

Deciphering the Relationship between SARS-CoV-2 and Cancer




Abstract
:1. Introduction
2. Oncogenic Viruses and Their Mechanisms
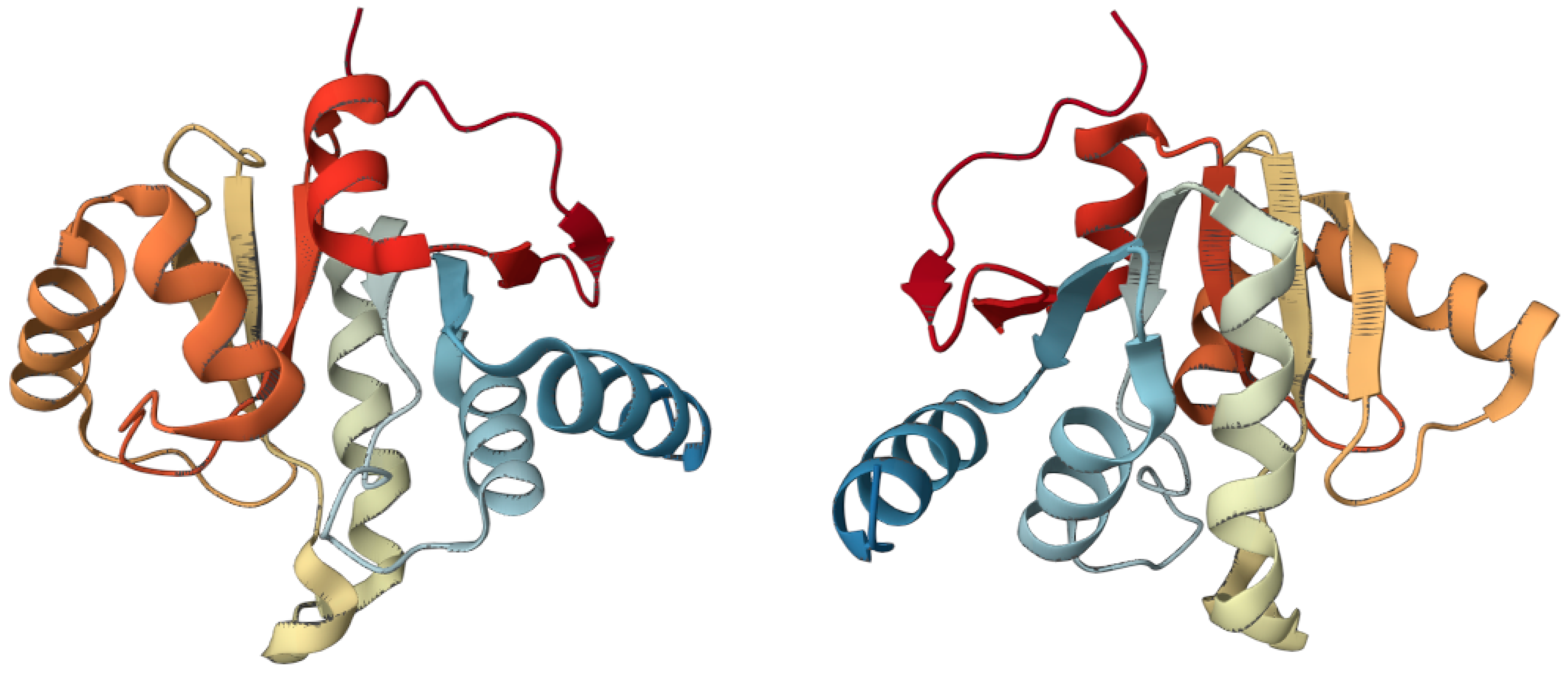
3. The Oncogenic Potential of SARS-CoV-2
4. The Oncolytic Potential of SARS-CoV-2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zaimy, M.A.; Saffarzadeh, N.; Mohammadi, A.; Pourghadamyari, H.; Izadi, P.; Sarli, A.; Moghaddam, L.K.; Paschepari, S.R.; Azizi, H.; Torkamandi, S.; et al. New methods in the diagnosis of cancer and gene therapy of cancer based on nanoparticles. Cancer Gene Ther. 2017, 24, 233–243. [Google Scholar] [CrossRef]
- Boopathi, E.; Thangavel, C. Dark Side of Cancer Therapy: Cancer Treatment-Induced Cardiopulmonary Inflammation, Fibrosis, and Immune Modulation. Int. J. Mol. Sci. 2021, 22, 10126. [Google Scholar] [CrossRef]
- Fotouhi, S.; Asadi, S.; Kattan, M.W. A comprehensive data level analysis for cancer diagnosis on imbalanced data. J. Biomed. Inform. 2019, 90, 103089. [Google Scholar] [CrossRef]
- Herbert, A.; Abel, G.A.; Winters, S.; McPhail, S.; Elliss-Brookes, L.; Lyratzopoulos, G. Are inequalities in cancer diagnosis through emergency presentation narrowing, widening or remaining unchanged? Longitudinal analysis of English population-based data 2006–2013. J. Epidemiol. Community Health 2019, 73, 3–10. [Google Scholar] [CrossRef]
- Raab, S.S.; Grzybicki, D.M. Quality in Cancer Diagnosis. CA Cancer J. Clin. 2010, 60, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Stingi, A.; Cirillo, L. SARS-CoV-2 infection and cancer. BioEssays 2021, 43, 2000289. [Google Scholar] [CrossRef]
- Arisi, M.; Zane, C.; Caravello, S.; Rovati, C.; Zanca, A.; Venturini, M.; Calzavara-Pinton, P. Sun Exposure and Melanoma, Certainties and Weaknesses of the Present Knowledge. Front. Med. 2018, 5, 235. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Rascovan, N.; Duraisamy, R.; Desnues, C. Metagenomics and the Human Virome in Asymptomatic Individuals. Annu. Rev. Microbiol. 2016, 70, 125–141. [Google Scholar] [CrossRef]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Schlecht-Louf, G.; Deback, C.; Bachelerie, F. The Chemokine System in Oncogenic Pathways Driven by Viruses: Perspectives for Cancer Immunotherapy. Cancers 2022, 14, 848. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- zur Hausen, H.; de Villiers, E.-M. Cancer “Causation” by Infections—Individual Contributions and Synergistic Networks. Semin. Oncol. 2014, 41, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Lieberman, P.M. Oncogenic Viruses as Entropic Drivers of Cancer Evolution. Front. Virol. 2021, 1, 753366. [Google Scholar] [CrossRef]
- Mui, U.; Haley, C.; Tyring, S. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, D. Searching for human oncoviruses: Histories, challenges, and opportunities. J. Cell. Biochem. 2018, 119, 4897–4906. [Google Scholar] [CrossRef]
- Mirzaei, H.; Khodadad, N.; Karami, C.; Pirmoradi, R.; Khanizadeh, S. The AP-1 pathway; A key regulator of cellular transformation modulated by oncogenic viruses. Rev. Med. Virol. 2019, 30, e2008. [Google Scholar] [CrossRef]
- Costanzo, M.; Fiocchetti, M.; Ascenzi, P.; Marino, M.; Caterino, M.; Ruoppolo, M. Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells. Molecules 2021, 26, 2397. [Google Scholar] [CrossRef]
- Costanzo, M.; Caterino, M.; Salvatori, I.; Manganelli, V.; Ferri, A.; Misasi, R.; Ruoppolo, M. Proteome data of neuroblastoma cells overexpressing Neuroglobin. Data Brief 2022, 41, 107843. [Google Scholar] [CrossRef]
- Manganelli, V.; Salvatori, I.; Costanzo, M.; Capozzi, A.; Caissutti, D.; Caterino, M.; Valle, C.; Ferri, A.; Sorice, M.; Ruoppolo, M.; et al. Overexpression of Neuroglobin Promotes Energy Metabolism and Autophagy Induction in Human Neuroblastoma SH-SY5Y Cells. Cells 2021, 10, 3394. [Google Scholar] [CrossRef]
- Rous, P. A Transmissible Avian Neoplasm. (Sarcoma of the Common Fowl.). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Cancers in Humans: A Lifelong Search for Contributions of Infectious Agents, Autobiographic Notes. Annu. Rev. Virol. 2019, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D. A sarcoma involving the jaws in african children. Br. J. Surg. 1958, 46, 218–223. [Google Scholar] [CrossRef]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and Biological Studies on a Virus in Cultured Lymphoblasts from Burkitt’s Lymphoma. J. Exp. Med. 1965, 121, 761–770. [Google Scholar] [CrossRef]
- Pipas, J.M.; Sullivan, C.S. DNA Tumor Viruses and Their Contributions to Molecular Biology. J. Virol. 2019, 93, e01524-18. [Google Scholar] [CrossRef]
- Lion, T. Adenovirus persistence, reactivation, and clinical management. FEBS Lett. 2019, 593, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-S.; Ren, H.-C.; Cao, J.-H. Correlation of SARS-CoV-2 to cancer: Carcinogenic or anticancer? (Review). Int. J. Oncol. 2022, 60, 42. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol. Oncol. 2010, 117, S5–S10. [Google Scholar] [CrossRef]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Chen, Z.; Burk, R.D. Human Papillomavirus Genomics: Past, Present and Future. In Human Papillomavirus; Karger: Basel, Switzerland, 2014; pp. 1–18. [Google Scholar]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef]
- Tu, T.; Bühler, S.; Bartenschlager, R. Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 2017, 398, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.F.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef]
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249. [Google Scholar] [CrossRef]
- Naseem, M.; Barzi, A.; Brezden-Masley, C.; Puccini, A.; Berger, M.D.; Tokunaga, R.; Battaglin, F.; Soni, S.; McSkane, M.; Zhang, W.; et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat. Rev. 2018, 66, 15–22. [Google Scholar] [CrossRef]
- Tagaya, Y.; Gallo, R.C. The Exceptional Oncogenicity of HTLV-1. Front. Microbiol. 2017, 8, 1425. [Google Scholar] [CrossRef]
- Brites, C.; Grassi, M.F.; Quaresma, J.A.S.; Ishak, R.; Vallinoto, A.C.R. Pathogenesis of HTLV-1 infection and progression biomarkers: An overview. Braz. J. Infect. Dis. 2021, 25, 101594. [Google Scholar] [CrossRef] [PubMed]
- Etta, E.; Alayande, D.; Mavhandu-Ramarumo, L.; Gachara, G.; Bessong, P. HHV-8 Seroprevalence and Genotype Distribution in Africa, 1998–2017: A Systematic Review. Viruses 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J. Human Herpesvirus 8-Encoded Proteins with Potential Roles in Virus-Associated Neoplasia. Front. Biosci. 2007, 12, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef] [PubMed]
- Krump, N.A.; You, J. From Merkel Cell Polyomavirus Infection to Merkel Cell Carcinoma Oncogenesis. Front. Microbiol. 2021, 12, 739695. [Google Scholar] [CrossRef] [PubMed]
- De Groof, T.W.M.; Elder, E.G.; Siderius, M.; Heukers, R.; Sinclair, J.H.; Smit, M.J.; Schulte, G. Viral G Protein–Coupled Receptors: Attractive Targets for Herpesvirus-Associated Diseases. Pharmacol. Rev. 2021, 73, 828–846. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Morales-Sánchez, A.; Fuentes-Pananá, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef]
- Adoue, V.; Joffre, O. Les rétrovirus endogènes. Med. Sci. 2020, 36, 253–260. [Google Scholar] [CrossRef]
- Tang, K.-W.; Larsson, E. Tumour virology in the era of high-throughput genomics. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160265. [Google Scholar] [CrossRef]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef]
- Elgui de Oliveira, D. DNA viruses in human cancer: An integrated overview on fundamental mechanisms of viral carcinogenesis. Cancer Lett. 2007, 247, 182–196. [Google Scholar] [CrossRef]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K–Akt–mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gaglia, M.M.; Munger, K. More than just oncogenes: Mechanisms of tumorigenesis by human viruses. Curr. Opin. Virol. 2018, 32, 48–59. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Münger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; Launoit, Y.d.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2006, 26, 1650–1655. [Google Scholar] [CrossRef]
- Costanzo, M.; De Giglio, M.A.; Roviello, G.N. SARS-CoV-2: Recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr. Med. Chem. 2020, 27, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, H.; Mashouri, L.; Okpechi, S.C.; Alahari, N.; Alahari, S.K. Repurposing existing drugs for the treatment of COVID-19/SARS-CoV-2 infection: A review describing drug mechanisms of action. Biochem. Pharmacol. 2021, 183, 114296. [Google Scholar] [CrossRef]
- Borbone, N.; Piccialli, I.; Falanga, A.P.; Piccialli, V.; Roviello, G.N.; Oliviero, G. Nucleic Acids as Biotools at the Interface between Chemistry and Nanomedicine in the COVID-19 Era. Int. J. Mol. Sci. 2022, 23, 4359. [Google Scholar] [CrossRef]
- Ricci, A.; Roviello, G.N. Exploring the Protective Effect of Food Drugs against Viral Diseases: Interaction of Functional Food Ingredients and SARS-CoV-2, Influenza Virus, and HSV. Life 2023, 13, 402. [Google Scholar] [CrossRef]
- Roviello, V.; Roviello, G.N. Less COVID-19 deaths in southern and insular Italy explained by forest bathing, Mediterranean environment, and antiviral plant volatile organic compounds. Environ. Chem. Lett. 2022, 20, 7–17. [Google Scholar] [CrossRef]
- Costanzo, M.; De Giglio, M.A.; Roviello, G.N. Anti-coronavirus vaccines: Past investigations on SARS-CoV-1 and MERS-CoV, the approved vaccines from BioNTech/Pfizer, Moderna, Oxford/AstraZeneca and others under Development Against SARSCoV-2 Infection. Curr. Med. Chem. 2022, 29, 4–18. [Google Scholar] [CrossRef]
- Damasceno, D.H.; Amaral, A.A.; Silva, C.A.; Simões e Silva, A.C. The impact of vaccination worldwide on SARS-CoV-2 infection: A review on vaccine mechanisms, results of clinical trials, vaccinal coverage and interactions with novel variants. Curr. Med. Chem. 2022, 29, 2673–2690. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, V.; Roviello, G.N. The Potential Role of Vaccines in Preventing Antimicrobial Resistance (AMR): An Update and Future Perspectives. Vaccines 2023, 11, 333. [Google Scholar] [CrossRef]
- Cosar, B.; Karagulleoglu, Z.Y.; Unal, S.; Ince, A.T.; Uncuoglu, D.B.; Tuncer, G.; Kilinc, B.R.; Ozkan, Y.E.; Ozkoc, H.C.; Demir, I.N. SARS-CoV-2 mutations and their viral variants. Cytokine Growth Factor Rev. 2022, 63, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and evolution of the SARS-CoV-2 spike protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Thieblemont, C.; Alran, S.; Faivre, S. Impact of the COVID-19 Outbreak on the Management of Patients with Cancer. Target. Oncol. 2020, 15, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Liu, D.; Liu, M.; Zhou, F.; Li, G.; Chen, Z.; Zhang, Z.; You, H.; Wu, M.; Zheng, Q.; et al. Patients with Cancer Appear More Vulnerable to SARS-CoV-2: A Multicenter Study during the COVID-19 Outbreak. Cancer Discov. 2020, 10, 783–791. [Google Scholar] [CrossRef]
- Li, F.; Fu, L.; Liu, X.; Liu, X.-a.; Liang, Y.; Lv, Y.; Yang, Z.; Guo, A.; Chen, Z.; Li, W.; et al. Serum metabolomic abnormalities in survivors of non-severe COVID-19. Heliyon 2022, 8, e10473. [Google Scholar] [CrossRef]
- Shen, T.; Wang, T. Metabolic Reprogramming in COVID-19. Int. J. Mol. Sci. 2021, 22, 11475. [Google Scholar] [CrossRef]
- Rudiansyah, M.; Jasim, S.A.; Mohammad Pour, Z.G.; Athar, S.S.; Jeda, A.S.; Doewes, R.I.; Jalil, A.T.; Bokov, D.O.; Mustafa, Y.F.; Noroozbeygi, M.; et al. Coronavirus disease 2019 (COVID-19) update: From metabolic reprogramming to immunometabolism. J. Med. Virol. 2022, 94, 4611–4627. [Google Scholar] [CrossRef]
- Li, S.; Ma, F.; Yokota, T.; Garcia, G.; Palermo, A.; Wang, Y.; Farrell, C.; Wang, Y.-C.; Wu, R.; Zhou, Z.; et al. Metabolic reprogramming and epigenetic changes of vital organs in SARS-CoV-2–induced systemic toxicity. JCI Insight 2021, 6, e145027. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Nie, M.; Pang, H.; Wang, B.; Hu, J.; Meng, X.; Li, K.; Ran, X.; Long, Q.; Deng, H.; et al. Integrated cytokine and metabolite analysis reveals immunometabolic reprogramming in COVID-19 patients with therapeutic implications. Nat. Commun. 2021, 12, 1618. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Caterino, M.; Fedele, R.; Cevenini, A.; Pontillo, M.; Barra, L.; Ruoppolo, M. COVIDomics: The Proteomic and Metabolomic Signatures of COVID-19. Int. J. Mol. Sci. 2022, 23, 2414. [Google Scholar] [CrossRef]
- Caterino, M.; Gelzo, M.; Sol, S.; Fedele, R.; Annunziata, A.; Calabrese, C.; Fiorentino, G.; D’Abbraccio, M.; Dell’Isola, C.; Fusco, F.M.; et al. Dysregulation of lipid metabolism and pathological inflammation in patients with COVID-19. Sci. Rep. 2021, 11, 2941. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wang, C.; Duan, B.; Yang, P.; Wu, J.; Zhang, Q. Altered Lipid Profile in COVID-19 Patients and Metabolic Reprogramming. Front. Microbiol. 2022, 13, 863802. [Google Scholar] [CrossRef]
- Borella, R.; De Biasi, S.; Paolini, A.; Boraldi, F.; Lo Tartaro, D.; Mattioli, M.; Fidanza, L.; Neroni, A.; Caro-Maldonado, A.; Meschiari, M.; et al. Metabolic reprograming shapes neutrophil functions in severe COVID-19. Eur. J. Immunol. 2021, 52, 484–502. [Google Scholar] [CrossRef]
- Moolamalla, S.T.R.; Balasubramanian, R.; Chauhan, R.; Priyakumar, U.D.; Vinod, P.K. Host metabolic reprogramming in response to SARS-CoV-2 infection: A systems biology approach. Microb. Pathog. 2021, 158, 105114. [Google Scholar] [CrossRef]
- Goubran, H.; Stakiw, J.; Seghatchian, J.; Ragab, G.; Burnouf, T. SARS-CoV-2 and cancer: The intriguing and informative cross-talk. Transfus. Apher. Sci. 2022, 61, 103488. [Google Scholar] [CrossRef]
- Kim, J.-M.; Kim, H.M.; Lee, E.J.; Jo, H.J.; Yoon, Y.; Lee, N.-J.; Son, J.; Lee, Y.-J.; Kim, M.S.; Lee, Y.-P.; et al. Detection and Isolation of SARS-CoV-2 in Serum, Urine, and Stool Specimens of COVID-19 Patients from the Republic of Korea. Osong Public Health Res. Perspect. 2020, 11, 112–117. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.d.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Policard, M.; Jain, S.; Rego, S.; Dakshanamurthy, S. Immune characterization and profiles of SARS-CoV-2 infected patients reveals potential host therapeutic targets and SARS-CoV-2 oncogenesis mechanism. Virus Res. 2021, 301, 198464. [Google Scholar] [CrossRef]
- Santorelli, L.; Caterino, M.; Costanzo, M. Dynamic Interactomics by Cross-Linking Mass Spectrometry: Mapping the Daily Cell Life in Postgenomic Era. OMICS J. Integr. Biol. 2022, 26, 633–649. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Liu, P.; Leibowitz, J.L.; Kao, C.C. The Coronavirus Endoribonuclease Nsp15 Interacts with Retinoblastoma Tumor Suppressor Protein. J. Virol. 2012, 86, 4294–4304. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Sheng, Y.; Laister, R.C.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.; Sunnerhagen, M.; Srisailam, S.; Karra, M.; et al. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 2008, 15, 1334–1342. [Google Scholar] [CrossRef]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-Induced Ubiquitin-Protein Ligase, Promotes p53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Frick, D.N.; Virdi, R.S.; Vuksanovic, N.; Dahal, N.; Silvaggi, N.R. Molecular basis for ADP-ribose binding to the Mac1 domain of SARS-CoV-2 nsp3. Biochemistry 2020, 59, 2608–2615. [Google Scholar] [CrossRef]
- Cardozo, C.M.; Hainaut, P. Viral strategies for circumventing p53: The case of severe acute respiratory syndrome coronavirus. Curr. Opin. Oncol. 2021, 33, 149–158. [Google Scholar] [CrossRef]
- Gómez-Carballa, A.; Martinón-Torres, F.; Salas, A. Is SARS-CoV-2 an oncogenic virus? J. Infect. 2022, 85, 573–607. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, X.; Kuang, E. Subversion of autophagy machinery and organelle-specific autophagy by SARS-CoV-2 and coronaviruses. Autophagy 2023, 19, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Cai, K.; Li, J.; Yuan, Z.; Chen, R.; Xiao, H.; Xu, C.; Hu, B.; Qin, Y.; Ding, B. Coronavirus subverts ER-phagy by hijacking FAM134B and ATL3 into p62 condensates to facilitate viral replication. Cell Rep. 2023, 42, 112286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-I. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Maiti, A.; Hait, N.C. Autophagy-mediated tumor cell survival and progression of breast cancer metastasis to the brain. J. Cancer 2021, 12, 954–964. [Google Scholar] [CrossRef]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Lu, W.; Liu, X.; Wang, T.; Liu, F.; Zhu, A.; Lin, Y.; Luo, J.; Ye, F.; He, J.; Zhao, J.; et al. Elevated MUC1 and MUC5AC mucin protein levels in airway mucus of critical ill COVID-19 patients. J. Med. Virol. 2021, 93, 582–584. [Google Scholar] [CrossRef]
- Knack, R.; Hanada, T.Z.B.; Knack, R.S.; Dana, S.; Afonso, G.L.; Omena, T.; Mayr, K.; Knack, R.S. SARS-CoV-2, a possible new oncovirus? Qeios, 2023; preprint. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, P.; Zhang, K.; Huang, C. The application of CA72-4 in the diagnosis, prognosis, and treatment of gastric cancer. Biochim. Biophys. Acta-Rev. Cancer 2021, 1876, 188634. [Google Scholar] [CrossRef]
- Saini, G.; Aneja, R. Cancer as a prospective sequela of long COVID-19. BioEssays 2021, 43, e2000331. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Neves, H.; Kwok, H.F. Recent advances in the field of anti-cancer immunotherapy. BBA Clin. 2015, 3, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular aspects and future perspectives of cytokine-based anti-cancer immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef]
- Krekorian, M.; Fruhwirth, G.O.; Srinivas, M.; Figdor, C.G.; Heskamp, S.; Witney, T.H.; Aarntzen, E.H. Imaging of T-cells and their responses during anti-cancer immunotherapy. Theranostics 2019, 9, 7924. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. Anti-cancer immunotherapy: Breakthroughs and future strategies. Semin. Immunopathol. 2019, 41, 1–3. [Google Scholar] [CrossRef]
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.-d.; He, X.-b.; Sun, Q.; Chen, S.; Wan, K.; Xu, X.; Feng, X.; Li, P.-p.; Chen, B.; Xiong, M.-m. The Oncolytic Virus in Cancer Diagnosis and Treatment. Front. Oncol. 2020, 10, 1786. [Google Scholar] [CrossRef]
- Ji, W.; Li, L.; Zhou, S.; Qiu, L.; Qian, Z.; Zhang, H.; Zhao, P. Combination immunotherapy of oncolytic virus nanovesicles and PD-1 blockade effectively enhances therapeutic effects and boosts antitumour immune response. J. Drug Target. 2020, 28, 982–990. [Google Scholar] [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Pasin, F.; Calveri, M.M.; Pizzarelli, G.; Calabrese, A.; Andreoli, M.; Bongiovanni, I.; Cattaneo, C.; Rignanese, G. Oncolytic effect of SARS-CoV2 in a patient with NK lymphoma. Acta Bio Med. Atenei Parm. 2020, 91, e2020047. [Google Scholar]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Duijf, P.H.G.; Flórez de Sessions, P. Low Baseline Pulmonary Levels of Cytotoxic Lymphocytes as a Predisposing Risk Factor for Severe COVID-19. mSystems 2020, 5, e00741-20. [Google Scholar] [CrossRef]
- Challenor, S.; Tucker, D. SARS-CoV-2-induced remission of Hodgkin lymphoma. Br. J. Haematol. 2021, 192, 415. [Google Scholar] [CrossRef]
- Gonzalez Melo, M.; Remacle, N.; Cudré-Cung, H.P.; Roux, C.; Poms, M.; Cudalbu, C.; Barroso, M.; Gersting, S.W.; Feichtinger, R.G.; Mayr, J.A.; et al. The first knock-in rat model for glutaric aciduria type I allows further insights into pathophysiology in brain and periphery. Mol. Genet. Metab. 2021, 133, 157–181. [Google Scholar] [CrossRef]
- Gonzalez Melo, M.; Fontana, A.O.; Viertl, D.; Allenbach, G.; Prior, J.O.; Rotman, S.; Feichtinger, R.G.; Mayr, J.A.; Costanzo, M.; Caterino, M.; et al. A knock-in rat model unravels acute and chronic renal toxicity in glutaric aciduria type I. Mol. Genet. Metab. 2021, 134, 287–300. [Google Scholar] [CrossRef]
- Liu, N.; Shi, F.; Yang, L.; Liao, W.; Cao, Y. Oncogenic viral infection and amino acid metabolism in cancer progression: Molecular insights and clinical implications. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2022, 1877, 188724. [Google Scholar] [CrossRef]
- Roviello, G.N.; Roviello, G.; Musumeci, D.; Capasso, D.; Di Gaetano, S.; Costanzo, M.; Pedone, C. Synthesis and supramolecular assembly of 1,3-bis(1’-uracilyl)-2-propanone. RSC Adv. 2014, 4, 28691–28698. [Google Scholar] [CrossRef]
- Costanzo, M.; Caterino, M.; Ruoppolo, M. Targeted metabolomics. In Metabolomics Perspectives; Troisi, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 219–236. [Google Scholar]
- Di Minno, A.; Gelzo, M.; Caterino, M.; Costanzo, M.; Ruoppolo, M.; Castaldo, G. Challenges in Metabolomics-Based Tests, Biomarkers Revealed by Metabolomic Analysis, and the Promise of the Application of Metabolomics in Precision Medicine. Int. J. Mol. Sci. 2022, 23, 5213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Sabatello, M.; Eyal, G.; Lee, S.S.-J.; Rowe, J.W.; Stiles, D.F.; Swanson, A.; Appelbaum, P.S. Is precision medicine relevant in the age of COVID-19? Genet. Med. 2021, 23, 999–1000. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Caterino, M.; Sotgiu, G.; Ruoppolo, M.; Franconi, F.; Campesi, I. Sex differences in the human metabolome. Biol. Sex Differ. 2022, 13, 30. [Google Scholar] [CrossRef]
- Tao, S.-L.; Wang, X.-M.; Feng, Y.-G.; Kang, P.-M.; Li, Q.-Y.; Sun, T.-Y.; Tan, Q.-Y.; Deng, B. Is the presence of lung injury in COVID-19 an independent risk factor for secondary lung cancer? Med. Hypotheses 2020, 143, 110074. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Virus | Cancer | Main Geographic Area |
|---|---|---|
| Human papillomavirus (HPV) |
| Asia Central America South America Sub-Saharan Africa |
| Hepatitis B virus (HBV) |
| Asia South America Sub-Saharan Africa |
| Hepatitis C virus (HCV) |
| America Asia North Africa South Europe |
| Epstein–Barr virus (EBV) |
| America East Asia East Africa |
| Human T-cell leukemia virus 1 (HTLV-1) |
| Africa Australia Japan Middle East South America |
| Kaposi sarcoma-associated herpesvirus (KSHV/HHV-8) |
| Europe Sub-Saharan Africa |
| Merkel cell polyomavirus (MCPyV) |
| Australia Europe North America |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. Deciphering the Relationship between SARS-CoV-2 and Cancer. Int. J. Mol. Sci. 2023, 24, 7803. https://doi.org/10.3390/ijms24097803
Costanzo M, De Giglio MAR, Roviello GN. Deciphering the Relationship between SARS-CoV-2 and Cancer. International Journal of Molecular Sciences. 2023; 24(9):7803. https://doi.org/10.3390/ijms24097803
Chicago/Turabian StyleCostanzo, Michele, Maria Anna Rachele De Giglio, and Giovanni Nicola Roviello. 2023. "Deciphering the Relationship between SARS-CoV-2 and Cancer" International Journal of Molecular Sciences 24, no. 9: 7803. https://doi.org/10.3390/ijms24097803
APA StyleCostanzo, M., De Giglio, M. A. R., & Roviello, G. N. (2023). Deciphering the Relationship between SARS-CoV-2 and Cancer. International Journal of Molecular Sciences, 24(9), 7803. https://doi.org/10.3390/ijms24097803