
The Role of NAD+, SIRTs Interactions in Stimulating and Counteracting Carcinogenesis

Abstract
:1. Introduction
2. Role of NAD+ in Cancer Metabolism
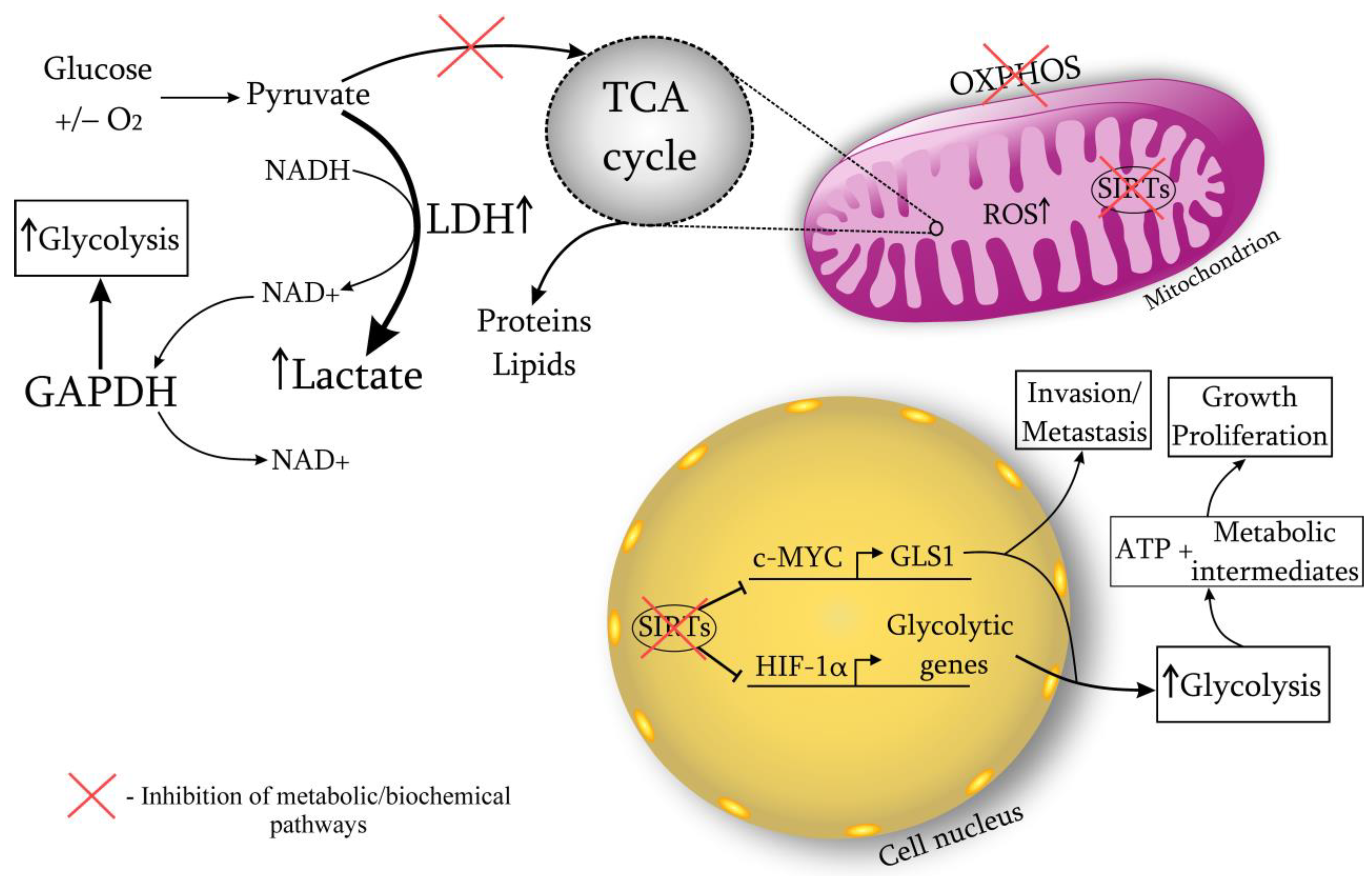
2.1. Warburg Effect
2.2. NAD+ Metabolism
2.3. Pro-Oncogenic Effects of NAD+
2.4. NAMPT Inhibitors

{kind=link}
{kind=link}
{kind=link}
| NAMPT Inhibitors | |||||
|---|---|---|---|---|---|
| Specific | Dual | ||||
| Name | References | Target Molecules | Name | References | Target Molecules |
| CHS-828 (GMX1778) | [55,56] | Anticancer activity in lung and breast cancer cell lines; causes NAD+ depletion | Chidamide (HBI-8000) | [67] | HDAC/NAMPT inhibitor; suppresses cyclin E1; causes NAD+ depletion and apoptosis |
| FK866 (APO866/WK175) | [57,58,59,60] | Anticancer activity in the treatment of a wide range of solid and hematological cancers; causes NAD+ depletion and apoptosis | STF-31 | [67] | NAMPT and GLUT1 inhibitor; impairs glucose uptake, lowers NAD+ levels and causes apoptosis |
| GMX1777 (EB1627) | [64] | Improved version of CHS-828 | KPT-9274 (ATG-019) | [66] | NADPH and PAK4 inhibitor; reduces viability, invasion/migration of cancer cells and induces apoptosis; suppresses c-MYC and cyclin D1; causes depletion of NAD+ and SIRT1 |
| OT-82 | [65] | Strong anticancer activity against hematopoietic malignancies; causes depletion of NAD+, ATP and apoptosis | |||
3. SIRTs/PARPs as Pro- and Anti-Oncogenic Targets
3.1. Sirtuins
3.1.1. SIRT1
3.1.2. SIRT2
3.1.3. SIRT3
3.1.4. SIRT4
3.1.5. SIRT5
3.1.6. SIRT6
3.1.7. SIRT7
3.2. PARPs
4. Modulation of the Ratio of NAD+, SIRTs and PARPs as a Pharmacological Effect on Cancer
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Moothart, D.R.; Lowy, D.R.; Qian, X. The Expression of Glyceraldehyde-3-Phosphate Dehydrogenase Associated Cell Cycle (GACC) Genes Correlates with Cancer Stage and Poor Survival in Patients with Solid Tumors. PLoS ONE 2013, 8, e61262. [Google Scholar] [CrossRef] [PubMed]
- Mehrmohamadi, M.; Liu, X.; Shestov, A.A.; Locasale, J.W. Characterization of the Usage of the Serine Metabolic Network in Human Cancer. Cell Rep. 2014, 9, 1507–1519. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Turner, C.E. Cytoplasm-Localized SIRT1 Enhances Apoptosis. J. Cell. Physiol. 2007, 211, 88–97. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1: Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Zu, Y.; Liu, L.; Lee, M.Y.K.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. SIRT1 is a class III histone deacetylase within the sirtuin family of related proteins that is uniquely dependent on NAD. Nat. Rev. Cancer 2009, 9, 123–128. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. Interference between PARPs and SIRT1: A novel approach to healthy ageing? Aging 2011, 3, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Von Heideman, A.; Berglund, Å.; Larsson, R.; Nygren, P.; Larsson, R. Safety and efficacy of NAD depleting cancer drugs: Results of a phase i clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef]
- Kanno, T.; Sudo, K.; Maekawa, M.; Nishimura, Y.; Ukita, M.; Fukutake, K. Lactate dehydrogenase M-subunit deficiency: A new type of hereditary exertional myopathy. Clin. Chim. Acta 1988, 173, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 287. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; Ralph, J.; Invest, J.C.; Hensley, C.T.; Wasti, A.T.; Deberardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities Find the latest version: Review series Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, V.A.; Belyaeva, L.E. Cancer cell metabolism as a therapeutic target. Vestn. Vitebsk State Med. Univ. 2016, 15, 7–20. [Google Scholar] [CrossRef]
- Knight, J.R.P.; Milner, J. SIRT1, metabolism and cancer. Curr. Opin. Oncol. 2012, 24, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhang, T.; Chang, C.Y.; Wang, J.; Bazile, L.; Zhang, L.; Haffty, B.G.; Hu, W.; Feng, Z. Metabolic enzyme LDHA activates Rac1 GTPase as a noncanonical mechanism to promote cancer. Nat. Metab. 2022, 4, 1830–1846. [Google Scholar] [CrossRef] [PubMed]
- Keshari, K.R.; Wilson, D.M.; Van Criekinge, M.; Sriram, R.; Koelsch, B.L.; Wang, Z.J.; Vanbrocklin, H.F.; Peehl, D.M.; O’Brien, T.; Sampath, D.; et al. Metabolic response of prostate cancer to nicotinamide phophoribosyltransferase inhibition in a hyperpolarized MR/PET compatible bioreactor. Prostate 2015, 75, 1601–1609. [Google Scholar] [CrossRef]
- Phadke, M.; Krynetskaia, N.; Mishra, A.; Krynetskiy, E. Accelerated cellular senescence phenotype of GAPDH-depleted human lung carcinoma cells. Biochem. Biophys. Res. Commun. 2011, 411, 409–415. [Google Scholar] [CrossRef]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, G.J. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009, 8, 100–112. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven glycolysis is a therapeutic target in glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- Evertts, A.G.; Zee, B.M.; Dimaggio, P.A.; Gonzales-Cope, M.; Coller, H.A.; Garcia, B.A. Quantitative dynamics of the link between cellular metabolism and histone acetylation. J. Biol. Chem. 2013, 288, 12142–12151. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Little, J.B.; Yuan, Z.M. Glycolytic metabolism influences global chromatin structure. Oncotarget 2015, 6, 4214–4225. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Huang, H.; Dai, L.; Liu, X.; Zhao, Y.; Locasale, J.W. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 2015, 3, 10. [Google Scholar] [CrossRef]
- Podyacheva, E.; Toropova, Y. Nicotinamide Riboside for the Prevention and Treatment of Doxorubicin Cardiomyopathy. Opportunities and Prospects. Nutrients 2021, 13, 3435. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef]
- Wei, Y.; Xiang, H.; Zhang, W. Review of various NAMPT inhibitors for the treatment of cancer. Front. Pharmacol. 2022, 13, 970553. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef]
- Carbone, F.; Liberale, L.; Bonaventura, A.; Vecchié, A.; Casula, M.; Cea, M.; Monacelli, F.; Caffa, I.; Bruzzone, S.; Montecucco, F.; et al. Regulation and function of extracellular nicotinamide phosphoribosyltransferase/visfatin. Compr. Physiol. 2017, 7, 603–621. [Google Scholar] [CrossRef]
- Managò, A.; Audrito, V.; Mazzola, F.; Sorci, L.; Gaudino, F.; Gizzi, K.; Vitale, N.; Incarnato, D.; Minazzato, G.; Ianniello, A.; et al. Extracellular nicotinate phosphoribosyltransferase binds Toll like receptor 4 and mediates inflammation. Nat. Commun. 2019, 10, 4116. [Google Scholar] [CrossRef]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Lüscher, B.; Larsson, L.G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Lydia, B.; Yina, Z.; Nina, K.; Andrea, S.; Boos, S.L.; Peter, J.; Florian, G.; Roland, R.; Menssen, A. Targeting c-MYC through Interference with NAMPT and SIRT1 and Their Association to Oncogenic Drivers in Murine Serrated Intestinal Tumorigenesis. Neoplasia 2019, 21, 974–988. [Google Scholar] [CrossRef]
- Brandl, L.; Kirstein, N.; Neumann, J.; Sendelhofert, A.; Vieth, M.; Kirchner, T.; Menssen, A. The c-MYC/NAMPT/SIRT1 feedback loop is activated in early classical and serrated route colorectal cancer and represents a therapeutic target. Med. Oncol. 2019, 36, 5. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD + metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.; Park, J.W.; Kim, J.G.; Lee, B.J. FOXO1 functions in the regulation of nicotinamide phosphoribosyltransferase (Nampt) expression. Biochem. Biophys. Res. Commun. 2019, 511, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Pour, Z.B.; Nourbakhsh, M.; Mousavizadeh, K.; Madjd, Z.; Ghorbanhosseini, S.S.; Abdolvahabi, Z.; Hesari, Z.; Mobaser, S.E. Up-regulation of MIR-381 inhibits NAD+ salvage pathway and promotes apoptosis in breast cancer cells. EXCLI J. 2019, 18, 683–696. [Google Scholar] [CrossRef]
- Bolandghamat Pour, Z.; Nourbakhsh, M.; Mousavizadeh, K.; Madjd, Z.; Ghorbanhosseini, S.S.; Abdolvahabi, Z.; Hesari, Z.; Ezzati Mobasser, S. Suppression of nicotinamide phosphoribosyltransferase expression by miR-154 reduces the viability of breast cancer cells and increases their susceptibility to doxorubicin. BMC Cancer 2019, 19, 1027. [Google Scholar] [CrossRef]
- Ghorbanhosseini, S.S.; Nourbakhsh, M.; Zangooei, M.; Abdolvahabi, Z.; Bolandghamtpour, Z.; Hesari, Z.; Yousefi, Z.; Panahi, G.; Meshkani, R. MicroRNA-494 induces breast cancer cell apoptosis and reduces cell viability by inhibition of nicotinamide phosphoribosyltransferase expression and activity. EXCLI J. 2019, 18, 838–851. [Google Scholar] [CrossRef]
- Ju, H.Q.; Zhuang, Z.N.; Li, H.; Tian, T.; Lu, Y.X.; Fan, X.Q.; Zhou, H.J.; Mo, H.Y.; Sheng, H.; Chiao, P.J.; et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016, 379, 1–11. [Google Scholar] [CrossRef]
- Lv, R.; Yu, J.; Sun, Q. Anti-angiogenic role of microRNA-23b in melanoma by disturbing NF-κB signaling pathway targeted inhibition of NAMPT. Futur. Oncol. 2020, 16, 541–558. [Google Scholar] [CrossRef]
- Zhang, C.; Tong, J.; Huang, G. Nicotinamide Phosphoribosyl Transferase (Nampt) Is a Target of MicroRNA-26b in Colorectal Cancer Cells. PLoS ONE 2013, 8, e69963. [Google Scholar] [CrossRef] [PubMed]
- Sociali, G.; Grozio, A.; Caffa, I.; Schuster, S.; Becherini, P.; Damonte, P.; Sturla, L.; Fresia, C.; Passalacqua, M.; Mazzola, F.; et al. SIRT6 deacetylase activity regulates NAMPT activity and NAD(P)(H) pools in cancer cells. FASEB J. 2019, 33, 3704–3717. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, L.J.Y.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2017, 2, e90019. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Pereira, S.; Pereira-Castro, I.; Silva, S.S.; Correia, M.G.; Neto, C.; da Costa, L.T.; Amorim, A.; Silva, R.M. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget 2016, 7, 1973–1983. [Google Scholar] [CrossRef]
- Olesen, U.H.; Christensen, M.K.; Björkling, F.; Jäättelä, M.; Jensen, P.B.; Sehested, M.; Nielsen, S.J. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 2008, 367, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Hjarnaa, P.J.; Jonsson, E.; Latini, S.; Dhar, S.; Larsson, R.; Bramm, E.; Skov, T.; Binderup, L. CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res 1999, 59, 5751–5757. [Google Scholar]
- Khan, J.A.; Tao, X.; Tong, L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Liu, L.Y.; Qie, L.L.; Ling, K.N.; Xu, L.H.; Wang, F.; Fang, S.H.; Lu, Y.B.; Hu, H.; Wei, E.Q.; et al. Anti-proliferation effect of APO866 on C6 glioblastoma cells by inhibiting nicotinamide phosphoribosyltransferase. Eur. J. Pharmacol. 2012, 674, 163–170. [Google Scholar] [CrossRef]
- Olesen, U.H.; Thougaard, A.V.; Jensen, P.B.; Sehested, M. A preclinical study on the rescue of normal tissue by nicotinic acid in high-dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol. Cancer Ther. 2010, 9, 1609–1617. [Google Scholar] [CrossRef]
- Wosikowski, K.; Mattern, K.; Schemainda, I.; Hasmann, M.; Rattel, B.; Löser, R. WK175, a novel antitumor agent, decreases the intracellular nicotinamide adenine dinucleotide concentration and induces the apoptotic cascade in human leukemia cells. Cancer Res. 2002, 15, 1057–1062. [Google Scholar]
- Van Horssen, R.; Willemse, M.; Haeger, A.; Attanasio, F.; Güneri, T.; Schwab, A.; Stock, C.M.; Buccione, R.; Fransen, J.A.M.; Wieringa, B. Intracellular NAD(H) levels control motility and invasion of glioma cells. Cell. Mol. Life Sci. 2013, 70, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef]
- Sociali, G.; Raffaghello, L.; Magnone, M.; Zamporlini, F.; Emionite, L.; Sturla, L.; Bianchi, G.; Vigliarolo, T.; Nahimana, A.; Nencioni, A.; et al. Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 2016, 7, 2968–2984. [Google Scholar] [CrossRef] [PubMed]
- Binderup, E.; Björkling, F.; Hjarnaa, P.V.; Latini, S.; Baltzer, B.; Carlsen, M.; Binderup, L. EB1627: A soluble prodrug of the potent anticancer cyanoguanidine CHS828. Bioorganic Med. Chem. Lett. 2005, 15, 2491–2494. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.; Kazyulkin, D.; Komarov, P.G.; Polinsky, A.; Andrianova, E.L.; Joshi, S.; Gupta, M.; Vujcic, S.; Kononov, E.; Toshkov, I.; et al. OT-82, a novel anticancer drug candidate that targets the strong dependence of hematological malignancies on NAD biosynthesis. Leukemia 2020, 34, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Aboud, O.A.; Chen, C.H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and specific inhibition of NAMPT and PAK4 by KPT-9274 decreases kidney cancer growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef]
- Gong, K.; Xie, J.; Yi, H.; Li, W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G 1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem. J. 2012, 443, 735–746. [Google Scholar] [CrossRef]
- Breton, C.S.; Aubry, D.; Ginet, V.; Puyal, J.; Heulot, M.; Widmann, C.; Duchosal, M.A.; Nahimana, A. Combinative effects of β-Lapachone and APO866 on pancreatic cancer cell death through reactive oxygen species production and PARP-1 activation. Biochimie 2015, 116, 141–153. [Google Scholar] [CrossRef]
- Cagnetta, A.; Caffa, I.; Acharya, C.; Soncini, D.; Acharya, P.; Adamia, S.; Pierri, I.; Bergamaschi, M.; Garuti, A.; Fraternali, G.; et al. APO866 increases antitumor activity of cyclosporin—A by inducing mitochondrial and endoplasmic reticulum stress in leukemia cells. Clin. Cancer Res. 2015, 21, 3934–3945. [Google Scholar] [CrossRef]
- Kennedy, B.E.; Sharif, T.; Martell, E.; Dai, C.; Kim, Y.; Lee, P.W.K.; Gujar, S.A. NAD+ salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 2016, 114, 274–283. [Google Scholar] [CrossRef]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates Reactive Oxygen Species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, I.; Bouchard, E.D.J.; Beiggi, S.; Poeppl, A.G.; Johnston, J.B.; Gibson, S.B.; Banerji, V. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2014, 20, 4861–4872. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Park, C.W.; Kim, S.W.; Nam, Y.J.; Yu, J.H.; Shin, J.H.; Yun, C.H.; Im, S.H.; Kim, K.T.; Sung, Y.C.; et al. NAMPT suppresses glucose deprivation-induced oxidative stress by increasing NADPH levels in breast cancer. Oncogene 2016, 35, 3544–3554. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Bułdak, R.J.; Bułdak, Ł.; Polaniak, R.; Kukla, M.; Birkner, E.; Kubina, R.; Kabała-Dzik, A.; Duława-Bułdak, A.; Zwirska-Korczala, K. Visfatin affects redox adaptative responses and proliferation in Me45 human malignant melanoma cells: An in vitro study. Oncol. Rep. 2013, 29, 771–778. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; Huang, R.; Fang, L.; Yu, S. SIRT4 and SIRT6 Serve as Novel Prognostic Biomarkers With Competitive Functions in Serous Ovarian Cancer. Front. Genet. 2021, 12, 666630. [Google Scholar] [CrossRef]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef]
- Costa-Machado, L.F.; Fernandez-Marcos, P.J. The sirtuin family in cancer. Cell Cycle 2019, 18, 2164–2196. [Google Scholar] [CrossRef]
- Mennella, M.R.F. A new facet of ADP-ribosylation reactions: SIRTs and PARPs interplay. Front. Biosci. Landmark 2015, 20, 458–473. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2α promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Ford, J.; Jiang, M.; Milner, J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005, 65, 10457–10463. [Google Scholar] [CrossRef]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef]
- Wen, Y.C.; Wang, D.H.; RayWhay, C.Y.; Luo, J.; Gu, W.; Baylin, S.B. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Pullmann, R.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 Regulates SIRT1 Expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef]
- Yamakuchi, M. MicroRNA regulation of SIRT1. Front. Physiol. 2012, 3, 68. [Google Scholar] [CrossRef]
- Saunders, L.R.; Verdin, E. Sirtuins: Critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar] [CrossRef]
- Olmos, Y.; Brosens, J.J.; Lam, E.W.F. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist. Updat. 2011, 14, 35–44. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsy, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Wang, R.H.; Zheng, Y.; Kim, H.S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef]
- Zheng, Z.; Bian, Y.; Zhang, Y.; Ren, G.; Li, G. Metformin activates AMPK/SIRT1/NF-κB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle 2020, 19, 1089–1104. [Google Scholar] [CrossRef]
- Yang, X.; Potts, P.R. CSAG2 is a cancer-specific activator of SIRT1. EMBO Rep. 2020, 21, e50912. [Google Scholar] [CrossRef]
- Leng, S.; Huang, W.; Chen, Y.; Yang, Y.; Feng, D.; Liu, W.; Gao, T.; Ren, Y.; Huo, M.; Zhang, J.; et al. SIRT1 coordinates with the CRL4B complex to regulate pancreatic cancer stem cells to promote tumorigenesis. Cell Death Differ. 2021, 28, 3329–3343. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wei, Y.; Liu, Y.; Lu, X.; Ding, F.; Wang, J.; Yang, S. Resveratrol promotes sensitization to Doxorubicin by inhibiting epithelial-mesenchymal transition and modulating SIRT1/β-catenin signaling pathway in breast cancer. Cancer Med. 2019, 8, 1246–1257. [Google Scholar] [CrossRef]
- Yousafzai, N.A.; Jin, H.; Ullah, M.; Wang, X. Recent advances of SIRT1 and implications in chemotherapeutics resistance in cancer. Am. J. Cancer Res. 2021, 11, 5233–5248. [Google Scholar]
- Zhang, S.; Yang, Y.; Huang, S.; Deng, C.; Zhou, S.; Yang, J.; Cao, Y.; Xu, L.; Yuan, Y.; Yang, J.; et al. SIRT1 inhibits gastric cancer proliferation and metastasis via STAT3/MMP-13 signaling. J. Cell. Physiol. 2019, 234, 15395–15406. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Ong, A.L.C.; Ramasamy, T.S. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res. Rev. 2018, 43, 64–80. [Google Scholar] [CrossRef]
- Li, K.; Luo, J. The role of SIRT1 in tumorigenesis. N. Am. J. Med. Sci. 2011, 4, 104–106. [Google Scholar] [CrossRef]
- Parsamanesh, N.; Asghari, A.; Sardari, S.; Tasbandi, A.; Jamialahmadi, T.; Xu, S.; Sahebkar, A. Resveratrol and endothelial function: A literature review. Pharmacol. Res. 2021, 170, 105725. [Google Scholar] [CrossRef]
- Cao, Y.W.; Li, W.Q.; Wan, G.X.; Li, Y.X.; Du, X.M.; Li, Y.C.; Li, F. Correlation and prognostic value of SIRT1 and Notch1 signaling in breast cancer. J. Exp. Clin. Cancer Res. 2014, 33, 97. [Google Scholar] [CrossRef] [PubMed]
- Perrini, S.; Porro, S.; Nigro, P.; Cignarelli, A.; Caccioppoli, C.; Genchi, V.A.; Martines, G.; De Fazio, M.; Capuano, P.; Natalicchio, A.; et al. Reduced SIRT1 and SIRT2 expression promotes adipogenesis of human visceral adipose stem cells and associates with accumulation of visceral fat in human obesity. Int. J. Obes. 2020, 44, 307–319. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Zhou, X.; Yan, Y.; Tang, J.; Tolbert, E.; Zhao, T.C.; Gong, R.; Zhuang, S. Blocking sirtuin 1 and 2 inhibits renal interstitial fibroblast activation and attenuates renal interstitial fibrosis in obstructive nephropathy. J. Pharmacol. Exp. Ther. 2014, 350, 243–256. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, Z.; Tang, D.; Zhou, Q.; Li, Y.; Zhou, L.; Yin, Y.; Wang, Y.; Pan, Y.; Dorfman, R.G.; et al. Downregulation of SIRT2 Inhibits Invasion of Hepatocellular Carcinoma by Inhibiting Energy Metabolism. Transl. Oncol. 2017, 10, 917–927. [Google Scholar] [CrossRef]
- Zhao, D.; Zou, S.W.; Liu, Y.; Zhou, X.; Mo, Y.; Wang, P.; Xu, Y.H.; Dong, B.; Xiong, Y.; Lei, Q.Y.; et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase a and is decreased in pancreatic cancer. Cancer Cell 2013, 23, 464–476. [Google Scholar] [CrossRef]
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218. [Google Scholar] [CrossRef]
- Chen, G.; Huang, P.; Hu, C. The role of SIRT2 in cancer: A novel therapeutic target. Int. J. Cancer 2020, 147, 3297–3304. [Google Scholar] [CrossRef]
- Sayd, S.; Thirant, C.; El-Habr, E.A.; Lipecka, J.; Dubois, L.G.; Bogeas, A.; Tahiri-Jouti, N.; Chneiweiss, H.; Junier, M.P. Sirtuin-2 Activity is Required for Glioma Stem Cell Proliferation Arrest but not Necrosis Induced by Resveratrol. Stem Cell Rev. Rep. 2014, 10, 103–113. [Google Scholar] [CrossRef]
- Li, Y.; Dai, D.; Lu, Q.; Fei, M.; Li, M.; Wu, X. Sirt2 suppresses glioma cell growth through targeting NF-κB-miR-21 axis. Biochem. Biophys. Res. Commun. 2013, 441, 661–667. [Google Scholar] [CrossRef]
- Bajpe, P.K.; Prahallad, A.; Horlings, H.; Nagtegaal, I.; Beijersbergen, R.; Bernards, R. A chromatin modifier genetic screen identifies SIRT2 as a modulator of response to targeted therapies through the regulation of MEK kinase activity. Oncogene 2015, 34, 531–536. [Google Scholar] [CrossRef]
- Wang, B.; Ye, Y.; Yang, X.; Liu, B.; Wang, Z.; Chen, S.; Jiang, K.; Zhang, W.; Jiang, H.; Mustonen, H.; et al. SIRT 2-dependent IDH 1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020, 21, e48183. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, Y.; Kim, J.T.; Sengoku, T.; Alstott, M.C.; Weiss, H.L.; Wang, Q.; Evers, B.M. Regulation of SIRT2 by Wnt/β-catenin signaling pathway in colorectal cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118966. [Google Scholar] [CrossRef]
- Wang, J.; Wu, J.; Wang, L.; Min, X.; Chen, Z. The LINC00152/miR-138 Axis Facilitates Gastric Cancer Progression by Mediating SIRT2. J. Oncol. 2021, 2021, 1173869. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Z.; Jiang, H.; Shi, F. The sirtuin 3 expression profile is associated with pathological and clinical outcomes in colon cancer patients. Biomed Res. Int. 2014, 2014, 871263. [Google Scholar] [CrossRef]
- Cui, Y.; Qin, L.; Wu, J.; Qu, X.; Hou, C.; Sun, W.; Li, S.; Vaughan, A.T.M.; Li, J.J.; Liu, J. SIRT3 enhances glycolysis and proliferation in SIRT3-expressing gastric cancer cells. PLoS ONE 2015, 10, e0129834. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, H.; Wang, X.; Zhang, R.; Wang, C.; Guo, Z. Sirtuin-3 (SIRT3) expression is associated with overall survival in esophageal cancer. Ann. Diagn. Pathol. 2013, 17, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Pons, D.G.; Sastre-Serra, J.; Oliver, J.; Roca, P. SIRT3 Silencing Sensitizes Breast Cancer Cells to Cytotoxic Treatments Through an Increment in ROS Production. J. Cell. Biochem. 2017, 118, 397–406. [Google Scholar] [CrossRef]
- Alhazzazi, T.Y.; Kamarajan, P.; Joo, N.; Huang, J.Y.; Verdin, E.; D’Silva, N.J.; Kapila, Y.L. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 2011, 117, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Nihal, M.; Singh, C.K.; Zhong, W.; Liu, X.; Ahmad, N. Pro-Proliferative Function of Mitochondrial Sirtuin Deacetylase SIRT3 in Human Melanoma. J. Investig. Dermatol. 2016, 136, 809–818. [Google Scholar] [CrossRef]
- Choi, J.; Koh, E.; Lee, Y.S.; Lee, H.W.; Kang, H.G.; Yoon, Y.E.; Han, W.K.; Choi, K.H.; Kim, K.S. Mitochondrial Sirt3 supports cell proliferation by regulating glutamine-dependent oxidation in renal cell carcinoma. Biochem. Biophys. Res. Commun. 2016, 474, 547–553. [Google Scholar] [CrossRef]
- Shackelford, R.; Hirsh, S.; Henry, K.; Abdel-Mageed, A.; Kandil, E.; Coppola, D. Nicotinamide phosphoribosyltransferase and SIRT3 expression are increased in well-differentiated thyroid carcinomas. Anticancer Res. 2013, 33, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Desouki, M.M.; Doubinskaia, I.; Gius, D.; Abdulkadir, S.A. Decreased mitochondrial SIRT3 expression is a potential molecular biomarker associated with poor outcome in breast cancer. Hum. Pathol. 2014, 45, 1071–1077. [Google Scholar] [CrossRef]
- Bai, Z.; Wang, Z. Genistein protects against doxorubicin-induced cardiotoxicity through Nrf-2/HO-1 signaling in mice model. Environ. Toxicol. 2019, 34, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-X.; Yi, Y.; Li, Y.-W.; Cai, X.-Y.; He, H.-W.; Ni, X.-C.; Zhou, J.; Cheng, Y.-F.; Jin, J.-J.; Fan, J.; et al. Down-regulation of sirtuin 3 is associated with poor prognosis in hepatocellular carcinoma after resection. BMC Cancer 2014, 14, 1331–1336. [Google Scholar] [CrossRef]
- Tao, N.N.; Zhou, H.Z.; Tang, H.; Cai, X.F.; Zhang, W.L.; Ren, J.H.; Zhou, L.; Chen, X.; Chen, K.; Li, W.Y.; et al. Sirtuin 3 enhanced drug sensitivity of human hepatoma cells through glutathione S-transferase pi 1/JNK signaling pathway. Oncotarget 2016, 7, 50117–50130. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, L.M.; McCluney, S.; Jamieson, N.B.; Thomson, J.; MacDonald, A.I.; Oien, K.; Dickson, E.J.; Carter, C.R.; McKay, C.J.; Shiels, P.G. SIRT3 & SIRT7: Potential novel biomarkers for determining outcome in pancreatic cancer patients. PLoS ONE 2015, 10, e0131344. [Google Scholar] [CrossRef]
- Temel, M.; Koç, M.N.; Ulutaş, S.; Göğebakan, B. The expression levels of the sirtuins in patients with BCC. Tumor Biol. 2016, 37, 6429–6435. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-C.; Jing, L.-M.; Wang, W.-X.; Gao, Y.-X. Down-regulation of SIRT3 promotes ovarian carcinoma metastasis. Biochem. Biophys. Res. Commun. 2016, 475, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhou, Y.; Qiao, C.; Ni, T.; Li, Z.; You, Q.; Guo, Q.; Lu, N. Oroxylin A inhibits glycolysis-dependent proliferation of human breast cancer via promoting SIRT3-mediated SOD2 transcription and HIF1α destabilization. Cell Death Dis. 2015, 6, e1714. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, M.; Zhao, J.; Han, Y.; Jia, L. Sirtuin 3: A Janus face in cancer (Review). Int. J. Oncol. 2016, 49, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.; Guan, K.; et al. SIRT 3-dependent GOT 2 acetylation status affects the malate–aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Shan, C.; Kang, H.B.; Elf, S.; Xie, J.; Tucker, M.; Gu, T.L.; Aguiar, M.; Lonning, S.; Chen, H.; et al. Tyr Phosphorylation of PDP1 Toggles Recruitment between ACAT1 and SIRT3 to Regulate the Pyruvate Dehydrogenase Complex. Mol. Cell 2014, 53, 534–548. [Google Scholar] [CrossRef]
- Torrens-Mas, M.; Oliver, J.; Roca, P.; Sastre-Serra, J. SIRT3: Oncogene and tumor suppressor in cancer. Cancers 2017, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, A.; Chen, Q. SirT3 and p53 Deacetylation in Aging and Cancer. J. Cell. Physiol. 2017, 232, 2308–2311. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef]
- Quan, Y.; Wang, N.; Chen, Q.; Xu, J.; Cheng, W.; Di, M.; Xia, W.; Gao, W.Q. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 2015, 6, 26494–26507. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhou, L.M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef]
- Wang, Z.; Inuzuka, H.; Zhong, J.; Liu, P.; Sarkar, F.H.; Sun, Y.; Wei, W. Identification of acetylation-dependent regulatory mechanisms that govern the oncogenic functions of Skp2. Oncotarget 2012, 3, 1294–1300. [Google Scholar] [CrossRef]
- Li, S.D.; Banck, M.; Mujtaba, S.; Zhou, M.M.; Sugrue, M.M.; Walsh, M.J. p53-Induced growth arrest is regulated by the Mitochondrial SirT3 deacetylase. PLoS ONE 2010, 5, e10486. [Google Scholar] [CrossRef]
- Dessai, A.S.; Dominguez, M.P.; Chen, U.I.; Hasper, J.; Prechtl, C.; Yu, C.; Katsuta, E.; Dai, T.; Zhu, B.; Jung, S.Y.; et al. Transcriptional Repression of SIRT3 Potentiates Mitochondrial Aconitase Activation to Drive Aggressive Prostate Cancer to the Bone. Cancer Res. 2021, 81, 50–63. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Cao, D.; Qiu, S.; Li, Y.; Jiang, C.; Bian, R.; Yang, Y.; Li, L.; Li, X.; et al. SIRT3 inhibits gallbladder cancer by induction of AKT-dependent ferroptosis and blockade of epithelial-mesenchymal transition. Cancer Lett. 2021, 510, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hao, B.; Li, D.; Reiter, R.J.; Bai, Y.; Abay, B.; Chen, G.; Lin, S.; Zheng, T.; Ren, Y.; et al. Melatonin inhibits lung cancer development by reversing the Warburg effect via stimulating the SIRT3/PDH axis. J. Pineal Res. 2021, 71, e12755. [Google Scholar] [CrossRef]
- Zhang, J.; Han, L.; Yu, J.; Li, H.; Li, Q. miR-224 aggravates cancer-associated fibroblast-induced progression of non-small cell lung cancer by modulating a positive loop of the SIRT3/AMPK/mTOR/HIF-1α axis. Aging 2021, 13, 10431–10449. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, G.; Du, J.; Li, J.; Lin, G.; Zhang, J. LncRNA FENDRR Inhibits Gastric Cancer Cell Proliferation and Invasion via the miR-421/SIRT3/ Notch-1 Axis. Cancer Manag. Res. 2021, 13, 9175–9187. [Google Scholar] [CrossRef]
- Chen, Z.; Lin, J.; Feng, S.; Chen, X.; Huang, H.; Wang, C.; Yu, Y.; He, Y.; Han, S.; Zheng, L.; et al. SIRT4 inhibits the proliferation, migration, and invasion abilities of thyroid cancer cells by inhibiting glutamine metabolism. Onco. Targets. Ther. 2019, 12, 2397–2408. [Google Scholar] [CrossRef]
- Jeong, S.M.; Lee, A.; Lee, J.; Haigis, M.C. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J. Biol. Chem. 2014, 289, 4135–4144. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, T.; Zhang, X.; Geng, J.; He, X.; Nu, M.; Pang, D. Decreased sirtuin 4 expression is associated with poor prognosis in patients with invasive breast cancer. Oncol. Lett. 2016, 12, 2606–2612. [Google Scholar] [CrossRef]
- Sun, H.; Huang, D.; Liu, G.; Jian, F.; Zhu, J.; Zhang, L. SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion. Onco. Targets. Ther. 2018, 11, 3959–3968. [Google Scholar] [CrossRef]
- Miyo, M.; Yamamoto, H.; Konno, M.; Colvin, H.; Nishida, N.; Koseki, J.; Kawamoto, K.; Ogawa, H.; Hamabe, A.; Uemura, M.; et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 2015, 113, 492–499. [Google Scholar] [CrossRef]
- Hu, Y.; Lin, J.; Lin, Y.A.O.; Chen, X.; Zhu, G.; Huang, G. Overexpression of SIRT4 inhibits the proliferation of gastric cancer cells through cell cycle arrest. Oncol. Lett. 2019, 17, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Y.; Zhu, Y.; Kong, C. Functions of mammalian Sirt4 in cellular metabolism and research progress in human cancer (Review). Oncol. Lett. 2020, 20, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhu, G. Sirtuin-4 (SIRT4), a therapeutic target with oncogenic and tumor-suppressive activity in cancer. Onco. Targets. Ther. 2018, 11, 3395–3400. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Bai, Y.; Yang, J.; Yao, Y.; Zhang, C.; Liu, C.; Shi, J.; Li, Q.W.; Zhang, J.; Lu, X.; et al. SIRT4 is the molecular switch mediating cellular proliferation in colorectal cancer through GLS mediated activation of AKT/GSK3β/CyclinD1 pathway. Carcinogenesis 2021, 42, 481–492. [Google Scholar] [CrossRef]
- Jeong, S.M.; Xiao, C.; Finley, L.W.S.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to dna damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef]
- Tong, Y.; Kai, J.; Wang, S.; Yu, Y.; Xie, S.; Zheng, H.; Wang, Y.; Liu, Y.; Zhu, K.; Guan, X.; et al. VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1α/HO-1 pathway. Cell Death Dis. 2021, 12, 621. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, H.L.; Xu, J.; Tan, J.; Fu, L.N.; Wang, J.L.; Zou, T.H.; Sun, D.F.; Gao, Q.Y.; Chen, Y.X.; et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 2018, 9, 545. [Google Scholar] [CrossRef]
- Chang, L.; Xi, L.; Liu, Y.; Liu, R.; Wu, Z.; Jian, Z. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol. Med. Rep. 2018, 17, 342–349. [Google Scholar] [CrossRef]
- Igci, M.; Kalender, M.E.; Borazan, E.; Bozgeyik, I.; Bayraktar, R.; Bozgeyik, E.; Camci, C.; Arslan, A. High-throughput screening of Sirtuin family of genes in breast cancer. Gene 2016, 586, 123–128. [Google Scholar] [CrossRef]
- Chen, X.; Tian, M.; Sun, R.; Zhang, M.; Zhou, L.; Jin, L.; Chen, L.; Zhou, W.; Duan, K.; Chen, Y.; et al. SIRT 5 inhibits peroxisomal ACOX 1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef]
- Lin, Z.F.; Xu, H.B.; Wang, J.Y.; Lin, Q.; Ruan, Z.; Liu, F.B.; Jin, W.; Huang, H.H.; Chen, X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef]
- Guan, D.; Lim, J.H.; Peng, L.; Liu, Y.; Lam, M.; Seto, E.; Kao, H.Y. Deacetylation of the tumor suppressor protein PML regulates hydrogen peroxide-Induced cell death. Cell Death Dis. 2014, 5, e1340-13. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP+-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Yihan, L.; Xiaojing, W.; Ao, L.; Chuanjie, Z.; Haofei, W.; Yan, S.; Hongchao, H. SIRT5 functions as a tumor suppressor in renal cell carcinoma by reversing the Warburg effect. J. Transl. Med. 2021, 19, 521. [Google Scholar] [CrossRef]
- Choi, S.Y.; Jeon, J.M.; Na, A.Y.; Kwon, O.K.; Bang, I.H.; Ha, Y.S.; Bae, E.J.; Park, B.H.; Lee, E.H.; Kwon, T.G.; et al. SIRT5 Directly Inhibits the PI3K/AKT Pathway in Prostate Cancer Cell Lines. Cancer Genom. Proteom. 2022, 19, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zuo, Y.; Feng, Y.; Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumor Biol. 2014, 35, 10699–10705. [Google Scholar] [CrossRef]
- Lv, X.B.; Liu, L.; Cheng, C.; Yu, B.; Xiong, L.; Hu, K.; Tang, J.; Zeng, L.; Sang, Y. SUN2 exerts tumor suppressor functions by suppressing the Warburg effect in lung cancer. Sci. Rep. 2015, 5, 17940. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Mu, X.; You, Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget 2017, 8, 53819–53828. [Google Scholar] [CrossRef]
- Ye, X.; Niu, X.; Gu, L.; Xu, Y.; Li, Z.; Yu, Y.; Chen, Z.; Lu, S. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 2017, 8, 6984–6993. [Google Scholar] [CrossRef]
- Du, Z.G.; Liu, X.J.; Chen, T.; Gao, W.C.; Wu, Z.M.; Hu, Z.Q.; Wei, D.; Gao, C.F.; Li, Q.Q. Targeting a Sirt5-Positive Subpopulation Overcomes Multidrug Resistance in Wild-Type Kras Colorectal Carcinomas. Cell Rep. 2018, 22, 2677–2689. [Google Scholar] [CrossRef]
- Jung, S.G.; Kwon, Y.D.; Song, J.A.; Back, M.J.; Lee, S.Y.; Lee, C.; Hwang, Y.Y.; An, H.J. Prognostic significance of Notch 3 gene expression in ovarian serous carcinoma. Cancer Sci. 2010, 101, 1977–1983. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Qin, C.Y. Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signal-regulated kinase signaling pathway. Mol. Med. Rep. 2014, 9, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Das, S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. USA 2016, 113, E538–E547. [Google Scholar] [CrossRef]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Lin, Z.; Yang, H.; Tan, C.; Li, J.; Liu, Z.; Quan, Q.; Kong, S.; Ye, J.; Gao, B.; Fang, D. USP10 Antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013, 5, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, Y.; Kang, M.; Feng, M.; Ren, Y.; Dai, H.; Wang, Y.; Wang, Y.; Tang, B. USP48 is upregulated by mettl14 to attenuate hepatocellular carcinoma via regulating SIRT6 stabilization. Cancer Res. 2021, 81, 3822–3834. [Google Scholar] [CrossRef]
- Li, N.; Mao, D.; Cao, Y.; Li, H.; Ren, F.; Li, K. Downregulation of SIRT6 by miR-34c-5p is associated with poor prognosis and promotes colon cancer proliferation through inhibiting apoptosis via the JAK2/STAT3 signaling pathway. Int. J. Oncol. 2018, 52, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Yi, L.; Li, J.; Yi, S.; Li, S.; Liu, P.; Yang, X. SIRT6 overexpression induces apoptosis of nasopharyngeal carcinoma by inhibiting Nf-κB signaling. Onco. Targets. Ther. 2018, 11, 7613–7624. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Wada-Hiraike, O.; Oda, K.; Tanikawa, M.; Makii, C.; Inaba, K.; Miyasaka, A.; Miyamoto, Y.; Yano, T.; Maeda, D.; et al. Putative tumor suppression function of SIRT6 in endometrial cancer. FEBS Lett. 2015, 589, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, J.; Li, L.; Tan, L.; Shi, P.; Zhang, J.; Zhong, X.; Ge, L.; Wu, Z.; Cui, H. FOXO3a-SIRT6 axis suppresses aerobic glycolysis in melanoma. Int. J. Oncol. 2020, 56, 728–742. [Google Scholar] [CrossRef]
- Fiorentino, F.; Carafa, V.; Favale, G.; Altucci, L.; Mai, A.; Rotili, D. The two-faced role of sirt6 in cancer. Cancers 2021, 13, 1156. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, M.; Zullo, A.; Servillo, L.; Mancini, F.P.; Borriello, A.; Giovane, A.; Della Ragione, F.; D’Onofrio, N.; Balestrieri, M.L. Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases. Ageing Res. Rev. 2017, 35, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, Y.; Huang, Q.; Tang, K. SIRT6 regulates the proliferation and apoptosis of hepatocellular carcinoma via the ERK1/2 signaling pathway. Mol. Med. Rep. 2019, 20, 1575–1582. [Google Scholar] [CrossRef]
- Zhou, H.Z.; Zeng, H.Q.; Yuan, D.; Ren, J.H.; Cheng, S.T.; Yu, H.B.; Ren, F.; Wang, Q.; Qin, Y.P.; Huang, A.L.; et al. NQO1 potentiates apoptosis evasion and upregulates XIAP via inhibiting proteasome-mediated degradation SIRT6 in hepatocellular carcinoma. Cell Commun. Signal. 2019, 17, 168. [Google Scholar] [CrossRef]
- Garcia-Peterson, L.M.; Guzmán-Pérez, G.; Krier, C.R.; Ahmad, N. The sirtuin 6: An overture in skin cancer. Exp. Dermatol. 2020, 29, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Zhang, Y.; Fang, X.; Chen, N.; Zhou, X.; Wang, X. Sirt6 promotes tumorigenesis and drug resistance of diffuse large B-cell lymphoma by mediating PI3K/Akt signaling. J. Exp. Clin. Cancer Res. 2020, 39, 142. [Google Scholar] [CrossRef]
- Tao, N.N.; Ren, J.H.; Tang, H.; Ran, L.K.; Zhou, H.Z.; Liu, B.; Huang, A.L.; Chen, J. Deacetylation of Ku70 by SIRT6 attenuates Bax-mediated apoptosis in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 485, 713–719. [Google Scholar] [CrossRef]
- Khongkow, M.; Olmos, Y.; Gong, C.; Gomesl, A.R.; Monteiro, L.J.; Yagüe, E.; Cavaco, T.B.; Khongkow, P.; Man, E.P.S.; Laohasinnarong, S.; et al. SIRT6 modulates paclitaxel and epirubicin resistance and survival in breast cancer. Carcinogenesis 2013, 34, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Han, W.; Zhao, B.; Sundaresan, N.R.; Deng, C.X.; Gupta, M.P.; He, Y.Y. SIRT6 promotes COX-2 expression and acts as an oncogene in skin cancer. Cancer Res. 2014, 74, 5925–5933. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, W.; Ma, J.; Dai, W.; Liu, L.; Guo, S.; Chen, J.; Wang, H.; Yang, Y.; Yi, X.; et al. Aberrant SIRT6 expression contributes to melanoma growth: Role of the autophagy paradox and IGF-AKT signaling. Autophagy 2018, 14, 518–533. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, R.; Song, L.D.; Zhu, L.F.; Zhan, J.F. SIRT6 Promotes the Progression of Prostate Cancer via Regulating the Wnt/ β -Catenin Signaling Pathway. J. Oncol. 2022, 2022, 2174758. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, X.; Chen, X.; Wang, Q.; Zhang, J.; Kalvakolanu, D.V.; Guo, B.; Zhang, L. GRIM-19 inhibits proliferation and induces apoptosis in a p53-dependent manner in colorectal cancer cells through the SIRT7/PCAF/MDM2 axis. Exp. Cell Res. 2021, 407, 112799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, P.; Huang, Z.; Hu, X.; Chen, M.; Hu, S.; Hu, Y.; Cai, T. Sirt7 promotes gastric cancer growth and inhibits apoptosis by epigenetically inhibiting miR-34a. Sci. Rep. 2015, 5, 9787. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Shi, L.; Xie, N.; Liu, Z.; Qian, M.; Meng, F.; Xu, Q.; Zhou, M.; Cao, X.; Zhu, W.G.; et al. SIRT7 antagonizes TGF-β signaling and inhibits breast cancer metastasis. Nat. Commun. 2017, 8, 318. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Shi, L.; Su, F.; Qian, M.; Liu, Z.; Meng, Y.; Sun, S.; Li, J.; Liu, B. Combined intermittent fasting and ERK inhibition enhance the anti-tumor effects of chemotherapy via the GSK3β-SIRT7 axis. Nat. Commun. 2021, 12, 5058. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.F.; Grummt, I. The seven faces of SIRT7. Transcription 2017, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Louise Pay, S.; Bhandare, S.B.; Arimpur, U.; Motea, E.A. Therapeutic strategies and biomarkers to modulate parp activity for targeted cancer therapy. Cancers 2020, 12, 972. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinforma. 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef]
- Doetsch, M.; Gluch, A.; Poznanović, G.; Bode, J.; Vidaković, M. YY1-Binding Sites Provide Central Switch Functions in the PARP-1 Gene Expression Network. PLoS ONE 2012, 7, e44125. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPs, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. PARP inhibitors in cancer diagnosis and therapy. Clin. Cancer Res. 2021, 27, 1585–1594. [Google Scholar] [CrossRef]
- Palazzo, L.; Andreja Mikoč, A.; Ahel, I. ADP-RIBOSYLATION: New facets of an ancient modification. FEBS J. 2017, 284, 2932–2946. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD metabolism in cancer therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Amé, J.C.; De Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Cantó, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [PubMed]
- Gil-Kulik, P.; Dudzińska, E.; Radzikowska-Büchner, E.; Wawer, J.; Jojczuk, M.; Nogalski, A.; Wawer, G.A.; Feldo, M.; Kocki, W.; Cioch, M.; et al. Different regulation of PARP1, PARP2, PARP3 and TRPM2 genes expression in acute myeloid leukemia cells. BMC Cancer 2020, 20, 435. [Google Scholar] [CrossRef]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Gozgit, J.M.; Vasbinder, M.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Molina, J.R.; Minissale, E.; Swinger, K.K.; et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 2021, 39, 1214–1226.e10. [Google Scholar] [CrossRef]
- Gozgit, J.; Vasbinder, M.; Abo, R.; Kunii, K.; Kuplast-Barr, K.; Gui, B.; Al, E. A Small-molecule PARP7 Inhibitor triggers Antitumor immunity. Cancer Discov. 2021, 11, 2125. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, H.; Long, Y.; Hua, H.; Jiang, Y.; Jing, J. PARP9 is overexpressed in human breast cancer and promotes cancer cell migration. Oncol. Lett. 2018, 16, 4073–4077. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, G.Z.; Westin, S.N.; Coleman, R.L. Rucaparib in ovarian cancer: Extending the use of PARP inhibitors in the recurrent disease. Futur. Oncol. 2018, 14, 3101–3110. [Google Scholar] [CrossRef]
- Haikarainen, T.; Krauss, S.; Lehtio, L. Tankyrases: Structure, Function and Therapeutic Implications in Cancer. Curr. Pharm. Des. 2014, 20, 6472–6488. [Google Scholar] [CrossRef]
- Prawira, A.; Munusamy, P.; Yuan, J.; Chan, C.H.T.; Koh, G.L.; Shuen, T.W.H.; Hu, J.; Yap, Y.S.; Tan, M.H.; Ang, P.; et al. Assessment of PARP4 as a candidate breast cancer susceptibility gene. Breast Cancer Res. Treat. 2019, 177, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Cirello, V.; Colombo, C.; Pogliaghi, G.; Proverbio, M.C.; Rossi, S.; Mussani, E.; Tosi, D.; Bulfamante, G.; Bonoldi, E.; Gherardi, G.; et al. Genetic variants of PARP4 gene and PARP4P2 pseudogene in patients with multiple primary tumors including thyroid cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2019, 816–818, 2–8. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Y.; Chu, M.; Wang, L.; Shen, H.; Zhang, Z.; Hu, J.; Yi, W.; Yang, W.; Ma, X. PARP6 acts as an oncogene and positively regulates Survivin in gastric cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 2364–2371. [Google Scholar] [PubMed]
- Qi, G.; Kudo, Y.; Tang, B.; Liu, T.; Jin, S.; Liu, J.; Zuo, X.; Mi, S.; Shao, W.; Ma, X.; et al. PARP6 acts as a tumor suppressor via downregulating Survivin expression in colorectal cancer. Oncotarget 2016, 7, 18812–18824. [Google Scholar] [CrossRef]
- Schleicher, E.M.; Galvan, A.M.; Imamura-Kawasawa, Y.; Moldovan, G.L.; Nicolae, C.M. PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 2018, 46, 8908–8916. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, P.; Tomar, V.S.; Chen, X.; Atherton, M.J.; Lu, Z.; Zhang, H.G.; Li, S.; Ortiz, A.; Gui, J.; et al. Targeting PARP11 to avert immunosuppression and improve CAR T therapy in solid tumors. Nat. Cancer 2022, 3, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, C.; Song, D.; Xia, R.; Yu, W.; Dang, Y.; Fei, Y.; Yu, L.; Wu, J. Epigallocatechin-3-gallate enhances ER stress-induced cancer cell apoptosis by directly targeting PARP16 activity. Cell Death Discov. 2017, 3, 17034. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Brunyánszki, A.; Huber, A.; Szántó, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.M.; et al. A phase I–II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Guney Eskiler, G. Talazoparib to treat BRCA-positive breast cancer. Drugs Today 2019, 55, 459–467. [Google Scholar] [CrossRef]
- Steffen, J.D.; Brody, J.R.; Armen, R.S.; Pascal, J.M. Structural implications for selective targeting of PARPs. Front. Oncol. 2013, 3, 301. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A.; Yano, T.; Miura, T.; Hisahara, S.; Ishikawa, S.; Shimamoto, K.; Horio, Y. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010, 285, 8375–8382. [Google Scholar] [CrossRef]
- Kwon, H.S.; Ott, M. The ups and downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef]
- Beltrà, M.; Pöllänen, N.; Fornelli, C.; Tonttila, K.; Hsu, M.Y.; Zampieri, S.; Moletta, L.; Porporato, P.E.; Kivelä, R.; Sandri, M.; et al. NAD+ repletion with niacin counteracts cancer cachexia. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]



| NAMPT-Regulating Pathways | Interaction of Essential Factors | References |
|---|---|---|
| c-MYC/SIRT1 | c-MYC → NAMPT → ↑NAD+ level → SIRT1 activation → ↑c-MYC transcriptional activity | [41,42,43] |
| HMGA1/NAD+ | HMGA1 → NF-κB activity → NAMPT expression → ↑NAD+ | [44] |
| FOXO1 | FOXO1 → ↓NAMPT expression | [45] |
| microRNA | miR-381, miR-206, miR-494, miR-154, miR23b, miR26b → ↓NAMPT expression | [46,47,48,49,50,51] |
| SIRT6/SIRT1 | SIRT6/SIRT1 → ↑NADPH | [52] |
| C/EBPβ | C/EBPβ → ↑NAMPT expression | [53] |
| SIRT Classes | Cellular Compartment | Dominant Functions |
|---|---|---|
| SIRT1 | Nucleus/Cytoplasm | Cell cycle regulation, cell proliferation, gene expression, chromatin modification, genomic stability, energy metabolism, stress response, cell survival |
| SIRT2 | Nucleus/Cytoplasm | Cell cycle regulation, cell survival |
| SIRT3 | Mitochondrion | Energy metabolism, stress response, cell survival |
| SIRT4 | Mitochondrion | Energy metabolism, cell cycle regulation, stress response, cell survival |
| SIRT5 | Mitochondrion | Energy metabolism, stress response, cell survival |
| SIRT6 | Nucleus | Energy metabolism, stress response, cell survival |
| SIRT7 | Nucleus | Energy metabolism, stress response, cell survival, genomic stability |
| Class of Regulatory Sirtuins | Molecular Factors | Changes in the Functioning of Molecular Markers |
|---|---|---|
| SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7 | NAD+ | ? |
| SIRT1, SIRT2, SIRT6, SIRT7 | p53 | Non-functional |
| SIRT1, SIRT5, SIRT6 | E2F1 | Inhibition |
| SIRT1 | HIC1 | Inhibition |
| SIRT1, SIRT3, SIRT6 | c-MYC | Activation |
| SIRT1, SIRT3, SIRT6 | KU70 | Activation |
| SIRT2 | α-tubulin | Activation |
| SIRT1, SIRT2, SIRT6 | β-catenin | Activation |
| SIRT2 | PEPCK1 (RAS/ERK/JNK/MMP-9) | Activation |
| SIRT5 | SUN2 | Inhibition |
| SIRT5 | NRF2 | Activation |
| SIRT5 | PKM2 | Inhibition |
| SIRT5 | SDHA | Inhibition |
| SIRT1 | miR-22, miR-93, miR-217, miR-449 | Inhibition |
| SIRT2 | miR-21, miR-138 | Inhibition |
| SIRT3 | miR-224 | Inhibition |
| SIRT1, SIRT6, SIRT7 | mir-34a | Inhibition |
| SIRT6 | mir-122 | Inhibition |
| Class of Regulatory Sirtuins | Molecular Factors | Changes in the Functioning of Molecular Markers |
|---|---|---|
| SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7 | NAD+ | ? |
| SIRT1, SIRT2, SIRT6, SIRT7 | p53 | Functioning |
| SIRT1, SIRT6, SIRT7 | MDM2 | Inhibition |
| SIRT1, SIRT2, SIRT3, SIRT6 | FOXO | Inhibition |
| SIRT1, SIRT6 | Notch | Inhibition |
| SIRT1, SIRT2 | VEGF/CTGF/ACLY | Inhibition |
| SIRT1, SIRT3, SIRT4, SIRT6 | HIF-1α | Inhibition |
| SIRT1, SIRT3, SIRT6, SIRT7 | AMPK | Activation |
| SIRT1, SIRT2, SIRT6 | NF-κB | Inhibition |
| SIRT2, SIRT3, SIRT4 | p38-MAPK/ERK | Inhibition |
| SIRT4 | N-cadherin/vimentin | Inhibition |
| SIRT4 | E-cadherin | Activation |
| SIRT4 | CyclinE | Inhibition |
| SIRT4, SIRT7 | AKT/GSK3β/CyclinD | Inhibition |
| SIRT5 | PI3K/AKT/NF-κB | Inhibition |
| SIRT6 | PI3K/AKT/mTOR | Inhibition |
| SIRT1 | BRCA1 | Deficiency |
| SIRT2 | APC/C | Inhibition |
| SIRT3 | HMG-CoA synthase | Activation |
| SIRT3, SIRT4 | GDH | Inhibition |
| SIRT3 | Akt/PKB | Inhibition |
| SIRT3 | IRP1 | Inhibition |
| SIRT2, SIRT3 | Skp2 | Inhibition |
| SIRT5 | ACOX1 | Inhibition |
| SIRT5 | SOD1 | Activation |
| SIRT7 | SMAD4 | Inhibition |
| SIRT6 | JAK2/STAT3 | Inhibition |
| Type of PARPs | Functional and Structural Distinctions | Participation in the Growth of Cancer Tissue |
|---|---|---|
| PARP1, PARP2, PARP3 | DNA-dependent | Overexpressed [213,214] |
| PARP7, PARP12, PARP13.1, PARP13.2 | Cys-Cys-Cys-His zinc finger | Overexpressed [215,216] |
| PARP9, PARP14, PARP15 | Macro-PARPs | Overexpressed [217,218] |
| Tankyrase 1 (PARP-5a), tankyrase 2 (PARP-5b) | Tankyrases | Overexpressed [219] |
| PARP4 | Lacks an N-terminal DNA binding domain (activated by protein-protein interaction) | Cancer susceptibility gene [220,221] |
| PARP6 | Less studied or not typical | Acts as an oncogene [222]; Acts as a tumor suppressor [223] |
| PARP8 | Less studied or not typical | - |
| PARP10 | Less studied or not typical | Overexpressed [224] |
| PARP11 | Less studied or not typical | Participates in the disorders of antitumor immunity [225] |
| PARP16 | Less studied or not typical | Acts as an oncogene [218,226] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podyacheva, E.; Toropova, Y. The Role of NAD+, SIRTs Interactions in Stimulating and Counteracting Carcinogenesis. Int. J. Mol. Sci. 2023, 24, 7925. https://doi.org/10.3390/ijms24097925
Podyacheva E, Toropova Y. The Role of NAD+, SIRTs Interactions in Stimulating and Counteracting Carcinogenesis. International Journal of Molecular Sciences. 2023; 24(9):7925. https://doi.org/10.3390/ijms24097925
Chicago/Turabian StylePodyacheva, Ekaterina, and Yana Toropova. 2023. "The Role of NAD+, SIRTs Interactions in Stimulating and Counteracting Carcinogenesis" International Journal of Molecular Sciences 24, no. 9: 7925. https://doi.org/10.3390/ijms24097925
APA StylePodyacheva, E., & Toropova, Y. (2023). The Role of NAD+, SIRTs Interactions in Stimulating and Counteracting Carcinogenesis. International Journal of Molecular Sciences, 24(9), 7925. https://doi.org/10.3390/ijms24097925