
The Function of ASK1 in Sepsis and Stress-Induced Disorders


{kind=link}
Abstract
:1. Introduction
2. ASK1
3. ASK1 Structure
4. ASK1 Activation
4.1. In Most Nucleated Cells
4.2. In Platelets
4.3. In Neurons
5. ASK1 Function
5.1. In Stress
5.2. In Sepsis
5.3. In Hemostasis and Thrombosis
5.4. In Heparin-Induced Thrombocytopenia
6. ASK1 and Platelet Clearance
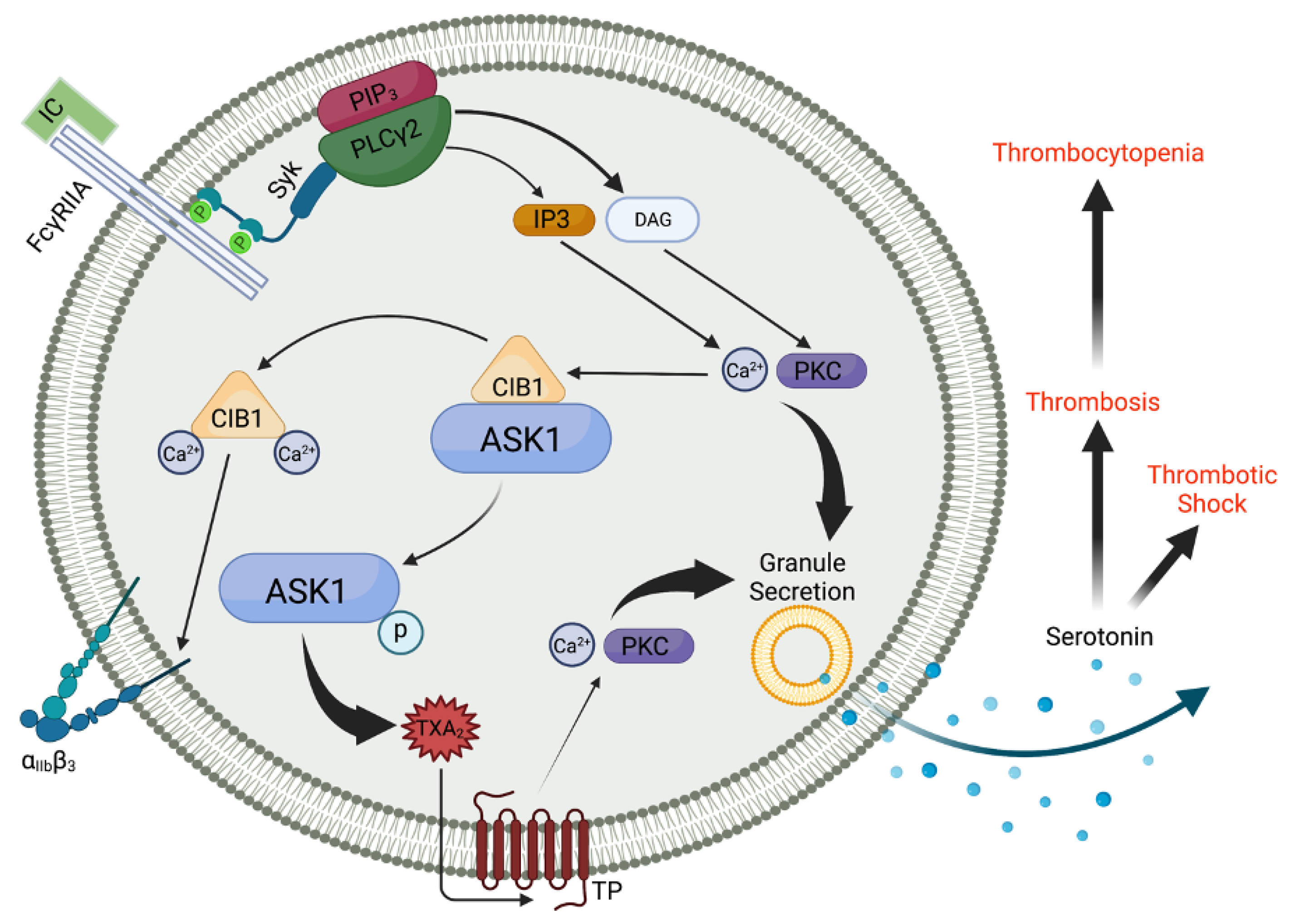
7. The Involvement of ASK1 in ITAM-Mediated Signaling
8. ASK1 and TxA2 Generation in Human Platelets
9. Conclusions
Funding
Conflicts of Interest
References
- Fan, X.; Wang, C.; Shi, P.; Gao, W.; Gu, J.; Geng, Y.; Yang, W.; Wu, N.; Wang, Y.; Xu, Y.; et al. Platelet MEKK3 regulates arterial thrombosis and myocardial infarct expansion in mice. Blood Adv. 2018, 2, 1439–1448. [Google Scholar] [CrossRef]
- Shi, P.; Zhang, L.; Zhang, M.; Yang, W.; Wang, K.; Zhang, J.; Otsu, K.; Huang, G.; Fan, X.; Liu, J. Platelet-Specific p38alpha Deficiency Improved Cardiac Function After Myocardial Infarction in Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e185–e196. [Google Scholar] [CrossRef]
- Patel, P.; Naik, U.P. Platelet MAPKs-a 20+ year history: What do we really know? J. Thromb. Haemost. 2020, 18, 2087–2102. [Google Scholar] [CrossRef]
- Adam, F.; Kauskot, A.; Rosa, J.P.; Bryckaert, M. Mitogen-activated protein kinases in hemostasis and thrombosis. J. Thromb. Haemost. 2008, 6, 2007–2016. [Google Scholar] [CrossRef]
- Federspiel, J.D.; Codreanu, S.G.; Palubinsky, A.M.; Winland, A.J.; Betanzos, C.M.; McLaughlin, B.; Liebler, D.C. Assembly Dynamics and Stoichiometry of the Apoptosis Signal-regulating Kinase (ASK) Signalosome in Response to Electrophile Stress. Mol. Cell. Proteom. 2016, 15, 1947–1961. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Dikshit, M.; Asthana, S. Crystallographic mining of ASK1 regulators to unravel the intricate PPI interfaces for the discovery of small molecule. Comput. Struct. Biotechnol. J. 2022, 20, 3734–3754. [Google Scholar] [CrossRef]
- Luyendyk, J.P.; Piper, J.D.; Tencati, M.; Reddy, K.V.; Holscher, T.; Zhang, R.; Luchoomun, J.; Chen, X.; Min, W.; Kunsch, C.; et al. A novel class of antioxidants inhibit LPS induction of tissue factor by selective inhibition of the activation of ASK1 and MAP kinases. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1857–1863. [Google Scholar] [CrossRef]
- Noh, K.T.; Park, Y.M.; Cho, S.G.; Choi, E.J. GSK-3beta-induced ASK1 stabilization is crucial in LPS-induced endotoxin shock. Exp. Cell Res. 2011, 317, 1663–1668. [Google Scholar] [CrossRef]
- Miller, M.R.; Koch, S.R.; Choi, H.; Lamb, F.S.; Stark, R.J. Apoptosis signal-regulating kinase 1 (ASK1) inhibition reduces endothelial cytokine production without improving permeability after toll-like receptor 4 (TLR4) challenge. Transl. Res. 2021, 235, 115–128. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Saegusa, K.; Noguchi, T.; Sadamitsu, C.; Nishitoh, H.; Nagai, S.; Koyasu, S.; Matsumoto, K.; Takeda, K.; Ichijo, H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 2005, 6, 587–592. [Google Scholar] [CrossRef]
- Liu, H.; Xu, R.; Feng, L.; Guo, W.; Cao, N.; Qian, C.; Teng, P.; Wang, L.; Wu, X.; Sun, Y.; et al. A novel chromone derivative with anti-inflammatory property via inhibition of ROS-dependent activation of TRAF6-ASK1-p38 pathway. PLoS ONE 2012, 7, e37168. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Nakao, K. To die or not to die: Death signaling in nonalcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Wang, P.X.; Wang, A.B.; Zhang, X.J.; Zhang, Y.; Zhang, P.; Mei, F.H.; Chen, M.H.; Li, H. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J. Hepatol. 2016, 64, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Demian, W.L.; Jacob, R.A.; Cormier, O.; Nazli, A.; Melki, M.; Asavajaru, A.; Baid, K.; Zhang, A.; Miller, M.S.; Kaushic, C.; et al. ASK1 inhibitors are potential pan-antiviral drugs, which dampen replication of diverse viruses including SARS-CoV2. Antivir. Res. 2023, 220, 105736. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Pang, L.; He, M.; Wang, Z. Small-molecule inhibitors targeting apoptosis signal-regulated kinase 1. Eur. J. Med. Chem. 2023, 262, 115889. [Google Scholar] [CrossRef] [PubMed]
- Obsilova, V.; Honzejkova, K.; Obsil, T. Structural Insights Support Targeting ASK1 Kinase for Therapeutic Interventions. Int. J. Mol. Sci. 2021, 22, 13395. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Patel, P.; Derstine, R.; Turaga, R.; Chen, X.; Golla, K.; Neeves, K.B.; Ichijo, H.; Naik, U.P. Ask1 regulates murine platelet granule secretion, thromboxane A(2) generation, and thrombus formation. Blood 2017, 129, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Shaik, N.F.; Zhou, Y.; Golla, K.; McKenzie, S.E.; Naik, U.P. Apoptosis signal-regulating kinase 1 regulates immune-mediated thrombocytopenia, thrombosis, and systemic shock. J. Thromb. Haemost. 2020, 18, 3013–3028. [Google Scholar] [CrossRef]
- Patel, P.; Naik, M.U.; Golla, K.; Shaik, N.F.; Naik, U.P. Calcium-induced dissociation of CIB1 from ASK1 regulates agonist-induced activation of the p38 MAPK pathway in platelets. Biochem. J. 2019, 476, 2835–2850. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Nishitoh, H.; Ichijo, H.; Kyriakis, J.M. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol. Cell. Biol. 2000, 20, 2198–2208. [Google Scholar] [CrossRef]
- Weijman, J.F.; Kumar, A.; Jamieson, S.A.; King, C.M.; Caradoc-Davies, T.T.; Ledgerwood, E.C.; Murphy, J.M.; Mace, P.D. Structural basis of autoregulatory scaffolding by apoptosis signal-regulating kinase 1. Proc. Natl. Acad. Sci. USA 2017, 114, E2096–E2105. [Google Scholar] [CrossRef] [PubMed]
- Bunkoczi, G.; Salah, E.; Filippakopoulos, P.; Fedorov, O.; Muller, S.; Sobott, F.; Parker, S.A.; Zhang, H.; Min, W.; Turk, B.E.; et al. Structural and functional characterization of the human protein kinase ASK1. Structure 2007, 15, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.W.; Cho, J.H.; Lee, J.K.; Kang, Y.H.; Chae, J.S.; Kim, Y.M.; Kim, J.; Kim, E.K.; Kim, S.E.; Baik, J.H.; et al. CIB1 functions as a Ca(2+)-sensitive modulator of stress-induced signaling by targeting ASK1. Proc. Natl. Acad. Sci. USA 2009, 106, 17389–17394. [Google Scholar] [CrossRef] [PubMed]
- Fujino, G.; Noguchi, T.; Matsuzawa, A.; Yamauchi, S.; Saitoh, M.; Takeda, K.; Ichijo, H. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol. 2007, 27, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Nadeau, P.J.; Charette, S.J.; Landry, J. REDOX reaction at ASK1-Cys250 is essential for activation of JNK and induction of apoptosis. Mol. Biol. Cell 2009, 20, 3628–3637. [Google Scholar] [CrossRef]
- Kekulandara, D.N.; Nagi, S.; Seo, H.; Chow, C.S.; Ahn, Y.H. Redox-Inactive Peptide Disrupting Trx1-Ask1 Interaction for Selective Activation of Stress Signaling. Biochemistry 2018, 57, 772–780. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, T.J.; Lee, K.Y. A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett. 2008, 582, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Lee, Y.J. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 2003, 373, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Min, W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ. Res. 2002, 90, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Hattori, K.; Watanabe, K. The regulatory and signaling mechanisms of the ASK family. Adv. Biol. Regul. 2017, 66, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Kylarova, S.; Kosek, D.; Petrvalska, O.; Psenakova, K.; Man, P.; Vecer, J.; Herman, P.; Obsilova, V.; Obsil, T. Cysteine residues mediate high-affinity binding of thioredoxin to ASK1. FEBS J. 2016, 283, 3821–3838. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wen, H.; Fu, J.; Cai, L.; Li, P.L.; Zhao, C.L.; Dong, Z.F.; Ma, J.P.; Wang, X.; Tian, H.; et al. Hepatocyte TNF Receptor-Associated Factor 6 Aggravates Hepatic Inflammation and Fibrosis by Promoting Lysine 6-Linked Polyubiquitination of Apoptosis Signal-Regulating Kinase 1. Hepatology 2020, 71, 93–111. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, P.X.; Zhao, L.P.; Zhang, X.; Ji, Y.X.; Zhang, X.J.; Fang, C.; Lu, Y.X.; Yang, X.; Gao, M.M.; et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 84–94. [Google Scholar] [CrossRef]
- Clemetson, K.J. The role of platelets in defence against pathogens. Hamostaseologie 2011, 31, 264–268. [Google Scholar] [CrossRef]
- Corken, A.; Russell, S.; Dent, J.; Post, S.R.; Ware, J. Platelet glycoprotein Ib-IX as a regulator of systemic inflammation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 996–1001. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Tucker, S.C.; Kopetz, S.; Sood, A.K.; Crissman, J.D.; Honn, K.V. Platelets and cancer: A casual or causal relationship: Revisited. Cancer Metastasis Rev. 2014, 33, 231–269. [Google Scholar] [CrossRef] [PubMed]
- Page, C.; Pitchford, S. Platelets and allergic inflammation. Clin. Exp. Allergy 2014, 44, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.E.; Andrews, R.K. Platelets: Envoys at the infection frontline. J. Infect. Dis. 2013, 208, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Pabinger, I.; Ay, C. Platelets in cancer and thrombosis. Hamostaseologie 2014, 34, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Packham, M.A. Role of platelets in thrombosis and hemostasis. Can. J. Physiol. Pharmacol. 1994, 72, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Brass, L. Understanding and evaluating platelet function. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nacher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Wong, C.H.; Jenne, C.N.; Petri, B.; Chrobok, N.L.; Kubes, P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 2013, 14, 785–792. [Google Scholar] [CrossRef]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382.e1323. [Google Scholar] [CrossRef]
- Ali, R.A.; Wuescher, L.M.; Dona, K.R.; Worth, R.G. Platelets Mediate Host Defense against Staphylococcus aureus through Direct Bactericidal Activity and by Enhancing Macrophage Activities. J. Immunol. 2017, 198, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Handtke, S.; Palankar, R.; Wesche, J.; Kohler, T.P.; Kohler, C.; Gruel, Y.; Hammerschmidt, S.; Greinacher, A. Activated platelets kill Staphylococcus aureus, but not Streptococcus pneumoniae-The role of FcgammaRIIa and platelet factor 4/heparinantibodies. J. Thromb. Haemost. 2020, 18, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Youssefian, T.; Drouin, A.; Masse, J.M.; Guichard, J.; Cramer, E.M. Host defense role of platelets: Engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 2002, 99, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fuhrken, P.G.; Huang, L.T.; Apostolidis, P.; Wang, M.; Paredes, C.J.; Miller, W.M.; Papoutsakis, E.T. A systems-biology analysis of isogenic megakaryocytic and granulocytic cultures identifies new molecular components of megakaryocytic apoptosis. BMC Genom. 2007, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.W.; Yang, H.S.; Kim, Y.M.; Kim, Y.; Kang, S.; Sun, W.; Naik, U.P.; Parise, L.V.; Choi, E.J. CIB1 protects against MPTP-induced neurotoxicity through inhibiting ASK1. Sci. Rep. 2017, 7, 12178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Seimiya, H.; Naito, M.; Mashima, T.; Kizaki, A.; Dan, S.; Imaizumi, M.; Ichijo, H.; Miyazono, K.; Tsuruo, T. ASK1 mediates apoptotic cell death induced by genotoxic stress. Oncogene 1999, 18, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Hatai, T.; Matsuzawa, A.; Inoshita, S.; Mochida, Y.; Kuroda, T.; Sakamaki, K.; Kuida, K.; Yonehara, S.; Ichijo, H.; Takeda, K. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 2000, 275, 26576–26581. [Google Scholar] [CrossRef]
- Faist, A.; Schloer, S.; Mecate-Zambrano, A.; Janowski, J.; Schreiber, A.; Boergeling, Y.; Conrad, B.C.G.; Kumar, S.; Toebben, L.; Schughart, K.; et al. Inhibition of p38 signaling curtails the SARS-CoV-2 induced inflammatory response but retains the IFN-dependent antiviral defense of the lung epithelial barrier. Antivir. Res. 2023, 209, 105475. [Google Scholar] [CrossRef]
- He, S.F.; Wang, W.; Ren, H.; Zhao, L.J.; Qi, Z.T. Interferon alpha and ribavirin collaboratively regulate p38 mitogen-activated protein kinase signaling in hepatoma cells. Cytokine 2013, 61, 801–807. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Castedo, M.; Nardacci, R.; Ciccosanti, F.; Boya, P.; Roumier, T.; Larochette, N.; Piacentini, M.; Kroemer, G. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J. Exp. Med. 2005, 201, 279–289. [Google Scholar] [CrossRef]
- Fujisawa, T.; Takahashi, M.; Tsukamoto, Y.; Yamaguchi, N.; Nakoji, M.; Endo, M.; Kodaira, H.; Hayashi, Y.; Nishitoh, H.; Naguro, I.; et al. The ASK1-specific inhibitors K811 and K812 prolong survival in a mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2016, 25, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Arning, L.; Monte, D.; Hansen, W.; Wieczorek, S.; Jagiello, P.; Akkad, D.A.; Andrich, J.; Kraus, P.H.; Saft, C.; Epplen, J.T. ASK1 and MAP2K6 as modifiers of age at onset in Huntington’s disease. J. Mol. Med. 2008, 86, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Lee, B.I.; Cheon, S.Y.; Kim, H.W.; Kim, H.J.; Kim, G.W. Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease. Neuroscience 2009, 163, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, H.; Zhang, B.; Xiong, R.; Zhang, Y.; Kang, W.Y.; Chen, W.; Zhao, Z.B.; Chen, S.D. Small peptide inhibitor of JNK3 protects dopaminergic neurons from MPTP induced injury via inhibiting the ASK1-JNK3 signaling pathway. PLoS ONE 2015, 10, e0119204. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Woo, J.M.; Im, J.Y.; Park, E.S.; He, L.; Ichijo, H.; Junn, E.; Mouradian, M.M. Apoptosis signal-regulating kinase 1 modulates the phenotype of alpha-synuclein transgenic mice. Neurobiol. Aging 2015, 36, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, H.; Wu, B. RIPK1 inhibitor ameliorates the MPP(+)/MPTP-induced Parkinson’s disease through the ASK1/JNK signalling pathway. Brain Res. 2021, 1757, 147310. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Toyama, K.; Uekawa, K.; Ichijo, H.; Kim-Mitsuyama, S. Role of ASK1/p38 Cascade in a Mouse Model of Alzheimer’s Disease and Brain Aging. J. Alzheimers Dis. 2018, 61, 259–263. [Google Scholar] [CrossRef]
- Arefian, H.; Heublein, S.; Scherag, A.; Brunkhorst, F.M.; Younis, M.Z.; Moerer, O.; Fischer, D.; Hartmann, M. Hospital-related cost of sepsis: A systematic review. J. Infect. 2017, 74, 107–117. [Google Scholar] [CrossRef]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit. Care Med. 2017, 45, 486–552. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.R.; Lei, C.Q. TAK1-TABs Complex: A Central Signalosome in Inflammatory Responses. Front. Immunol. 2020, 11, 608976. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.; van Vught, L.A.; Scicluna, B.P.; Wiewel, M.A.; Klein Klouwenberg, P.M.; Hoogendijk, A.J.; Ong, D.S.; Cremer, O.L.; Horn, J.; Franitza, M.; et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood 2016, 127, 3062–3072. [Google Scholar] [CrossRef] [PubMed]
- Arman, M.; Krauel, K.; Tilley, D.O.; Weber, C.; Cox, D.; Greinacher, A.; Kerrigan, S.W.; Watson, S.P. Amplification of bacteria-induced platelet activation is triggered by FcgammaRIIA, integrin alphaIIbbeta3, and platelet factor 4. Blood 2014, 123, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Shannon, O.; Hertzen, E.; Norrby-Teglund, A.; Morgelin, M.; Sjobring, U.; Bjorck, L. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol. Microbiol. 2007, 65, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Svensson, L.; Roach, M.; Hambor, J.; McNeish, J.; Gabel, C.A. Deficiency of the stress kinase p38alpha results in embryonic lethality: Characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J. Exp. Med. 2000, 191, 859–870. [Google Scholar] [CrossRef]
- Sakurai, K.; Matsuo, Y.; Sudo, T.; Takuwa, Y.; Kimura, S.; Kasuya, Y. Role of p38 mitogen-activated protein kinase in thrombus formation. J. Recept. Signal Transduct. Res. 2004, 24, 283–296. [Google Scholar] [CrossRef]
- Clark, J.D.; Lin, L.L.; Kriz, R.W.; Ramesha, C.S.; Sultzman, L.A.; Lin, A.Y.; Milona, N.; Knopf, J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell 1991, 65, 1043–1051. [Google Scholar] [CrossRef]
- Suurmond, J.; Zou, Y.R.; Kim, S.J.; Diamond, B. Therapeutics to block autoantibody initiation and propagation in systemic lupus erythematosus and rheumatoid arthritis. Sci. Transl. Med. 2015, 7, 280ps5. [Google Scholar] [CrossRef]
- Watson, C.N.; Kerrigan, S.W.; Cox, D.; Henderson, I.R.; Watson, S.P.; Arman, M. Human platelet activation by Escherichia coli: Roles for FcgammaRIIA and integrin alphaIIbbeta3. Platelets 2016, 27, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Rauova, L.; Zhai, L.; Kowalska, M.A.; Arepally, G.M.; Cines, D.B.; Poncz, M. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: Diagnostic and therapeutic implications. Blood 2006, 107, 2346–2353. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.; Kreuziger, L.B.; Rein, L.; Kleman, A.; Fraser, R.; Aster, R.H.; Hari, P.; Padmanabhan, A. Disease burden, complication rates, and health-care costs of heparin-induced thrombocytopenia in the USA: A population-based study. Lancet Haematol. 2018, 5, e220–e231. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.S.; Pasquet, J.M.; Hers, I.; Cornall, R.; Knight, G.; Barnes, M.; Hibbs, M.L.; Dunn, A.R.; Lowell, C.A.; Watson, S.P. Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood 2000, 96, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, J.M.; Okuma, M.; Farndale, R.; Barnes, M.; Watson, S.P. Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor gamma-chain. FEBS Lett. 1997, 413, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.P.; Gibbins, J. Collagen receptor signalling in platelets: Extending the role of the ITAM. Immunol. Today 1998, 19, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Nakajima, H.; Suzuki, H.; Oda, A.; Matsubara, Y.; Moroi, M.; Terauchi, Y.; Kadowaki, T.; Suzuki, H.; Koyasu, S.; et al. Functional phenotype of phosphoinositide 3-kinase p85alpha-null platelets characterized by an impaired response to GP VI stimulation. Blood 2003, 102, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Mangin, P.; Dangelmaier, C.; Lillian, R.; Jackson, S.P.; Daniel, J.L.; Kunapuli, S.P. Role of phosphoinositide 3-kinase beta in glycoprotein VI-mediated Akt activation in platelets. J. Biol. Chem. 2009, 284, 33763–33772. [Google Scholar] [CrossRef]
- McKenzie, S.E.; Taylor, S.M.; Malladi, P.; Yuhan, H.; Cassel, D.L.; Chien, P.; Schwartz, E.; Schreiber, A.D.; Surrey, S.; Reilly, M.P. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: A transgenic mouse model. J. Immunol. 1999, 162, 4311–4318. [Google Scholar] [CrossRef]
- Huynh, A.; Arnold, D.M.; Michael, J.V.; Clare, R.; Smith, J.W.; Daka, M.; Ianosi-Irimie, M.; McKenzie, S.E.; Kelton, J.G.; Nazy, I. Characteristics of VITT antibodies in patients vaccinated with Ad26.COV2.S. Blood Adv. 2023, 7, 246–250. [Google Scholar] [CrossRef]
- Reilly, M.P.; Taylor, S.M.; Hartman, N.K.; Arepally, G.M.; Sachais, B.S.; Cines, D.B.; Poncz, M.; McKenzie, S.E. Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcgammaRIIA. Blood 2001, 98, 2442–2447. [Google Scholar] [CrossRef]
- Iba, T.; Levi, M.; Levy, J.H. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 2020, 46, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Lin, Y.; Tang, S.; Yu, L.; Zhang, H.; Wan, T.; Luhn, T.; Fu, H.; Chen, H. AIP1 recruits phosphatase PP2A to ASK1 in tumor necrosis factor-induced ASK1-JNK activation. Circ. Res. 2008, 102, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Tan, R.; Ye, B.; Yan, S.; Sui, M.; Zhao, W.; Zhang, L.; Zhu, Y.; Zeng, L. SHP-1 inhibits renal ischemia reperfusion injury via dephosphorylating ASK1 and suppressing apoptosis. Biochem. Biophys. Res. Commun. 2019, 513, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Korporaal, S.J.; Koekman, C.A.; Verhoef, S.; van der Wal, D.E.; Bezemer, M.; Van Eck, M.; Akkerman, J.W. Downregulation of platelet responsiveness upon contact with LDL by the protein-tyrosine phosphatases SHP-1 and SHP-2. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Makhoul, S.; Kumm, E.; Zhang, P.; Walter, U.; Jurk, K. The Serine/Threonine Protein Phosphatase 2A (PP2A) Regulates Syk Activity in Human Platelets. Int. J. Mol. Sci. 2020, 21, 8939. [Google Scholar] [CrossRef] [PubMed]
- Moscardo, A.; Valles, J.; Pinon, M.; Aznar, J.; Martinez-Sales, V.; Santos, M.T. Regulation of cytosolic PlA2 activity by PP1/PP2A serine/threonine phosphatases in human platelets. Platelets 2006, 17, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Sledz, K.M.; Moore, S.F.; Vijayaragavan, V.; Mallah, S.; Goudswaard, L.J.; Williams, C.M.; Hunter, R.W.; Hers, I. Redundant role of ASK1-mediated p38MAPK activation in human platelet function. Cell. Signal. 2020, 68, 109528. [Google Scholar] [CrossRef]
- Reines, H.D.; Halushka, P.V.; Cook, J.A.; Wise, W.C.; Rambo, W. Plasma thromboxane concentrations are raised in patients dying with septic shock. Lancet 1982, 2, 174–175. [Google Scholar] [CrossRef]
- Stolla, M.; Stefanini, L.; Andre, P.; Ouellette, T.D.; Reilly, M.P.; McKenzie, S.E.; Bergmeier, W. CalDAG-GEFI deficiency protects mice in a novel model of Fcgamma RIIA-mediated thrombosis and thrombocytopenia. Blood 2011, 118, 1113–1120. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostyak, J.C.; McKenzie, S.E.; Naik, U.P. The Function of ASK1 in Sepsis and Stress-Induced Disorders. Int. J. Mol. Sci. 2024, 25, 213. https://doi.org/10.3390/ijms25010213
Kostyak JC, McKenzie SE, Naik UP. The Function of ASK1 in Sepsis and Stress-Induced Disorders. International Journal of Molecular Sciences. 2024; 25(1):213. https://doi.org/10.3390/ijms25010213
Chicago/Turabian StyleKostyak, John C., Steven E. McKenzie, and Ulhas P. Naik. 2024. "The Function of ASK1 in Sepsis and Stress-Induced Disorders" International Journal of Molecular Sciences 25, no. 1: 213. https://doi.org/10.3390/ijms25010213
APA StyleKostyak, J. C., McKenzie, S. E., & Naik, U. P. (2024). The Function of ASK1 in Sepsis and Stress-Induced Disorders. International Journal of Molecular Sciences, 25(1), 213. https://doi.org/10.3390/ijms25010213