
Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer



Abstract
:1. Introduction
2. Genetic Mutation Patterns in Sporadic CRC and CAC
3. Role of Inflammation and Immune System in CRC Oncogenesis
3.1. Inflammation Preceding Carcinogenesis
3.2. Tumor-Associated Inflammation
3.3. Inflammation Induced by CRC Treatment
4. G-Protein Coupled Receptors
5. Selected GPCR in IBD and Their Connections to Carcinogenesis
6. Regulators of G-Protein Signaling and AXIN
7. RGS in IBD, CAC and Sporadic CRC Carcinogenesis
7.1. R4 Family: RGS1, RGS2, RGS4, RGS13 and RGS16
7.2. R7 Family: RGS6, RGS7, RGS9-2 and RGS11
7.3. R12 Family—RGS10
7.4. RZ Family: RGS17, RGS19, RGS20
7.5. Atypical RGS—AXIN
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society Key Statistics for Colorectal Cancer. Available online: https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html (accessed on 31 October 2023).
- International Agency for Research on Cancer; World Health Organization. CANCER TODAY. Data Visualization Tools for Exploring the Global Cancer Burden in 2020. Available online: https://gco.iarc.fr/today/home (accessed on 31 October 2023).
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, M.; Szymaszkiewicz, A.; Jacenik, D.; Schodel, L.; Sałaga, M.; Zatorski, H.; Kordek, R.; Becker, C.; Krajewska, W.M.; Fichna, J. Cyclic Derivative of Morphiceptin Dmt-Cyclo-(D-Lys-Phe-D-Pro-Asp)-NH2(P-317), a Mixed Agonist of MOP and KOP Opioid Receptors, Exerts Anti-Inflammatory and Anti-Tumor Activity in Colitis and Colitis-Associated Colorectal Cancer in Mice. Eur. J. Pharmacol. 2020, 885, 173463. [Google Scholar] [CrossRef] [PubMed]
- Andersen, N.N.; Jess, T. Has the Risk of Colorectal Cancer in Inflammatory Bowel Disease Decreased? World J. Gastroenterol. 2013, 19, 7561–7568. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; Depinho, R.A. Genetic and Biological Hallmarks of Colorectal Cancer. Genes Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. WNT Signaling and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Świerczyński, M.; Szymaszkiewicz, A.; Fichna, J.; Zielińska, M. New Insights into Molecular Pathways in Colorectal Cancer: Adiponectin, Interleukin-6 and Opioid Signaling. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2021, 1875, 188460. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal Cancer and Dysplasia in Inflammatory Bowel Disease: A Review of Disease Epidemiology, Pathophysiology, and Management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef]
- Schmitt, M.; Greten, F.R. The Inflammatory Pathogenesis of Colorectal Cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121. [Google Scholar] [CrossRef]
- Świerczyński, M.; Fichna, J. Inflamasom Jako Czynnik Sprawczy i Ochronny w Patogenezie Nieswoistych Chorób Zapalnych Jelit. Postep. Biochem. 2021, 67, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B. Inflammatory Bowel Disease: Epidemiology, Pathogenesis, and Therapeutic Opportunities. Inflamm. Bowel Dis. 2006, 12, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and Risk Factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Tang, Z.-H.; Liu, S.; Guo, S.-S. Clinicopathological Significance of Overexpression of Interleukin-6 in Colorectal Cancer. World J. Gastroenterol. 2017, 23, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-Linked Barrier Defects and Microbial Products Drive IL-23/IL-17-Mediated Tumour Growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef]
- Dong, S.; Liang, S.; Cheng, Z.; Zhang, X.; Luo, L.; Li, L.; Zhang, W.; Li, S.; Xu, Q.; Zhong, M.; et al. ROS/PI3K/Akt and Wnt/β-Catenin Signalings Activate HIF-1α-Induced Metabolic Reprogramming to Impart 5-Fluorouracil Resistance in Colorectal Cancer. J. Exp. Clin. Cancer Res. 2022, 41, 15. [Google Scholar] [CrossRef]
- Roth, M.T.; Das, S. Pembrolizumab in Unresectable or Metastatic MSI-High Colorectal Cancer: Safety and Efficacy. Expert Rev. Anticancer Ther. 2021, 21, 229–238. [Google Scholar] [CrossRef]
- Hill, M.; Segovia, M.; Russo, S.; Girotti, M.R.; Rabinovich, G.A. The Paradoxical Roles of Inflammation during PD-1 Blockade in Cancer. Trends Immunol. 2020, 41, 982–993. [Google Scholar] [CrossRef]
- Lee, L.; Gupta, M.; Sahasranaman, S. Immune Checkpoint Inhibitors: An Introduction to the next-Generation Cancer Immunotherapy. J. Clin. Pharmacol. 2016, 56, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. An Overview on GPCRs and Drug Discovery: Structure-Based Drug Design and Structural Biology on GPCRs. In G Protein-Coupled Receptors in Drug Discovery; Leifert, W.R., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 51–66. ISBN 978-1-60327-317-6. [Google Scholar]
- Kobilka, B.K. G Protein Coupled Receptor Structure and Activation. Biochim. Biophys. Acta 2007, 1768, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Sałaga, M.; Storr, M.A.; Fichna, J. Physiology, Signaling, and Pharmacology of Opioid Receptors and Their Ligands in the Gastrointestinal Tract: Current Concepts and Future Perspectives. J. Gastroenterol. 2014, 49, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Balasubramanian, S.; Sumandeep, S.; Charboneau, R.; Wang, J.; Melnyk, D.; Beilman, G.J.; Vatassery, R.; Barke, R.A. Morphine Directs T Cells toward T(H2) Differentiation. Surgery 2001, 130, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Plein, L.M.; Rittner, H.L. Opioids and the Immune System—Friend or Foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef]
- Ninković, J.; Roy, S. Role of the Mu-Opioid Receptor in Opioid Modulation of Immune Function. Amino Acids 2013, 45, 9–24. [Google Scholar] [CrossRef]
- Pol, O.; Palacio, J.R.; Puig, M.M. The Expression of Delta- and Kappa-Opioid Receptor Is Enhanced during Intestinal Inflammation in Mice. J. Pharmacol. Exp. Ther. 2003, 306, 455–462. [Google Scholar] [CrossRef]
- Salaga, M.; Storr, M.; Martemyanov, K.A.; Fichna, J. RGS Proteins as Targets in the Treatment of Intestinal Inflammation and Visceral Pain: New Insights and Future Perspectives. BioEssays 2016, 38, 344–354. [Google Scholar] [CrossRef]
- Sacerdote, P. Effects of in Vitro and in Vivo Opioids on the Production of IL-12 and IL-10 by Murine Macrophages. Ann. N. Y. Acad. Sci. 2003, 992, 129–140. [Google Scholar] [CrossRef]
- Walker, J.S. Anti-Inflammatory Effects of Opioids. Adv. Exp. Med. Biol. 2003, 521, 148–160. [Google Scholar] [PubMed]
- Keränen, U.; Kiviluoto, T.; Järvinen, H.; Bäck, N.; Kivilaakso, E.; Soinila, S. Changes in Substance P-Immunoreactive Innervation of Human Colon Associated with Ulcerative Colitis. Dig. Dis. Sci. 1995, 40, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Valaiyaduppu Subas, S.; Ghani, M.R.; Busa, V.; Dardeir, A.; Marudhai, S.; Cancarevic, I. Role of Substance P in the Pathophysiology of Inflammatory Bowel Disease and Its Correlation With the Degree of Inflammation. Cureus 2020, 12, e11027. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Nagase, H.; Okuzumi, A.; Nishiyama, C. Delta Opioid Receptor Agonists Ameliorate Colonic Inflammation by Modulating Immune Responses. Front. Immunol. 2021, 12, 730706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rajput, A.; Jin, N.; Wang, J. Mechanisms of Immunosuppression in Colorectal Cancer. Cancers 2020, 12, 3850. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, X.; Wang, J.; Sun, P.; Weng, M.; Chen, W.; Sun, Z.; Zhu, M.; Miao, C. Nalmefene Attenuates Malignant Potential in Colorectal Cancer Cell via Inhibition of Opioid Receptor. Acta Biochim. Biophys. Sin. 2018, 50, 156–163. [Google Scholar] [CrossRef]
- Howlett, A.C. The Cannabinoid Receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 619–631. [Google Scholar] [CrossRef]
- Singh, U.P.; Singh, N.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Cannabinoid Receptor-2 (CB2) Agonist Ameliorates Colitis in IL-10(-/-) Mice by Attenuating the Activation of T Cells and Promoting Their Apoptosis. Toxicol. Appl. Pharmacol. 2012, 258, 256–267. [Google Scholar] [CrossRef]
- Massa, F.; Marsicano, G.; Hermann, H.; Cannich, A.; Monory, K.; Cravatt, B.F.; Ferri, G.-L.; Sibaev, A.; Storr, M.; Lutz, B. The Endogenous Cannabinoid System Protects against Colonic Inflammation. J. Clin. Investig. 2004, 113, 1202–1209. [Google Scholar] [CrossRef]
- D’Argenio, G.; Valenti, M.; Scaglione, G.; Cosenza, V.; Sorrentini, I.; Di Marzo, V. Up-Regulation of Anandamide Levels as an Endogenous Mechanism and a Pharmacological Strategy to Limit Colon Inflammation. FASEB J. 2006, 20, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Sałaga, M.; Mokrowiecka, A.; Zakrzewski, P.K.; Cygankiewicz, A.; Leishman, E.; Sobczak, M.; Zatorski, H.; Małecka-Panas, E.; Kordek, R.; Storr, M.; et al. Experimental Colitis in Mice Is Attenuated by Changes in the Levels of Endocannabinoid Metabolites Induced by Selective Inhibition of Fatty Acid Amide Hydrolase (FAAH). J. Crohns Colitis 2014, 8, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Ciaglia, E.; Gangemi, G.; Gazzerro, P.; Laezza, C.; Bifulco, M. Update on the Endocannabinoid System as an Anticancer Target. Expert Opin. Ther. Targets 2011, 15, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; DuBois, R.N. Loss of Cannabinoid Receptor 1 Accelerates Intestinal Tumor Growth. Cancer Res. 2008, 68, 6468–6476. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased Endocannabinoid Levels Reduce the Development of Precancerous Lesions in the Mouse Colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol Lipase Regulates a Fatty Acid Network That Promotes Cancer Pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef]
- Roberto, D.; Klotz, L.H.; Venkateswaran, V. Cannabinoid WIN 55,212-2 Induces Cell Cycle Arrest and Apoptosis, and Inhibits Proliferation, Migration, Invasion, and Tumor Growth in Prostate Cancer in a Cannabinoid-Receptor 2 Dependent Manner. Prostate 2019, 79, 151–159. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Roth, B.L. Structure and Function of Serotonin G Protein-Coupled Receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef]
- Salaga, M.; Binienda, A.; Piscitelli, F.; Mokrowiecka, A.; Cygankiewicz, A.I.; Verde, R.; Malecka-Panas, E.; Kordek, R.; Krajewska, W.M.; Di Marzo, V.; et al. Systemic Administration of Serotonin Exacerbates Abdominal Pain and Colitis via Interaction with the Endocannabinoid System. Biochem. Pharmacol. 2019, 161, 37–51. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, F. Lower Level of Bacteroides in the Gut Microbiota Is Associated with Inflammatory Bowel Disease: A Meta-Analysis. Biomed Res. Int. 2016, 5828959. [Google Scholar] [CrossRef] [PubMed]
- Margolis, K.G.; Stevanovic, K.; Li, Z.; Yang, Q.M.; Oravecz, T.; Zambrowicz, B.; Jhaver, K.G.; Diacou, A.; Gershon, M.D. Pharmacological Reduction of Mucosal but Not Neuronal Serotonin Opposes Inflammation in Mouse Intestine. Gut 2014, 63, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Wang, H.; Denou, E.; Ghia, J.-E.; Rossi, L.; Fontes, M.E.; Bernier, S.P.; Shajib, M.S.; Banskota, S.; Collins, S.M.; et al. Modulation of Gut Microbiota Composition by Serotonin Signaling Influences Intestinal Immune Response and Susceptibility to Colitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 709–728. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Gustafsson, B.I.; Drozdov, I.; Modlin, I.M. IL1beta- and LPS-Induced Serotonin Secretion Is Increased in EC Cells Derived from Crohn’s Disease. Neurogastroenterol. Motil. 2009, 21, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Manzella, C.R.; Jayawardena, D.; Pagani, W.; Li, Y.; Alrefai, W.A.; Bauer, J.; Jung, B.; Weber, C.R.; Gill, R.K. Serum Serotonin Differentiates Between Disease Activity States in Crohn’s Patients. Inflamm. Bowel Dis. 2020, 26, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Tryptophan Metabolites Modulate Inflammatory Bowel Disease and Colorectal Cancer by Affecting Immune System. Int. Rev. Immunol. 2022, 41, 326–345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Lu, T.; Chen, Z.; Liu, B.; Fan, D.; Li, C.; Wu, J.; He, L.; Zhu, X.; Du, Y.; et al. 5-Hydroxytryptamine Produced by Enteric Serotonergic Neurons Initiates Colorectal Cancer Stem Cell Self-Renewal and Tumorigenesis. Neuron 2022, 110, 2268–2282.e4. [Google Scholar] [CrossRef]
- Ahlers-Dannen, K.E.; Alqinyah, M.; Bodle, C.; Bou Dagher, J.; Chakravarti, B.; Choudhuri, S.P.; Druey, K.M.; Fisher, R.A.; Gerber, K.J.; Hepler, J.R.; et al. Regulators of G Protein Signaling (RGS) Proteins in GtoPdb v.2021.2. IUPHAR/BPS Guide Pharmacol. CITE 2021, 2020. [Google Scholar] [CrossRef]
- Verstockt, B.; Verstockt, S.; Veny, M.; Dehairs, J.; Arnauts, K.; Van Assche, G.; De Hertogh, G.; Vermeire, S.; Salas, A.; Ferrante, M. Expression Levels of 4 Genes in Colon Tissue Might Be Used to Predict Which Patients Will Enter Endoscopic Remission After Vedolizumab Therapy for Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 1142–1151.e10. [Google Scholar] [CrossRef]
- Zhu, F.; Qin, Y.; Wang, Y.; Zhang, F.; Xu, Z.; Dai, F.; Chu, W.; Wang, Y.; Zhou, G. Critical Roles of RGS16 in the Mucosal Inflammation of Ulcerative Colitis. Eur. J. Gastroenterol. Hepatol. 2022, 34, 993–999. [Google Scholar] [CrossRef]
- Alam, N.A.; Gorman, P.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.M.; Ratnavel, R.; Murdoch, M.E.; Houlston, R.S.; Aaltonen, L.A.; Roylance, R.R.; et al. Germline Deletions of EXO1 Do Not Cause Colorectal Tumors and Lesions Which Are Null for EXO1 Do Not Have Microsatellite Instability. Cancer Genet. Cytogenet. 2003, 147, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Houser, M.C.; Caudle, W.M.; Chang, J.; Kannarkat, G.T.; Yang, Y.; Kelly, S.D.; Oliver, D.; Joers, V.; Shannon, K.M.; Keshavarzian, A.; et al. Experimental Colitis Promotes Sustained, Sex-Dependent, T-Cell-Associated Neuroinflammation and Parkinsonian Neuropathology. Acta Neuropathol. Commun. 2021, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kakuta, Y.; Naito, T.; Takagawa, T.; Hanai, H.; Araki, H.; Sasaki, Y.; Sakuraba, H.; Sasaki, M.; Hisamatsu, T.; et al. Genetic Background of Mesalamine-Induced Fever and Diarrhea in Japanese Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Dhukhwa, A.; Al Aameri, R.F.H.; Sheth, S.; Mukherjea, D.; Rybak, L.; Ramkumar, V. Regulator of G Protein Signaling 17 Represents a Novel Target for Treating Cisplatin Induced Hearing Loss. Sci. Rep. 2021, 11, 8116. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Jerchow, B.-A.; Würtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kühl, M.; Wedlich, D.; Birchmeier, W. Functional Interaction of an Axin Homolog, Conductin, with β-Catenin, APC, and GSK3β. Science 1998, 280, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Li, Z.; Guo, L.; Ye, C.; Li, J.; Yu, X.; Yang, H.; Wang, Y.; Chen, C.; Zhang, D.; et al. Regulator of G Protein Signaling Proteins Differentially Modulate Signaling of Mu and Delta Opioid Receptors. Eur. J. Pharmacol. 2007, 565, 45–53. [Google Scholar] [CrossRef]
- Kim, Y.; Ghil, S. Regulators of G-Protein Signaling, RGS2 and RGS4, Inhibit Protease-Activated Receptor 4-Mediated Signaling by Forming a Complex with the Receptor and Gα in Live Cells. Cell Commun. Signal. 2020, 18, 86. [Google Scholar] [CrossRef]
- Hu, W.; Li, F.; Mahavadi, S.; Murthy, K.S. Upregulation of RGS4 Expression by IL-1beta in Colonic Smooth Muscle Is Enhanced by ERK1/2 and P38 MAPK and Inhibited by the PI3K/Akt/GSK3beta Pathway. Am. J. Physiol. Cell Physiol. 2009, 296, C1310–C1320. [Google Scholar] [CrossRef]
- Georgoussi, Z.; Leontiadis, L.; Mazarakou, G.; Merkouris, M.; Hyde, K.; Hamm, H. Selective Interactions between G Protein Subunits and RGS4 with the C-Terminal Domains of the μ- and δ-Opioid Receptors Regulate Opioid Receptor Signaling. Cell Signal. 2006, 18, 771–782. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Woo, J.; Park, J.O.; Choi, E.J.; Shin, H.S.; Roh, D.H.; Kim, K.S. Intrathecal RGS4 Inhibitor, CCG50014, Reduces Nociceptive Responses and Enhances Opioid-Mediated Analgesic Effects in the Mouse Formalin Test. Anesth. Analg. 2015, 120, 671–677. [Google Scholar] [CrossRef]
- Sutor, S.; Heilmann, J.; Seifert, R. Impact of Fusion to Gα(I2) and Co-Expression with RGS Proteins on Pharmacological Properties of Human Cannabinoid Receptors CB1R and CB2R. J. Pharm. Pharmacol. 2011, 63, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Li, F.; Mahavadi, S.; Murthy, K.S. Interleukin-1β up-Regulates RGS4 through the Canonical IKK2/IκBα/NF-ΚB Pathway in Rabbit Colonic Smooth Muscle. Biochem. J. 2008, 412, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, P.; Zhang, C.; Lee, S.; Wang, W.; Zou, H. PAR4 Overexpression Promotes Colorectal Cancer Cell Proliferation and Migration. Oncol. Lett. 2018, 16, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, Z.; Xu, Y.; Wang, B.; Huang, W.; Cai, S. Analysis of RGS2 Expression and Prognostic Significance in Stage II and III Colorectal Cancer. Biosci. Rep. 2010, 30, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Weisshaar, N.; Wu, J.; Ming, Y.; Madi, A.; Hotz-Wagenblatt, A.; Ma, S.; Mieg, A.; Hering, M.; Zettl, F.; Mohr, K.; et al. Rgs16 Promotes Antitumor CD8+ T Cell Exhaustion. Sci. Immunol. 2022, 7, eabh1873. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef]
- Miyoshi, N.; Ishii, H.; Sekimoto, M.; Doki, Y.; Mori, M. RGS16 Is a Marker for Prognosis in Colorectal Cancer. Ann. Surg. Oncol. 2009, 16, 3507–3514. [Google Scholar] [CrossRef]
- Hwang, I.-Y.; Hwang, K.-S.; Park, C.; Harrison, K.A.; Kehrl, J.H. Rgs13 Constrains Early B Cell Responses and Limits Germinal Center Sizes. PLoS ONE 2013, 8, e60139. [Google Scholar] [CrossRef]
- Luo, Y.; Qin, S.L.; Yu, M.H.; Mu, Y.F.; Wang, Z.S.; Zhong, M. Prognostic Value of Regulator of G-Protein Signaling 6 in Colorectal Cancer. Biomed. Pharmacother. 2015, 76, 147–152. [Google Scholar] [CrossRef]
- Huang, J.; Stewart, A.; Maity, B.; Hagen, J.; Fagan, R.L.; Yang, J.; Quelle, D.E.; Brenner, C.; Fisher, R.A. RGS6 Suppresses Ras-Induced Cellular Transformation by Facilitating Tip60-Mediated Dnmt1 Degradation and Promoting Apoptosis. Oncogene 2014, 33, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Saeed, O.; Lopez-Beltran, A.; Fisher, K.W.; Scarpelli, M.; Montironi, R.; Cimadamore, A.; Massari, F.; Santoni, M.; Cheng, L. RAS Genes in Colorectal Carcinoma: Pathogenesis, Testing Guidelines and Treatment Implications. J. Clin. Pathol. 2019, 72, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cardús, A.; Martinez-Balibrea, E.; Bandrés, E.; Malumbres, R.; Ginés, A.; Manzano, J.L.; Taron, M.; Garcia-Foncillas, J.; Abad, A. Pharmacogenomic Approach for the Identification of Novel Determinants of Acquired Resistance to Oxaliplatin in Colorectal Cancer. Mol. Cancer Ther. 2009, 8, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-F.; Huang, Y.-Q.; Wu, K.-M.; Jou, A.F.-J.; Shih, N.-Y.; Ho, J.-A.A. Diagnosing the RGS11 Lung Cancer Biomarker: The Integration of Competitive Immunoassay and Isothermal Nucleic Acid Exponential Amplification Reaction. Anal. Chem. 2019, 91, 3327–3335. [Google Scholar] [CrossRef]
- Ghavami, A.; Hunt, R.A.; Olsen, M.A.; Zhang, J.; Smith, D.L.; Kalgaonkar, S.; Rahman, Z.; Young, K.H. Differential Effects of Regulator of G Protein Signaling (RGS) Proteins on Serotonin 5-HT1A, 5-HT2A, and Dopamine D2 Receptor-Mediated Signaling and Adenylyl Cyclase Activity. Cell Signal. 2004, 16, 711–721. [Google Scholar] [CrossRef]
- Caldiran, F.Y.; Cacan, E. RGS10 Suppression by DNA Methylation Is Associated with Low Survival Rates in Colorectal Carcinoma. Pathol. Res. Pract. 2022, 236, 154007. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; de la Torre-Madrid, E.; Sánchez-Blázquez, P.; Wang, J.B.; Garzón, J. NMDAR-NNOS Generated Zinc Recruits PKCgamma to the HINT1-RGS17 Complex Bound to the C Terminus of Mu-Opioid Receptors. Cell Signal. 2008, 20, 1855–1864. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Y.; Tso, P.H.; Wong, Y.H. Regulator of G Protein Signaling 19 Suppresses Ras-Induced Neoplastic Transformation and Tumorigenesis. Cancer Lett. 2013, 339, 33–41. [Google Scholar] [CrossRef]
- Katoh, M. Functional Proteomics, Human Genetics and Cancer Biology of GIPC Family Members. Exp. Mol. Med. 2013, 45, e26. [Google Scholar] [CrossRef]
- Gao, H.; Ma, L.; Zou, Q.; Hu, B.; Cai, K.; Sun, Y.; Lu, L.; Ren, D. Unraveling Dynamic Interactions between Tumor-Associated Macrophages and Consensus Molecular Subtypes in Colorectal Cancer: An Integrative Analysis of Single-Cell and Bulk RNA Transcriptome. Heliyon 2023, 9, e19224. [Google Scholar] [CrossRef]
- Yang, L.; Lee, M.M.K.; Leung, M.M.H.; Wong, Y.H. Regulator of G Protein Signaling 20 Enhances Cancer Cell Aggregation, Migration, Invasion and Adhesion. Cell Signal. 2016, 28, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Dong, X.W.; Zhao, F.; Li, C.X. MiR-203 Inhibits Cell Proliferation, Invasion, and Migration of Ovarian Cancer through Regulating RGS17. J. Biol. Regul. Homeost. Agents 2021, 35, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, L.; Lin, J.; Hu, X.; Li, B.; Xue, A.; Shen, Y.; Jiang, J.; Zhang, M.; Xie, J.; et al. Deregulation of RGS17 Expression Promotes Breast Cancer Progression. J. Cancer 2015, 6, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-Z.; Ren, L.-F. MiRNA-199 Inhibits Malignant Progression of Lung Cancer through Mediating RGS17. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3390–3400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-S.; Ma, H.-G.; Sun, F.-H.; Zhao, W.-C.; Li, G. MiR-203 Inhibits the Malignant Behavior of Prostate Cancer Cells by Targeting RGS17. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5667–5674. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhang, N.; Jiang, Y.; Huang, Y.; Lian, Y.-Y.; Liu, T.; Li, N.; Guan, G. RGS17 Inhibits Tumorigenesis and Improves 5-Fluorouracil Sensitivity in Nasopharyngeal Carcinoma. Onco Targets Ther. 2018, 11, 7591–7600. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 Variants in Gastrointestinal Cancers. Cancer Lett. 2014, 355, 1–8. [Google Scholar] [CrossRef]
- Cha, P.H.; Cho, Y.H.; Lee, S.K.; Lee, J.; Jeong, W.J.; Moon, B.S.; Yun, J.H.; Yang, J.S.; Choi, S.; Yoon, J.; et al. Small-Molecule Binding of the Axin RGS Domain Promotes β-Catenin and Ras Degradation. Nat. Chem. Biol. 2016, 12, 593–600. [Google Scholar] [CrossRef]
- Li, W.; Zhang, N.; Jin, C.; Long, M.D.; Rajabi, H.; Yasumizu, Y.; Fushimi, A.; Yamashita, N.; Hagiwara, M.; Zheng, R.; et al. MUC1-C Drives Stemness in Progression of Colitis to Colorectal Cancer. JCI Insight 2020, 5, e137112. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C in Chronic Inflammation and Carcinogenesis; Emergence as a Target for Cancer Treatment. Carcinogenesis 2020, 41, 1173–1183. [Google Scholar] [CrossRef]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The Intestinal Stem Cell Markers Bmi1 and Lgr5 Identify Two Functionally Distinct Populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A Distinct Role for Lgr5+ Stem Cells in Primary and Metastatic Colon Cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E. Epigenetic Regulation of RGS2 (Regulator of G-Protein Signaling 2) in Chemoresistant Ovarian Cancer Cells. J. Chemother. 2017, 29, 173–178. [Google Scholar] [CrossRef]
- Tso, P.H.; Yung, L.Y.; Wang, Y.; Wong, Y.H. RGS19 Stimulates Cell Proliferation by Deregulating Cell Cycle Control and Enhancing Akt Signaling. Cancer Lett. 2011, 309, 199–208. [Google Scholar] [CrossRef]
- Steffan, J.J.; Dykes, S.S.; Coleman, D.T.; Adams, L.K.; Rogers, D.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. Supporting a Role for the GTPase Rab7 in Prostate Cancer Progression. PLoS ONE 2014, 9, e87882. [Google Scholar] [CrossRef]



{kind=link}
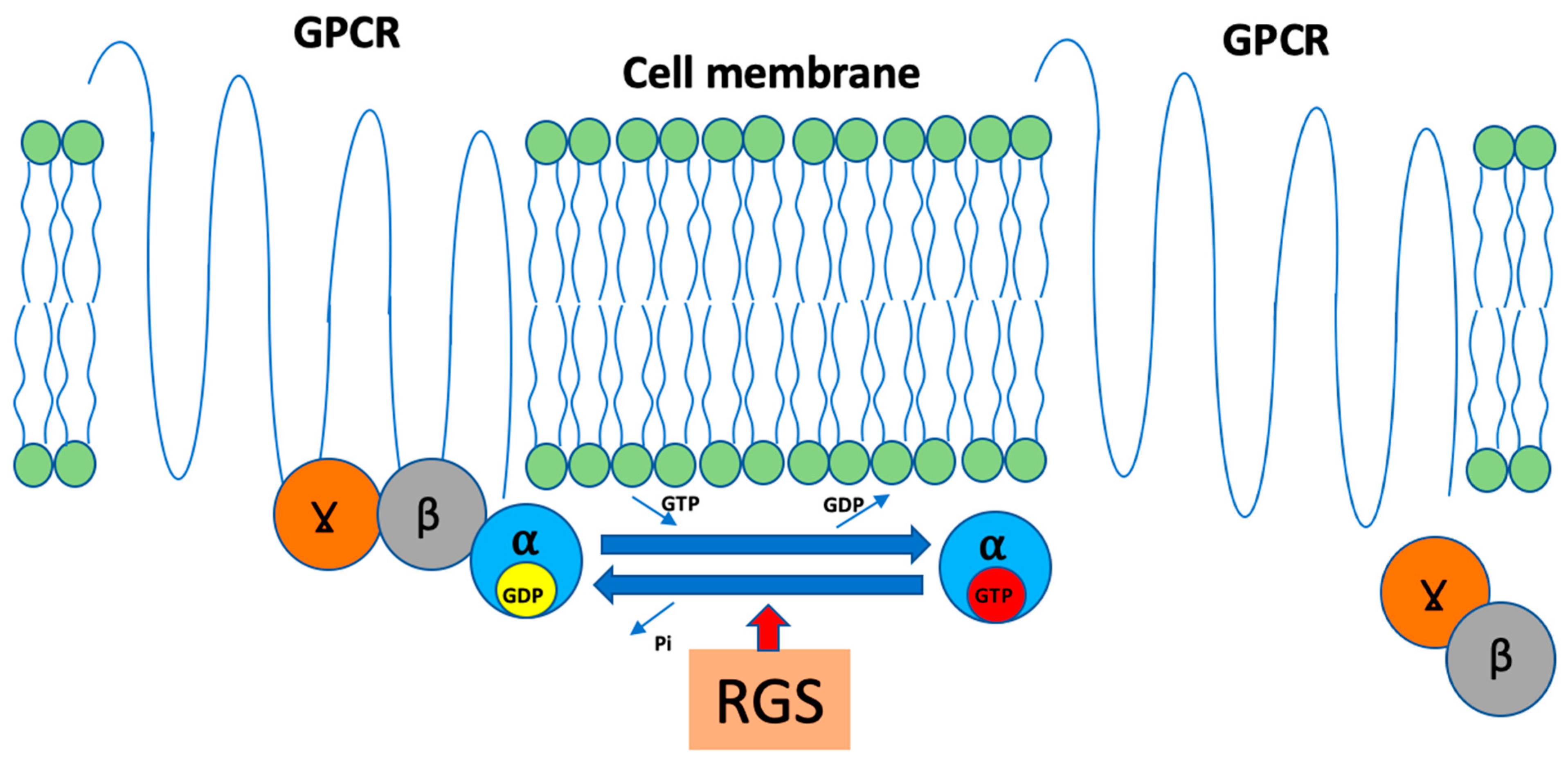{kind=link}
| Feature | UC | CD | Reference |
|---|---|---|---|
| Anatomical location | Large intestine, from left-sided colitis to pancolitis, with the possibility of backwash ileitis | Any part of the GI tract may be involved with ‘skip areas’ between inflammatory sites | [14,15,16] |
| Depth | Limited to mucosa and submucosa | The whole width of the GI tract wall can be affected by fistulae formation | |
| Immune cells phenotype and key cytokines | Th2 IL-4, IL-5, IL-10, IL-13 | Th1/Th17 IL-2, TNF, IFN-γ, IL-17, IL-23 | |
| CAC relative risk ratio | RR = 2.75 (95% CI: 1.91–3.97) Well-studied, generally considered higher than in CD | RR = 2.5 (95% CI: 1.3–4.7) Less studied, generally considered lower than in UC | [5] |
| RGS Family | Representatives | Connection with IBD and IBD-Related GPCR | References |
|---|---|---|---|
| R4 | RGS: 1, 2, 3, 4, 5, 8, 13, 16, 18, 21 | OR: RGS1, RGS2, RGS4 CB: RGS4 5-HTR: RGS4 Other links with IBD: RGS13, RGS16 | [32,62,63] [32,64] [32,65] |
| R7 | RGS: 6, 7, 9–2, 11 | OR: RGS6, RGS7, RGS11 CB: RGS7 5-HTR: RGS6 Other links with CRC: RGS7 | |
| R12 | RGS: 10, 12, 14 | OR: RGS10 5-HTR: RGS10, RGS12 Other links with IBD: RGS10 | |
| RZ | RGS: 17, 19, 20 | OR: RGS17, RGS19 RGS20 CB: RGS17 Other links with IBD: RGS17 | [32,66,67] |
| Atypical RGS | AXIN1, AXIN2 | Prevents pro-inflammatory effects of Wnt/β-catenin pathway | [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swierczynski, M.; Kasprzak, Z.; Makaro, A.; Salaga, M. Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 577. https://doi.org/10.3390/ijms25010577
Swierczynski M, Kasprzak Z, Makaro A, Salaga M. Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(1):577. https://doi.org/10.3390/ijms25010577
Chicago/Turabian StyleSwierczynski, Mikolaj, Zuzanna Kasprzak, Adam Makaro, and Maciej Salaga. 2024. "Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer" International Journal of Molecular Sciences 25, no. 1: 577. https://doi.org/10.3390/ijms25010577
APA StyleSwierczynski, M., Kasprzak, Z., Makaro, A., & Salaga, M. (2024). Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer. International Journal of Molecular Sciences, 25(1), 577. https://doi.org/10.3390/ijms25010577