
Molecular Evolution of Histone Methylation Modification Families in the Plant Kingdom and Their Genome-Wide Analysis in Barley

Abstract
:1. Introduction
2. Results
2.1. Origin and Evolution of HMT and HDM in Green Plants
2.2. Number of HMTs and HDMs in Barley
2.3. Variation in HMT and HDM Family Members in Barley
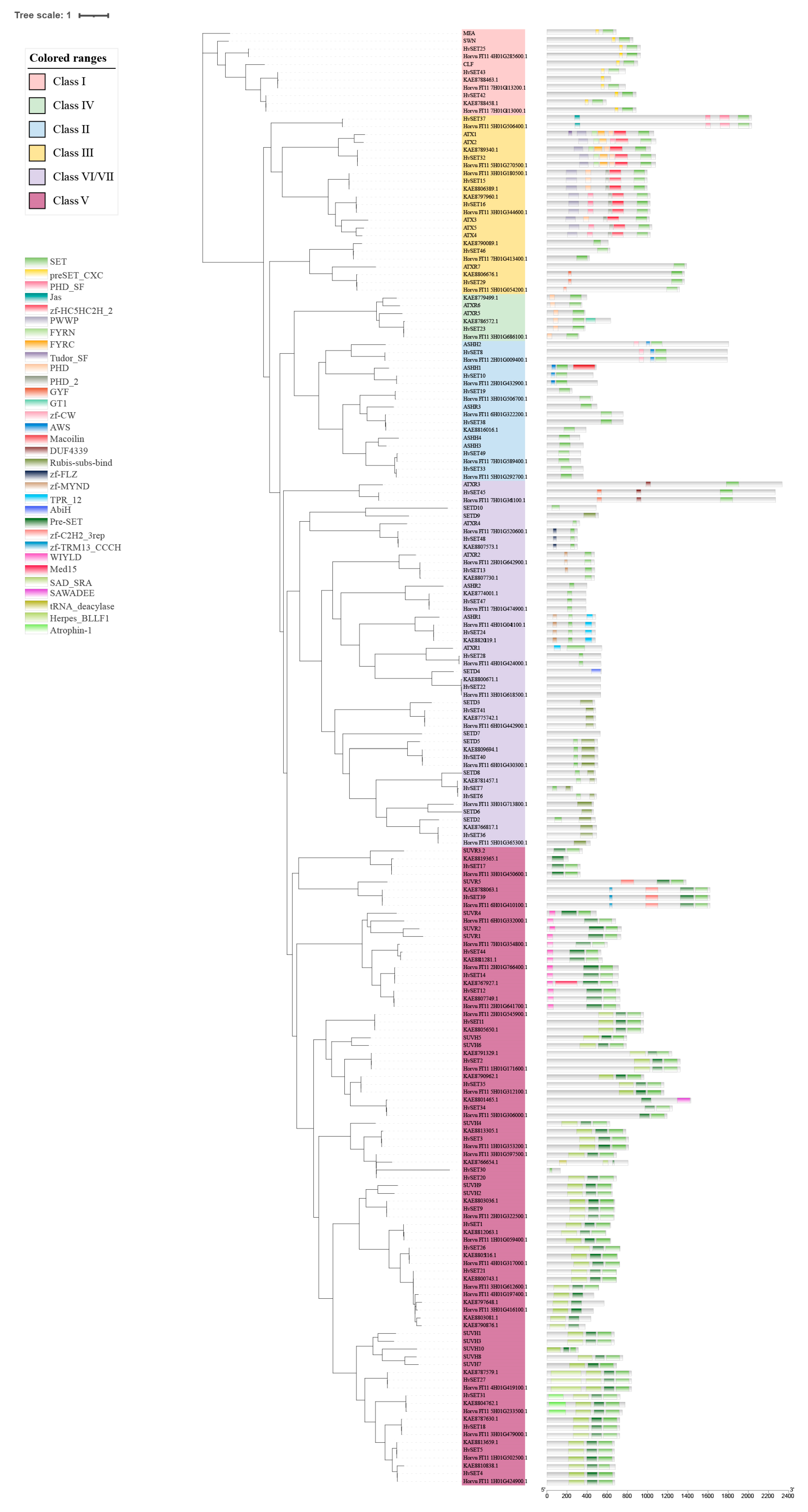
2.4. Phylogenetic, Conserved Domain and Motif Analysis of HMT and HDM in Barley
2.5. Chromosomal Location Analysis of HvHMTs and HvHDMs
2.6. Analysis of Cis-Acting Elements of HvHMTs and HvHDMs
2.7. HvHMTs and HvHDMs Expression Patterns in Different Organs
2.8. HvHMTs and HvHDMs Expression Profiling under Abiotic Stresses
3. Discussion
4. Materials and Methods
4.1. Evolutionary Analysis
4.2. Biogenic Analysis of HMT and HDM in Barley
4.3. Plant Material, Stress Treatment, RNA Extraction, and qRT–PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, J.; Jiang, X.; Bai, H.; Liu, C. Genome-wide identification, classification and expression analysis of the JmjC domain-containing histone demethylase gene family in Jatropha curcas L. Sci. Rep. 2022, 12, 6543. [Google Scholar] [CrossRef]
- Hernando, C.E.; Sanchez, S.E.; Mancini, E.; Yanovsky, M.J. Genome wide comparative analysis of the effects of PRMT5 and PRMT4/CARM1 arginine methyltransferases on the Arabidopsis thaliana transcriptome. BMC Genom. 2015, 16, 192. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ali, S.; Zhang, X.; Zhang, Y.; Wang, M.; Zhang, Q.; Xie, L. Genome-wide identification, classification, and expression analysis of the JmjC domain-containing histone demethylase gene family in birch. BMC Genom. 2021, 22, 6543. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Yang, J.; Yi, J.; An, G. Controlling flowering time by histone methylation and acetylation in Arabidopsis and rice. J. Plant Biol. 2015, 58, 203–210. [Google Scholar] [CrossRef]
- Sehrish, S.; Sumbal, W.; Xie, M.; Zhao, C.; Zuo, R.; Gao, F.; Liu, S. Genome-Wide Identification and Characterization of SET Domain Family Genes in Brassica napus L. Int. J. Mol. Sci. 2022, 23, 1936. [Google Scholar] [CrossRef]
- Chen, D.-H.; Qiu, H.-L.; Huang, Y.; Zhang, L.; Si, J.-P. Genome-wide identification and expression profiling of SET DOMAIN GROUP family in Dendrobium catenatum. BMC Plant Biol. 2020, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Sethunath, V.; Metaferia, N.Y.; Nogueira, M.F.; Gallant, D.S.; Garner, E.R.; Lairson, L.A.; Penney, C.M.; Li, J.; Gelbard, M.K. A genome-scale CRISPR screen reveals PRMT1 as a critical regulator of androgen receptor signaling in prostate cancer. Cell Rep. 2022, 38, 110417. [Google Scholar] [CrossRef]
- Lopez, L.; Perrella, G.; Calderini, O.; Porceddu, A.; Panara, F. Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules. Plants 2022, 11, 322. [Google Scholar] [CrossRef]
- Kronholm, I.; Bassett, A.; Baulcombe, D.; Collins, S. Epigenetic and genetic contributions to adaptation in Chlamydomonas. Mol. Biol. Evol. 2017, 34, 2285–2306. [Google Scholar] [CrossRef]
- Day, T.; Bonduriansky, R. A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am. Nat. 2011, 178, E18–E36. [Google Scholar] [CrossRef]
- Klironomos, F.D.; Berg, J.; Collins, S. How epigenetic mutations can affect genetic evolution: Model and mechanism. BioEssays 2013, 35, 571–578. [Google Scholar] [CrossRef]
- Kronholm, I.; Collins, S. Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 2016, 25, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Miryeganeh, M. Plants’ epigenetic mechanisms and abiotic stress. Genes 2021, 12, 1106. [Google Scholar] [CrossRef]
- Yaish, M.W. Epigenetic modifications associated with abiotic and biotic stresses in plants: An implication for understanding plant evolution. Front. Plant Sci. 2017, 8, 1983. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, X.; Luo, M.; Yang, S.; Wu, K. Involvement of histone modifications in plant abiotic stress responses. J. Integr. Plant Biol. 2013, 55, 892–901. [Google Scholar] [CrossRef]
- Kong, L.; Liu, Y.; Wang, X.; Chang, C. Insight into the role of epigenetic processes in abiotic and biotic stress response in wheat and barley. Int. J. Mol. Sci. 2020, 21, 1480. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, A.; Yin, H.; Meng, Q.; Yu, X.; Huang, S.; Wang, J.; Ahmad, R.; Liu, B.; Xu, Z.Y. Trithorax-group proteins ARABIDOPSIS TRITHORAX4 (ATX4) and ATX 5 function in abscisic acid and dehydration stress responses. New Phytol. 2018, 217, 1582–1597. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.S.; Lee, D.; Choi, G.; Chung, W.I. Histone occupancy-dependent and-independent removal of H3K27 trimethylation at cold-responsive genes in Arabidopsis. Plant J. 2009, 60, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Zhang, Y.; Wang, X.; Li, D.; Li, Q.; Yue, M.; Li, Q.; Zhang, Y.-E.; Xu, Y. Arabidopsis floral initiator SKB1 confers high salt tolerance by regulating transcription and pre-mRNA splicing through altering histone H4R3 and small nuclear ribonucleoprotein LSM4 methylation. Plant Cell 2011, 23, 396–411. [Google Scholar] [CrossRef]
- Bourdareau, S.; Tirichine, L.; Lombard, B.; Loew, D.; Scornet, D.; Wu, Y.; Coelho, S.M.; Cock, J.M. Histone modifications during the life cycle of the brown alga Ectocarpus. Genome Biol. 2021, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Bacova, R.; Kolackova, M.; Klejdus, B.; Adam, V.; Huska, D. Epigenetic mechanisms leading to genetic flexibility during abiotic stress responses in microalgae: A review. Algal Res. 2020, 50, 101999. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Chen, Y.; Wang, S.; Fang, Y.; Zhang, X.; Wu, Y.; Xue, D. Genome-wide identification of the SOD gene family and expression analysis under drought and salt stress in barley. Plant Growth Regul. 2021, 94, 49–60. [Google Scholar] [CrossRef]
- Cai, K.; Zeng, F.; Wang, J.; Zhang, G. Identification and characterization of HAK/KUP/KT potassium transporter gene family in barley and their expression under abiotic stress. BMC Genom. 2021, 22, 317. [Google Scholar] [CrossRef]
- She, K.; Pan, W.; Yan, Y.; Shi, T.; Chu, Y.; Cheng, Y.; Ma, B.; Song, W. Genome-wide identification, evolution and expressional analysis of OSCA gene family in barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2022, 23, 13027. [Google Scholar] [CrossRef]
- Sharma, N.; Geuten, K.; Giri, B.S.; Varma, A. The molecular mechanism of vernalization in Arabidopsis and cereals: Role of Flowering Locus C and its homologs. Physiol. Plant. 2020, 170, 373–383. [Google Scholar] [CrossRef]
- Yuzhen, B.; Sang, Z.; Mu, W.; Yu, M.; Wang, Y.; Yuan, H.; Xu, Q. Whole-genome analysis of the trimethylation of histone H3 lysine 4 and lysine 27 in two contrasting Tibetan hulless barley genotypes under salinity stress. Acta Physiol. Plant. 2021, 43, 89. [Google Scholar] [CrossRef]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Stein, N. The barley pangenome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, T.; Ling, Z.; Wang, Y.; Li, X.; Xu, S.; Xu, Q.; Zha, S.; Qimei, W.; Basang, Y. An improved high-quality genome assembly and annotation of Tibetan hulless barley. Sci. Data 2020, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Buitrago, S.; Peng, Y.; Abou-Elwafa, S.F.; Wan, K.; Liu, Y.; Wang, R.; Yang, X.; Zhang, W. Genome-wide identification of cold-tolerance genes and functional analysis of IbbHLH116 gene in sweet potato. Gene 2022, 837, 146690. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An expanded resource for species divergence times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Ai, Q.; Pan, W.; Zeng, Y.; Li, Y.; Cui, L. CCCH Zinc finger genes in Barley: Genome-wide identification, evolution, expression and haplotype analysis. BMC Plant Biol. 2022, 22, 117. [Google Scholar]
- Ammar, R.; Torti, D.; Tsui, K.; Gebbia, M.; Durbic, T.; Bader, G.D.; Giaever, G.; Nislow, C. Chromatin is an ancient innovation conserved between Archaea and Eukarya. eLife 2012, 1, e00078. [Google Scholar] [CrossRef]
- Nalabothula, N.; Xi, L.; Bhattacharyya, S.; Widom, J.; Wang, J.-P.; Reeve, J.N.; Santangelo, T.J.; Fondufe-Mittendorf, Y.N. Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genom. 2013, 14, 391. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Lin, S.; Guo, Y.; Deng, W.; Zhang, Y.; Xue, Y. WERAM: A database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 2016, 45, gkw1011. [Google Scholar]
- Rastogi, A.; Lin, X.; Lombard, B.; Loew, D.; Tirichine, L. Probing the evolutionary history of epigenetic mechanisms: What can we learn from marine diatoms. Aims Genet. 2015, 2, 173–191. [Google Scholar] [CrossRef]
- Becker, B.; Feng, X.; Yin, Y.; Holzinger, A. Desiccation tolerance in streptophyte algae and the algae to land plant transition: Evolution of LEA and MIP protein families within the Viridiplantae. J. Exp. Bot. 2020, 71, 3270–3278. [Google Scholar] [CrossRef]
- Vigneau, J.; Borg, M. The epigenetic origin of life history transitions in plants and algae. Plant Reprod. 2021, 34, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Casas-Mollano, J.A.; van Dijk, K.; Eisenhart, J.; Cerutti, H. SET3p monomethylates histone H3 on lysine 9 and is required for the silencing of tandemly repeated transgenes in Chlamydomonas. Nucleic Acids Res. 2007, 35, 939–950. [Google Scholar] [CrossRef]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pangenomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Zhang, H.; Mittal, N.; Leamy, L.J.; Barazani, O.; Song, B.H. Back into the wild-Apply untapped genetic diversity of wild relatives for crop improvement. Evol. Appl. 2017, 10, 5–24. [Google Scholar] [CrossRef]
- Hyten, D.L.; Song, Q.; Zhu, Y.; Choi, I.-Y.; Nelson, R.L.; Costa, J.M.; Specht, J.E.; Shoemaker, R.C.; Cregan, P.B. Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl. Acad. Sci. USA 2006, 103, 16666–16671. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.I.; Bi, I.V.; Schroeder, S.G.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012, 30, 105–111. [Google Scholar] [CrossRef]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, J.; Li, T.; Yang, H.; Wang, P.; Su, L.; Zheng, Y.; Bao, C.; Zhou, C. SDG711 is involved in rice seed development through regulation of starch metabolism gene expression in coordination with other histone modifications. Rice 2021, 14, 25. [Google Scholar] [CrossRef]
- Zheng, L.; Ma, S.; Shen, D.; Fu, H.; Wang, Y.; Liu, Y.; Shah, K.; Yue, C.; Huang, J. Genome-wide identification of Gramineae histone modification genes and their potential roles in regulating wheat and maize growth and stress responses. BMC Plant Biol. 2021, 21, 543. [Google Scholar] [CrossRef]
- Ay, N.; Irmler, K.; Fischer, A.; Uhlemann, R.; Reuter, G.; Humbeck, K. Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J. 2009, 58, 333–346. [Google Scholar] [CrossRef]
- Plett, K.L.; Raposo, A.E.; Bullivant, S.; Anderson, I.C.; Piller, S.C.; Plett, J.M. Root morphogenic pathways in Eucalyptus grandis are modified by the activity of protein arginine methyltransferases. BMC Plant Biol. 2017, 17, 62. [Google Scholar] [CrossRef]
- Begcy, K.; Dresselhaus, T. Epigenetic responses to abiotic stresses during reproductive development in cereals. Plant Reprod. 2018, 31, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Liang, Y.; Zhou, D.; Li, S.; Lin, S.; Dong, H.; Huang, L. The function of histone lysine methylation related SET domain group proteins in plants. Protein Sci. 2020, 29, 1120–1137. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Avramova, Z.; Fromm, M. The Arabidopsis trithorax-like factor ATX1 functions in dehydration stress responses via ABA-dependent and ABA-independent pathways. Plant J. 2011, 66, 735–744. [Google Scholar] [CrossRef]
- Qiao, W.; Fan, L. Epigenetics, a mode for plants to respond to abiotic stresses. Front. Cell Dev. Biol. 2011, 6, 477–481. [Google Scholar] [CrossRef]
- Monat, C.; Schreiber, M.; Stein, N.; Mascher, M. Prospects of pan-genomics in barley. Theor. Appl. Genet. 2019, 132, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Tong, T.; Li, Q.; Jiang, W.; Chen, G.; Xue, D.; Deng, F.; Zeng, F.; Chen, Z.-H. Molecular evolution of calcium signaling and transport in plant adaptation to abiotic stress. Int. J. Mol. Sci. 2021, 22, 12308. [Google Scholar] [CrossRef]
- Jiang, W.; Tong, T.; Chen, X.; Deng, F.; Zeng, F.; Pan, R.; Zhang, W.; Chen, G.; Chen, Z.-H. Molecular response and evolution of plant anion transport systems to abiotic stress. Plant Mol. Biol. 2021, 110, 397–412. [Google Scholar] [CrossRef]
- Xu, Z.; Shen, Q.; Zhang, G. The mechanisms for the difference in waterlogging tolerance among sea barley, wheat and barley. Plant Growth Regul. 2022, 96, 431–441. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Germplasm | Number of Family Members | |||
|---|---|---|---|---|
| HMT | HDM | |||
| SDG | PRMT | JMJ | HDMA | |
| Akashinriki | 51 | 7 | 18 | 4 |
| B1K-04-12 | 50 | 7 | 18 | 4 |
| Barke | 52 | 7 | 18 | 4 |
| Golden_Promise | 51 | 7 | 17 | 4 |
| Hockett | 53 | 7 | 18 | 4 |
| HOR_10350 | 51 | 7 | 18 | 4 |
| HOR_13821 | 51 | 7 | 18 | 4 |
| HOR_13942 | 51 | 7 | 18 | 4 |
| HOR_21599 | 51 | 7 | 18 | 4 |
| HOR_3081 | 50 | 7 | 18 | 4 |
| HOR_3365 | 50 | 7 | 18 | 4 |
| HOR_7552 | 52 | 7 | 18 | 4 |
| HOR_8148 | 52 | 7 | 18 | 4 |
| HOR_9043 | 51 | 7 | 18 | 4 |
| Igri | 51 | 7 | 18 | 4 |
| Morex | 49 | 7 | 19 | 4 |
| OUN333 | 53 | 7 | 17 | 4 |
| RGT_Planet | 51 | 7 | 18 | 4 |
| ZDM01467 | 50 | 7 | 18 | 4 |
| ZDM02064 | 52 | 7 | 18 | 4 |
| Lasa Goumang | 42 | 7 | 17 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, B.; Cai, H.; Li, B.; Zhang, S.; He, Y.; Wang, R.; Jiao, C.; Guo, Y.; Xu, L.; Xu, Y. Molecular Evolution of Histone Methylation Modification Families in the Plant Kingdom and Their Genome-Wide Analysis in Barley. Int. J. Mol. Sci. 2023, 24, 8043. https://doi.org/10.3390/ijms24098043
An B, Cai H, Li B, Zhang S, He Y, Wang R, Jiao C, Guo Y, Xu L, Xu Y. Molecular Evolution of Histone Methylation Modification Families in the Plant Kingdom and Their Genome-Wide Analysis in Barley. International Journal of Molecular Sciences. 2023; 24(9):8043. https://doi.org/10.3390/ijms24098043
Chicago/Turabian StyleAn, Bingzhuang, Haiya Cai, Bo Li, Shuo Zhang, Yonggang He, Rong Wang, Chunhai Jiao, Ying Guo, Le Xu, and Yanhao Xu. 2023. "Molecular Evolution of Histone Methylation Modification Families in the Plant Kingdom and Their Genome-Wide Analysis in Barley" International Journal of Molecular Sciences 24, no. 9: 8043. https://doi.org/10.3390/ijms24098043
APA StyleAn, B., Cai, H., Li, B., Zhang, S., He, Y., Wang, R., Jiao, C., Guo, Y., Xu, L., & Xu, Y. (2023). Molecular Evolution of Histone Methylation Modification Families in the Plant Kingdom and Their Genome-Wide Analysis in Barley. International Journal of Molecular Sciences, 24(9), 8043. https://doi.org/10.3390/ijms24098043