14.28 ± 1.88 * | 14.20 ± 0.34 | 13.85 ± 0.31 | 13.85 ± 0.53 | 14.48 ± 0.95 * |
| aPTT (second) | 26.00 ± 7.22 | 25.06 ± 9.43 | 24.37 ± 4.74 | 24.58 ± 5.99 | 27.00 ± 4.38 | 35.89 ± 13.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simovic, M.O.; Yang, Z.; Jordan, B.S.; Fraker, T.L.; Cancio, T.S.; Lucas, M.L.; Cancio, L.C.; Li, Y. Immunopathological Alterations after Blast Injury and Hemorrhage in a Swine Model of Prolonged Damage Control Resuscitation. Int. J. Mol. Sci. 2023, 24, 7494. https://doi.org/10.3390/ijms24087494
Simovic MO, Yang Z, Jordan BS, Fraker TL, Cancio TS, Lucas ML, Cancio LC, Li Y. Immunopathological Alterations after Blast Injury and Hemorrhage in a Swine Model of Prolonged Damage Control Resuscitation. International Journal of Molecular Sciences. 2023; 24(8):7494. https://doi.org/10.3390/ijms24087494
Chicago/Turabian StyleSimovic, Milomir O., Zhangsheng Yang, Bryan S. Jordan, Tamara L. Fraker, Tomas S. Cancio, Michael L. Lucas, Leopoldo C. Cancio, and Yansong Li. 2023. "Immunopathological Alterations after Blast Injury and Hemorrhage in a Swine Model of Prolonged Damage Control Resuscitation" International Journal of Molecular Sciences 24, no. 8: 7494. https://doi.org/10.3390/ijms24087494
APA StyleSimovic, M. O., Yang, Z., Jordan, B. S., Fraker, T. L., Cancio, T. S., Lucas, M. L., Cancio, L. C., & Li, Y. (2023). Immunopathological Alterations after Blast Injury and Hemorrhage in a Swine Model of Prolonged Damage Control Resuscitation. International Journal of Molecular Sciences, 24(8), 7494. https://doi.org/10.3390/ijms24087494