
High-Density Lipoproteins at the Interface between the NLRP3 Inflammasome and Myocardial Infarction
 , , ,
, , ,  ,
,  and
and {kind=link}
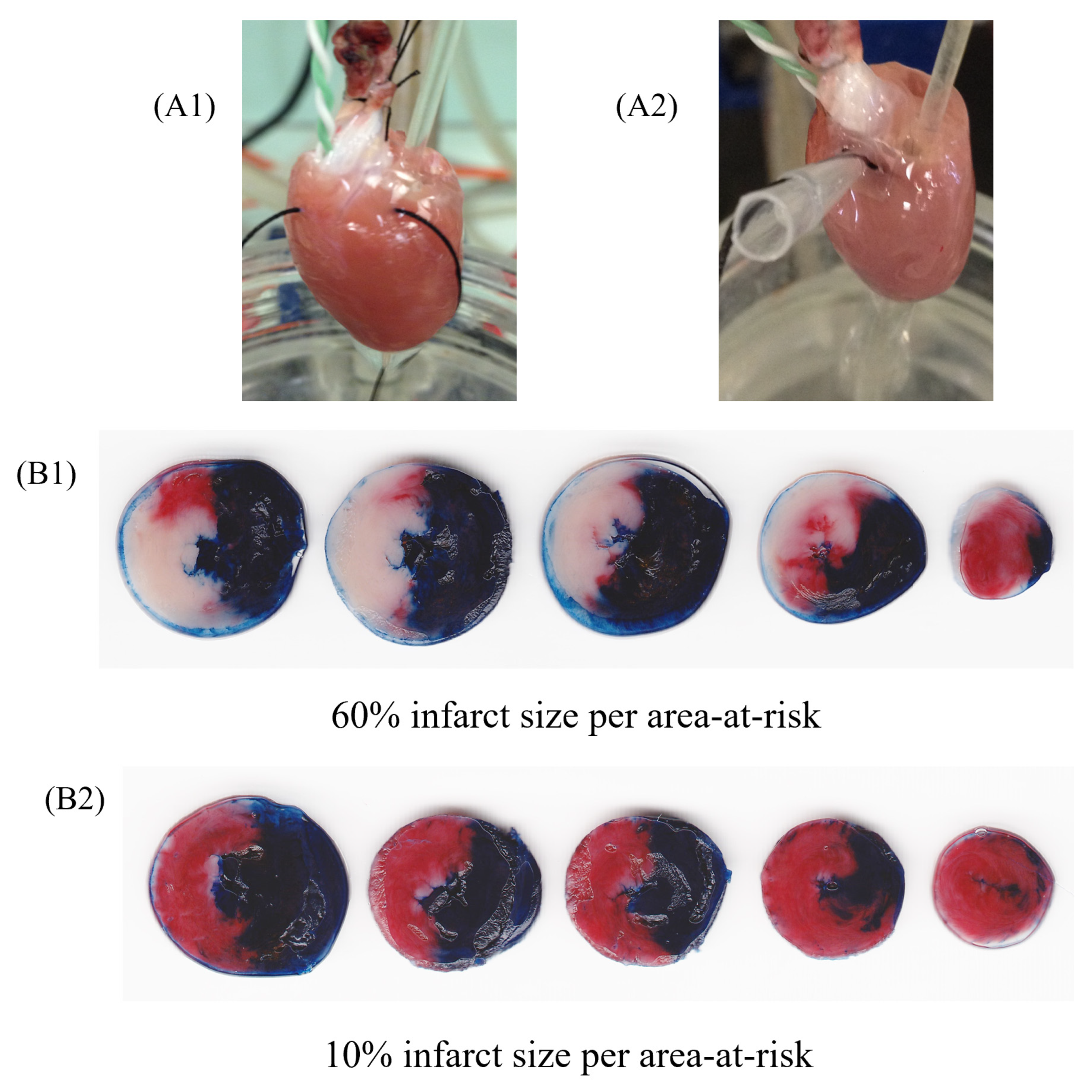{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Mechanisms Underlying the MI-Induced Activation of NLRP3 Inflammasome Signaling
3. The Mechanistic Basis of NLRP3 Inflammasome Priming and Activation
4. NLRP3 Inflammasome Activation and Pyroptosis after Coronary Reperfusion
5. The Non-Canonical Activation of Pyroptosis during MI
6. The Double-Edged Effects of HDL Particles in MI
7. The Protective Mechanisms of HDL on Cardiomyocyte Pyroptosis
8. The Molecular Changes and Pathogenic Effects Produced by HDL during MI
9. Oxidation in HDL Components during MI and Inflammasome Activation
10. HDL Lipid and Protein Composition Changes during MI and Inflammasome Activation
11. HDL microRNA Composition Change during MI
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Schelbert, E.B.; Cao, J.J.; Sigurdsson, S.; Aspelund, T.; Kellman, P.; Aletras, A.H.; Dyke, C.K.; Thorgeirsson, G.; Eiriksdottir, G.; Launer, L.J.; et al. Prevalence and prognosis of unrecognized myocardial infarction determined by cardiac magnetic resonance in older adults. JAMA 2012, 308, 890–896. [Google Scholar] [CrossRef] [PubMed]
- El Aidi, H.; Adams, A.; Moons, K.G.M.; Den Ruijter, H.M.; Mali, W.P.T.M.; Doevendans, P.A.; Nagel, E.; Schalla, S.; Bots, M.L.; Leiner, T. Cardiac Magnetic Resonance Imaging Findings and the Risk of Cardiovascular Events in Patients with Recent Myocardial Infarction or Suspected or Known Coronary Artery Disease. J. Am. Coll. Cardiol. 2014, 63, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.; Ortiz, J.T.; Tejedor, P.; Lee, D.C.; Bucciarelli-Ducci, C.; Kansal, P.; Carr, J.C.; Holly, T.A.; Lloyd-Jones, D.; Klocke, F.J.; et al. Infarct size by contrast enhanced cardiac magnetic resonance is a stronger predictor of outcomes than left ventricular ejection fraction or end-systolic volume index: Prospective cohort study. Heart 2008, 94, 730–736. [Google Scholar] [CrossRef]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship Between Infarct Size and Outcomes Following Primary PCI: Patient-Level Analysis From 10 Randomized Trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef]
- Redfors, B.; Mohebi, R.; Giustino, G.; Chen, S.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Eitel, I.; et al. Time Delay, Infarct Size, and Microvascular Obstruction After Primary Percutaneous Coronary Intervention for ST-Segment-Elevation Myocardial Infarction. Circ. Cardiovasc. Interv. 2021, 14, e009879. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. Cell-Specific Roles of NLRP3 Inflammasome in Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The IL-1 family and inflammatory diseases. Clin. Exp. Rheumatol. 2002, 20 (Suppl. S27), S1–S13. [Google Scholar] [PubMed]
- Braddock, M.; Quinn, A. Targeting IL-1 in inflammatory disease: New opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 2004, 3, 330–339. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200. [Google Scholar] [CrossRef]
- Do Carmo, H.; Arjun, S.; Petrucci, O.; Yellon, D.M.; Davidson, S.M. The Caspase 1 Inhibitor VX-765 Protects the Isolated Rat Heart via the RISK Pathway. Cardiovasc. Drugs Ther. 2018, 32, 165–168. [Google Scholar] [CrossRef]
- Frias, M.A.; Pedretti, S.; Hacking, D.; Somers, S.; Lacerda, L.; Opie, L.H.; James, R.W.; Lecour, S. HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis 2013, 228, 110–116. [Google Scholar] [CrossRef]
- Gomaraschi, M.; Calabresi, L.; Franceschini, G. Protective Effects of HDL against Ischemia/Reperfusion Injury. Front. Pharmacol. 2016, 7, 2. [Google Scholar] [CrossRef]
- Pedretti, S.; Brulhart-Meynet, M.C.; Montecucco, F.; Lecour, S.; James, R.W.; Frias, M.A. HDL protects against myocardial ischemia reperfusion injury via miR-34b and miR-337 expression which requires STAT3. PLoS ONE 2019, 14, e0218432. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.A.S.; Carvalho, L.S.F.; Bonilha, I.; Virginio, V.W.; Nadruz Junior, W.; Coelho-Filho, O.R.; e Silva, J.C.Q.; Junior, O.P.; Sposito, A.C. Adverse interaction between HDL and the mass of myocardial infarction. Atherosclerosis 2019, 281, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gardner, S.E.; Clarke, M.C. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2781–2786. [Google Scholar] [CrossRef]
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563. [Google Scholar] [CrossRef] [PubMed]
- Durrington, P.N.; Bashir, B.; Soran, H. Paraoxonase 1 and atherosclerosis. Front. Cardiovasc. Med. 2023, 10, 1065967. [Google Scholar] [CrossRef]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef]
- Land, W.G. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170. [Google Scholar]
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 2015, 4, e001993. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef]
- Bayer, A.L.; Alcaide, P. MyD88: At the heart of inflammatory signaling and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Jaen, R.I.; Val-Blasco, A.; Prieto, P.; Gil-Fernandez, M.; Smani, T.; Lopez-Sendon, J.L.; Delgado, C.; Bosca, L.; Fernandez-Velasco, M. Innate Immune Receptors, Key Actors in Cardiovascular Diseases. JACC Basic Transl. Sci. 2020, 5, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Maltez, V.I.; Miao, E.A. Reassessing the Evolutionary Importance of Inflammasomes. J. Immunol. 2016, 196, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed]
- Merkle, S.; Frantz, S.; Schon, M.P.; Bauersachs, J.; Buitrago, M.; Frost, R.J.; Schmitteckert, E.M.; Lohse, M.J.; Engelhardt, S. A role for caspase-1 in heart failure. Circ. Res. 2007, 100, 645–653. [Google Scholar] [CrossRef]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Yin, H.; Zhang, X.; Li, Y. NLRP3 Inflammasome: A Potential Alternative Therapy Target for Atherosclerosis. Evid. Based Complement. Altern. Med. 2020, 2020, 1561342. [Google Scholar] [CrossRef]
- Yoshioka, J.; Chutkow, W.A.; Lee, S.; Kim, J.B.; Yan, J.; Tian, R.; Lindsey, M.L.; Feener, E.P.; Seidman, C.E.; Seidman, J.G.; et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J. Clin. Investig. 2012, 122, 267–279. [Google Scholar] [CrossRef]
- Kinnunen, K.; Piippo, N.; Loukovaara, S.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. Lysosomal destabilization activates the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs). J. Cell Commun. Signal. 2017, 11, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; Van Tassell, B.W.; Abbate, A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox Signal. 2015, 22, 1146–1161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zeng, W.; Zhang, Y.; Yu, Q.; Zeng, M.; Gan, J.; Zhang, W.; Jiang, X.; Li, H. Focus on the role of mitochondria in NLRP3 inflammasome activation: A prospective target for the treatment of ischemic stroke (Review). Int. J. Mol. Med. 2022, 49, 74. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Dubyak, G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004, 286, C1100–C1108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, Z.; Liu, X.; Yin, H.Y.; Tang, Y.; Cao, X. P2X7 Receptor-Mediated Inflammation in Cardiovascular Disease. Front. Pharmacol. 2021, 12, 654425. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.; Liu, X.; Li, J.; Li, X.; Wang, G.; Liu, Y.; Lu, X.; Cui, L.; Shao, M.; et al. P2X7R-NEK7-NLRP3 Inflammasome Activation: A Novel Therapeutic Pathway of Qishen Granule in the Treatment of Acute Myocardial Ischemia. J. Inflamm. Res. 2022, 15, 5309–5326. [Google Scholar] [CrossRef]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Mathur, S.; Wang, Y.; Bateman, R.M.; Walley, K.R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc. Res. 2006, 72, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Song, J.; Li, W.; Yang, T.; Yue, X.; Lin, X.; Yang, X.; Luo, W.; Guo, J.; Wang, X.; et al. RhoE Fine-Tunes Inflammatory Response in Myocardial Infarction. Circulation 2019, 139, 1185–1198. [Google Scholar] [CrossRef]
- Sandanger, O.; Ranheim, T.; Vinge, L.E.; Bliksoen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, X.; Kao, R.; Mele, T.; Kvietys, P.; Martin, C.M.; Rui, T. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: The role of NLRP3 inflammasome activation. PLoS ONE 2014, 9, e107639. [Google Scholar] [CrossRef]
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, J.; et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 2016, 118, 1525–1539. [Google Scholar] [CrossRef]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575. [Google Scholar] [CrossRef]
- Rauf, A.; Shah, M.; Yellon, D.M.; Davidson, S.M. Role of Caspase 1 in Ischemia/Reperfusion Injury of the Myocardium. J. Cardiovasc. Pharmacol. 2019, 74, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases. J. Inflamm. Res. 2020, 13, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Ackermann, K.; Buss, S.; Lehmann, L.; Schnabel, P.A.; Haunstetter, A.; Katus, H.A.; Hardt, S.E. Inhibition of apoptosis by the intrinsic but not the extrinsic apoptotic pathway in myocardial ischemia-reperfusion. Cardiovasc. Pathol. 2013, 22, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Xu, Y.J.; Zheng, L.; Hu, Y.W.; Wang, Q. Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 2018, 476, 28–37. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar] [PubMed]
- Liu, X.; Lieberman, J. A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. Adv. Immunol. 2017, 135, 81–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.M.; Downey, J.M.; Cohen, M.V.; Housley, N.A.; Alvarez, D.F.; Audia, J.P. The Highly Selective Caspase-1 Inhibitor VX-765 Provides Additive Protection Against Myocardial Infarction in Rat Hearts When Combined with a Platelet Inhibitor. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26, 976. [Google Scholar] [CrossRef] [PubMed]
- Vigano, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [PubMed]
- Lagrange, B.; Benaoudia, S.; Wallet, P.; Magnotti, F.; Provost, A.; Michal, F.; Martin, A.; Di Lorenzo, F.; Py, B.F.; Molinaro, A.; et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat. Commun. 2018, 9, 242. [Google Scholar] [CrossRef]
- Yanpiset, P.; Maneechote, C.; Sriwichaiin, S.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Gasdermin D-mediated pyroptosis in myocardial ischemia and reperfusion injury: Cumulative evidence for future cardioprotective strategies. Acta Pharm. Sin. B 2022, 13, 29–53. [Google Scholar] [CrossRef]
- Jun, H.K.; Jung, Y.J.; Ji, S.; An, S.J.; Choi, B.K. Caspase-4 activation by a bacterial surface protein is mediated by cathepsin G in human gingival fibroblasts. Cell Death Differ. 2018, 25, 380–391. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef]
- Sun, W.; Lu, H.; Dong, S.; Li, R.; Chu, Y.; Wang, N.; Zhao, Y.; Zhang, Y.; Wang, L.; Sun, L.; et al. Beclin1 controls caspase-4 inflammsome activation and pyroptosis in mouse myocardial reperfusion-induced microvascular injury. Cell Commun. Signal. 2021, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Zhang, Z.; Hitomi, E.; Lee, Y.C.; Mukherjee, A.B. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 2006, 15, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Mo, G.; Liu, X.; Zhong, Y.; Mo, J.; Li, Z.; Li, D.; Zhang, L.; Liu, Y. IP3R1 regulates Ca2+ transport and pyroptosis through the NLRP3/Caspase-1 pathway in myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, Z.; Sun, H.; Ma, L. Role of NLRP3 Inflammasome in Myocardial Ischemia-Reperfusion Injury and Ventricular Remodeling. Med. Sci. Monit. 2022, 28, e934255. [Google Scholar] [CrossRef]
- Tall, A.R. HDL in Morbidity and Mortality: A 40+ Year Perspective. Clin. Chem. 2021, 67, 19–23. [Google Scholar] [CrossRef]
- Sposito, A.C.; Carmo, H.R.; Barreto, J.; Sun, L.; Carvalho, L.S.F.; Feinstein, S.B.; Zanotti, I.; Kontush, A.; Remaley, A. HDL-Targeted Therapies During Myocardial Infarction. Cardiovasc. Drugs Ther. 2019, 33, 371–381. [Google Scholar] [CrossRef]
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of microRNAs. Biochim. Biophys. Acta 2016, 1861, 2069–2074. [Google Scholar] [CrossRef]
- Richart, A.L.; Calkin, A.C.; Kingwell, B.A. Apolipoprotein A-I for Cardiac Recovery Post-Myocardial Infarction. JACC Basic Transl. Sci. 2021, 6, 768–771. [Google Scholar] [CrossRef]
- Thacker, S.G.; Zarzour, A.; Chen, Y.; Alcicek, M.S.; Freeman, L.A.; Sviridov, D.O.; Demosky, S.J., Jr.; Remaley, A.T. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology 2016, 149, 306–319. [Google Scholar] [CrossRef]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2019, 598, 2923–2939. [Google Scholar] [CrossRef]
- Durham, K.K.; Chathely, K.M.; Trigatti, B.L. High-density lipoprotein protects cardiomyocytes against necrosis induced by oxygen and glucose deprivation through SR-B1, PI3K, and AKT1 and 2. Biochem. J. 2018, 475, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Osto, E.; von Eckardstein, A. The Endothelium Is Both a Target and a Barrier of HDL’s Protective Functions. Cells 2021, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.R. Signal transduction by HDL: Agonists, receptors, and signaling cascades. Handb. Exp. Pharmacol. 2015, 224, 229–256. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Beikoghli Kalkhoran, S.; Davidson, S.M. The RISK pathway leading to mitochondria and cardioprotection: How everything started. Basic Res. Cardiol. 2023, 118, 22. [Google Scholar] [CrossRef] [PubMed]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2017, 113, 2. [Google Scholar] [CrossRef]
- White, C.R.; Giordano, S.; Anantharamaiah, G.M. High-density lipoprotein, mitochondrial dysfunction and cell survival mechanisms. Chem. Phys. Lipids 2016, 199, 161–169. [Google Scholar] [CrossRef]
- Heusch, G. Treatment of Myocardial Ischemia/Reperfusion Injury by Ischemic and Pharmacological Postconditioning. Compr. Physiol. 2015, 5, 1123–1145. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef]
- Gurel, E.; Ustunova, S.; Kapucu, A.; Yilmazer, N.; Eerbeek, O.; Nederlof, R.; Hollmann, M.W.; Demirci-Tansel, C.; Zuurbier, C.J. Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol. Biol. Rep. 2013, 40, 4153–4160. [Google Scholar] [CrossRef]
- McCommis, K.S.; Douglas, D.L.; Krenz, M.; Baines, C.P. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J. Am. Heart Assoc. 2013, 2, e000355. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E. Disorders due to enzyme defects in the red blood cell. Adv. Metab. Disord. 1972, 60, 131–160. [Google Scholar] [CrossRef] [PubMed]
- Sposito, A.C.; de Lima-Junior, J.C.; Moura, F.A.; Barreto, J.; Bonilha, I.; Santana, M.; Virginio, V.W.; Sun, L.; Carvalho, L.S.F.; Soares, A.A.S.; et al. Reciprocal Multifaceted Interaction Between HDL (High-Density Lipoprotein) and Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1550–1564. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femmino, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid. Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, O.; Gao, E.; Ranheim, T.; Bliksoen, M.; Kaasboll, O.J.; Alfsnes, K.; Nymo, S.H.; Rashidi, A.; Ohm, I.K.; Attramadal, H.; et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem. Biophys. Res. Commun. 2016, 469, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, H.; Yao, L.; Tang, H.; Dong, H.; Wu, Y.; Liu, L.; Zou, F.; Cai, S. Phosphatidylinositol 3-kinases pathway mediates lung caspase-1 activation and high mobility group box 1 production in a toluene-diisocyanate induced murine asthma model. Toxicol. Lett. 2015, 236, 25–33. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef]
- Garg, M.; Johri, S.; Chakraborty, K. Immunomodulatory role of mitochondrial DAMPs: A missing link in pathology? FEBS J. 2022, 290, 4395–4418. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, C.S.; Harrison, K.; Hwang, I.Y.; Nabar, N.R.; Wang, M.; Kehrl, J.H. AKT Regulates NLRP3 Inflammasome Activation by Phosphorylating NLRP3 Serine 5. J. Immunol. 2020, 205, 2255–2264. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1α-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357. [Google Scholar] [CrossRef]
- Nam, B.Y.; Jhee, J.H.; Park, J.; Kim, S.; Kim, G.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Yu, J.W.; Han, S.H. PGC-1α inhibits the NLRP3 inflammasome via preserving mitochondrial viability to protect kidney fibrosis. Cell Death Dis. 2022, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, H.; Zhao, Y.; Wang, Y.; Wang, W.; He, Y.; Zhang, W.; Zhang, W.; Zhu, X.; Zhou, Y.; et al. Telomere Dysfunction Disturbs Macrophage Mitochondrial Metabolism and the NLRP3 Inflammasome through the PGC-1α/TNFAIP3 Axis. Cell Rep. 2018, 22, 3493–3506. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Zeng, J.; Yang, Y.X.; Qi, C.L.; Xiong, T.; Wu, G.Z.; Zeng, C.Y.; Wang, D.X. DRD4 Mitigates Myocardial Ischemia/Reperfusion Injury in Association with PI3K/AKT Mediated Glucose Metabolism. Front. Pharmacol. 2020, 11, 619426. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Liu, X.; Miao, H.; Huang, B.; Liu, Z.; Chen, J.; Quan, X.; Zhu, L.; Dong, H.; Zhang, Z. PEDF increases GLUT4-mediated glucose uptake in rat ischemic myocardium via PI3K/AKT pathway in a PEDFR-dependent manner. Int. J. Cardiol. 2019, 283, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Lecour, S.; James, R.W.; Pedretti, S. High density lipoprotein/sphingosine-1-phosphate-induced cardioprotection: Role of STAT3 as part of the SAFE pathway. JAKSTAT 2012, 1, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Somers, S.J.; Frias, M.; Lacerda, L.; Opie, L.H.; Lecour, S. Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc. Drugs Ther. 2012, 26, 227–237. [Google Scholar] [CrossRef]
- Kalakech, H.; Hibert, P.; Prunier-Mirebeau, D.; Tamareille, S.; Letournel, F.; Macchi, L.; Pinet, F.; Furber, A.; Prunier, F. RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS ONE 2014, 9, e107950. [Google Scholar] [CrossRef]
- Feuerborn, R.; Becker, S.; Poti, F.; Nagel, P.; Brodde, M.; Schmidt, H.; Christoffersen, C.; Ceglarek, U.; Burkhardt, R.; Nofer, J.R. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis 2017, 257, 29–37. [Google Scholar] [CrossRef]
- Kang, J.-H.; Lee, S.-B.; Seok, J.; Kim, D.-H.; Ma, G.; Park, J.; Jeong, A.J.; Ye, S.-K.; Kang, T.-B. Novel Activity of ODZ10117, a STAT3 Inhibitor, for Regulation of NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2023, 24, 6079. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Z.; Sun, X.; Yu, J.; Li, T.; Zhao, H.; Ji, Y.; Peng, B.; Du, M. STAT3/Mitophagy Axis Coordinates Macrophage NLRP3 Inflammasome Activation and Inflammatory Bone Loss. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2023, 38, 335–353. [Google Scholar] [CrossRef]
- Bai, H.; Zhang, Q.F.; Duan, J.J.; Yu, D.J.; Liu, L.J. Downregulation of signal transduction and STAT3 expression exacerbates oxidative stress mediated by NLRP3 inflammasome. Neural Regen. Res. 2018, 13, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Abe, J.I. Enigma of Inflammasome Activation by Kinases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1501–1503. [Google Scholar] [CrossRef] [PubMed]
- Abderrahmani, A.; Niederhauser, G.; Favre, D.; Abdelli, S.; Ferdaoussi, M.; Yang, J.Y.; Regazzi, R.; Widmann, C.; Waeber, G. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 2007, 50, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Salvayre, R.; Nègre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011, 18, 817–828. [Google Scholar] [CrossRef]
- Sandall, C.F.; MacDonald, J.A. Effects of phosphorylation on the NLRP3 inflammasome. Arch. Biochem. Biophys. 2019, 670, 43–57. [Google Scholar] [CrossRef]
- Mulay, V.; Wood, P.; Rentero, C.; Enrich, C.; Grewal, T. Signal transduction pathways provide opportunities to enhance HDL and apoAI-dependent reverse cholesterol transport. Curr. Pharm. Biotechnol. 2012, 13, 352–364. [Google Scholar] [CrossRef]
- Pulli, I.; Asghar, M.Y.; Kemppainen, K.; Tornquist, K. Sphingolipid-mediated calcium signaling and its pathological effects. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1668–1677. [Google Scholar] [CrossRef]
- Ke, M.; Tang, Q.; Pan, Z.; Yin, Y.; Zhang, L.; Wen, K. Sphingosine-1-phosphate attenuates hypoxia/reoxygenation-induced cardiomyocyte injury via a mitochondrial pathway. Biochem. Biophys. Res. Commun. 2019, 510, 142–148. [Google Scholar] [CrossRef]
- Kalayci, A.; Gibson, C.M.; Ridker, P.M.; Wright, S.D.; Kingwell, B.A.; Korjian, S.; Chi, G.; Lee, J.J.; Tricoci, P.; Kazmi, S.H.; et al. ApoA-I Infusion Therapies Following Acute Coronary Syndrome: Past, Present, and Future. Curr. Atheroscler. Rep. 2022, 24, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Misharin, A.; Tertov, V.V.; Khashimov Kh, A.; Pokrovsky, S.N.; Repin, V.S.; Smirnov, V.N. Artificial HDL as an anti-atherosclerotic drug. Lancet 1984, 324, 1149–1150. [Google Scholar] [CrossRef] [PubMed]
- Krause, B.R.; Remaley, A.T. Reconstituted HDL for the acute treatment of acute coronary syndrome. Curr. Opin. Lipidol. 2013, 24, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Hubsch, A.P.; Casas, A.T.; Doran, J.E. Protective effects of reconstituted high-density lipoprotein in rabbit gram-negative bacteremia models. J. Lab. Clin. Med. 1995, 126, 548–558. [Google Scholar] [PubMed]
- Casas, A.T.; Hubsch, A.P.; Rogers, B.C.; Doran, J.E. Reconstituted high-density lipoprotein reduces LPS-stimulated TNF alpha. J. Surg. Res. 1995, 59, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Pajkrt, D.; Doran, J.E.; Koster, F.; Lerch, P.G.; Arnet, B.; van der Poll, T.; ten Cate, J.W.; van Deventer, S.J. Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J. Exp. Med. 1996, 184, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Didichenko, S.A.; Navdaev, A.V.; Cukier, A.M.; Gille, A.; Schuetz, P.; Spycher, M.O.; Therond, P.; Chapman, M.J.; Kontush, A.; Wright, S.D. Enhanced HDL Functionality in Small HDL Species Produced Upon Remodeling of HDL by Reconstituted HDL, CSL112: Effects on Cholesterol Efflux, Anti-Inflammatory and Antioxidative Activity. Circ. Res. 2016, 119, 751–763. [Google Scholar] [CrossRef]
- Morin, E.E.; Guo, L.; Schwendeman, A.; Li, X.A. HDL in sepsis—Risk factor and therapeutic approach. Front. Pharmacol. 2015, 6, 244. [Google Scholar] [CrossRef]
- Tardif, J.C.; Gregoire, J.; L’Allier, P.L.; Ibrahim, R.; Lesperance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.A.; et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. Jama 2007, 297, 1675–1682. [Google Scholar] [CrossRef]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Bandhuvula, P.; Honbo, N.; Wang, G.Y.; Jin, Z.Q.; Fyrst, H.; Zhang, M.; Borowsky, A.D.; Dillard, L.; Karliner, J.S.; Saba, J.D. S1P lyase: A novel therapeutic target for ischemia-reperfusion injury of the heart. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1753–H1761. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef] [PubMed]
- Marsolais, D.; Rosen, H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discov. 2009, 8, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Kobayashi, N.; Hisano, Y.; Kawahara, A.; Yamaguchi, A. Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochim. Et Biophys. Acta 2014, 1841, 759–765. [Google Scholar] [CrossRef]
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H250–H261. [Google Scholar] [CrossRef]
- Meshcheryakova, A.; Svoboda, M.; Tahir, A.; Köfeler, H.C.; Triebl, A.; Mungenast, F.; Heinze, G.; Gerner, C.; Zimmermann, P.; Jaritz, M.; et al. Exploring the role of sphingolipid machinery during the epithelial to mesenchymal transition program using an integrative approach. Oncotarget 2016, 7, 22295–22323. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Chui, L.G.; Moser, A.; Khovidhunkit, W.; Grunfeld, C. Infection and inflammation decrease apolipoprotein M expression. Atherosclerosis 2008, 199, 19–26. [Google Scholar] [CrossRef]
- Brulhart-Meynet, M.C.; Braunersreuther, V.; Brinck, J.; Montecucco, F.; Prost, J.C.; Thomas, A.; Galan, K.; Pelli, G.; Pedretti, S.; Vuilleumier, N.; et al. Improving Reconstituted HDL Composition for Efficient Post-Ischemic Reduction of Ischemia Reperfusion Injury. PLoS ONE 2015, 10, e0119664. [Google Scholar] [CrossRef]
- Morel, S.; Christoffersen, C.; Axelsen, L.N.; Montecucco, F.; Rochemont, V.; Frias, M.A.; Mach, F.; James, R.W.; Naus, C.C.; Chanson, M.; et al. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc. Res. 2016, 109, 385–396. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Dev, K.K. The structure and function of the S1P1 receptor. Trends Pharmacol. Sci. 2013, 34, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules 2017, 22, 344. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Cremer, S.; Schmidt, M.V.; von Knethen, A.; Angioni, C.; Geisslinger, G.; Brune, B. Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood 2010, 115, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Datta, G.; Giordano, S. High-Density Lipoprotein Regulation of Mitochondrial Function. Adv. Exp. Med. Biol. 2017, 982, 407–429. [Google Scholar] [CrossRef]
- Traxler, D.; Spannbauer, A.; Einzinger, P.; Mester-Tonczar, J.; Lukovic, D.; Winkler, J.; Zlabinger, K.; Gugerell, A.; Mandic, L.; Gyöngyösi, M.; et al. Early Elevation of Systemic Plasma Clusterin after Reperfused Acute Myocardial Infarction in a Preclinical Porcine Model of Ischemic Heart Disease. Int. J. Mol. Sci. 2020, 21, 4591. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.O.; Kim, D.H.; Lee, S.W.; Lee, H.S.; Seo, J.H.; Kim, J.H.; Kim, J.H.; Yu, Y.S.; Min, B.H.; Kim, K.W. Clusterin protects H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3β signaling pathway. Exp. Mol. Med. 2011, 43, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rivera, C.; Garcia, M.M.; Molina-Alvarez, M.; Gonzalez-Martin, C.; Goicoechea, C. Clusterin: Always protecting. Synthesis, function and potential issues. Biomed. Pharmacother. 2021, 134, 111174. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation—A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Ikeda, S.; Zablocki, D.; Sadoshima, J. The role of autophagy in death of cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 165, 1–8. [Google Scholar] [CrossRef]
- Carmo, H.R.P.; Yoshinaga, M.Y.; Castillo, A.R.; Britto Chaves-Filho, A.; Bonilha, I.; Barreto, J.; Muraro, S.P.; de Souza, G.F.; Davanzo, G.G.; Perroud, M.W., Jr.; et al. Phenotypic changes in low-density lipoprotein particles as markers of adverse clinical outcomes in COVID-19. Mol. Genet. Metab. 2023, 138, 107552. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Schoch, L.; Badimon, L.; Vilahur, G. Unraveling the Complexity of HDL Remodeling: On the Hunt to Restore HDL Quality. Biomedicines 2021, 9, 805. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Virginio, V.W.; Panzoldo, N.B.; Figueiredo, V.N.; Santos, S.N.; Modolo, R.G.; Andrade, J.M.; Quinaglia, E.S.J.C.; Nadruz-Junior, W.; de Faria, E.C.; et al. Elevated CETP activity during acute phase of myocardial infarction is independently associated with endothelial dysfunction and adverse clinical outcome. Atherosclerosis 2014, 237, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Panzoldo, N.; Santos, S.N.; Modolo, R.; Almeida, B.; e Silva, J.C.Q.; Nadruz-Jr, W.; de Faria, E.C.; Sposito, A.C.; Brasilia Heart Study Group. HDL levels and oxidizability during myocardial infarction are associated with reduced endothelial-mediated vasodilation and nitric oxide bioavailability. Atherosclerosis 2014, 237, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Bindu, G.H.; Rao, V.S.; Kakkar, V.V. Friend Turns Foe: Transformation of Anti-Inflammatory HDL to Proinflammatory HDL during Acute-Phase Response. Cholesterol 2011, 2011, 274629. [Google Scholar] [CrossRef]
- Annema, W.; Willemsen, H.M.; de Boer, J.F.; Dikkers, A.; van der Giet, M.; Nieuwland, W.; Muller Kobold, A.C.; van Pelt, L.J.; Slart, R.H.; van der Horst, I.C.; et al. HDL function is impaired in acute myocardial infarction independent of plasma HDL cholesterol levels. J. Clin. Lipidol. 2016, 10, 1318–1328. [Google Scholar] [CrossRef]
- Vyletelová, V.; Nováková, M.; Pašková, Ľ. Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies. Pharmaceuticals 2022, 15, 1278. [Google Scholar] [CrossRef]
- Jaimes, E.A.; Sweeney, C.; Raij, L. Effects of the reactive oxygen species hydrogen peroxide and hypochlorite on endothelial nitric oxide production. Hypertension 2001, 38, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, A.; Carta, S.; Lu, R.; Yokoyama, S.; Rubartelli, A.; Cavigiolio, G. Oxidation of methionine residues in human apolipoprotein A-I generates a potent pro-inflammatory molecule. J. Biol. Chem. 2019, 294, 3634–3646. [Google Scholar] [CrossRef] [PubMed]
- Sutter, I.; Klingenberg, R.; Othman, A.; Rohrer, L.; Landmesser, U.; Heg, D.; Rodondi, N.; Mach, F.; Windecker, S.; Matter, C.M.; et al. Decreased phosphatidylcholine plasmalogens—A putative novel lipid signature in patients with stable coronary artery disease and acute myocardial infarction. Atherosclerosis 2016, 246, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Moxon, J.V.; Jones, R.E.; Wong, G.; Weir, J.M.; Mellett, N.A.; Kingwell, B.A.; Meikle, P.J.; Golledge, J. Baseline serum phosphatidylcholine plasmalogen concentrations are inversely associated with incident myocardial infarction in patients with mixed peripheral artery disease presentations. Atherosclerosis 2017, 263, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G. High-density lipoprotein: A double-edged sword in cardiovascular physiology and pathophysiology. J. Lab. Precis. Med. 2021, 6, 28. [Google Scholar] [CrossRef]
- Meikle, P.J.; Formosa, M.F.; Mellett, N.A.; Jayawardana, K.S.; Giles, C.; Bertovic, D.A.; Jennings, G.L.; Childs, W.; Reddy, M.; Carey, A.L.; et al. HDL Phospholipids, but Not Cholesterol Distinguish Acute Coronary Syndrome from Stable Coronary Artery Disease. J. Am. Heart Assoc. 2019, 8, e011792. [Google Scholar] [CrossRef]
- Bozelli, J.C., Jr.; Azher, S.; Epand, R.M. Plasmalogens and Chronic Inflammatory Diseases. Front. Physiol. 2021, 12, 730829. [Google Scholar] [CrossRef]
- Rasmiena, A.A.; Barlow, C.K.; Stefanovic, N.; Huynh, K.; Tan, R.; Sharma, A.; Tull, D.; de Haan, J.B.; Meikle, P.J. Plasmalogen modulation attenuates atherosclerosis in ApoE- and ApoE/GPx1-deficient mice. Atherosclerosis 2015, 243, 598–608. [Google Scholar] [CrossRef]
- Yang, T.X.; Zhu, Y.F.; Wang, C.C.; Yang, J.Y.; Xue, C.H.; Huang, Q.R.; Wang, Y.M.; Zhang, T.T. EPA-enriched plasmalogen attenuates the cytotoxic effects of LPS-stimulated microglia on the SH-SY5Y neuronal cell line. Brain Res. Bull. 2022, 186, 143–152. [Google Scholar] [CrossRef]
- Correia, L.C.; Magalhães, L.P.; Braga, J.C.; Rocha, M.S.; Lima, J.C.; Passos, L.C.; D’Oliveira, A.; Péricles Esteves, J.; Spósito, A.C. Decrease of plasma triglycerides during the acute phase of unstable angina or non-ST elevation myocardial infarction is a marker of recurrent ischemia. Atherosclerosis 2004, 177, 71–76. [Google Scholar] [CrossRef]
- Cho, K.H.; Shin, D.G.; Baek, S.H.; Kim, J.R. Myocardial infarction patients show altered lipoprotein properties and functions when compared with stable angina pectoris patients. Exp. Mol. Med. 2009, 41, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R. High-Density Lipoproteins and Serum Amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yu, Y.; Zhu, I.; Cheng, Y.; Sun, P.D. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5189–5194. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, F.; Lamon-Fava, S.; Horvath, K.V.; Schaefer, E.J.; Asztalos, B.F. Comparing fluorescence-based cell-free assays for the assessment of antioxidative capacity of high-density lipoproteins. Lipids Health Dis. 2016, 15, 163. [Google Scholar] [CrossRef]
- Wilson, P.G.; Thompson, J.C.; Shridas, P.; McNamara, P.J.; de Beer, M.C.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum Amyloid A Is an Exchangeable Apolipoprotein. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Benditt, E.P.; Eriksen, N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. USA 1977, 74, 4025–4028. [Google Scholar] [CrossRef]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef]
- Phillips, M.C. New insights into the determination of HDL structure by apolipoproteins: Thematic review series: High density lipoprotein structure, function, and metabolism. J. Lipid Res. 2013, 54, 2034–2048. [Google Scholar] [CrossRef]
- Heywood, S.E.; Richart, A.L.; Henstridge, D.C.; Alt, K.; Kiriazis, H.; Zammit, C.; Carey, A.L.; Kammoun, H.L.; Delbridge, L.M.; Reddy, M.; et al. High-density lipoprotein delivered after myocardial infarction increases cardiac glucose uptake and function in mice. Sci. Transl. Med. 2017, 9, 6084. [Google Scholar] [CrossRef]
- Dadabayev, A.R.; Yin, G.; Latchoumycandane, C.; McIntyre, T.M.; Lesnefsky, E.J.; Penn, M.S. Apolipoprotein A1 regulates coenzyme Q10 absorption, mitochondrial function, and infarct size in a mouse model of myocardial infarction. J. Nutr. 2014, 144, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Richart, A.L.; Reddy, M.; Khalaji, M.; Natoli, A.L.; Heywood, S.E.; Siebel, A.L.; Lancaster, G.L.; Murphy, A.J.; Carey, A.L.; Drew, B.G.; et al. Apo AI Nanoparticles Delivered Post Myocardial Infarction Moderate Inflammation. Circ. Res. 2020, 127, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Michell, D.L.; Vickers, K.C. HDL and microRNA therapeutics in cardiovascular disease. Pharmacol. Ther. 2016, 168, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Dullaart, R.P.; Annema, W.; Tio, R.A.; Tietge, U.J. The HDL anti-inflammatory function is impaired in myocardial infarction and may predict new cardiac events independent of HDL cholesterol. Clin. Chim. Acta 2014, 433, 34–38. [Google Scholar] [CrossRef]
- Liu, J.; Xing, J.; Li, Y.; Liu, J.; Tian, M.; Xu, Z.-S. Protective effects of miR-34b and miR-337 against myocardial ischemia/reperfusion injury in rats by liposomal nano-delivery system. Mater. Express 2021, 11, 1147–1153. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef]
- D’Alessandra, Y.; Carena, M.C.; Spazzafumo, L.; Martinelli, F.; Bassetti, B.; Devanna, P.; Rubino, M.; Marenzi, G.; Colombo, G.I.; Achilli, F.; et al. Diagnostic potential of plasmatic MicroRNA signatures in stable and unstable angina. PLoS ONE 2013, 8, e80345. [Google Scholar] [CrossRef]
- Gao, J.; Chen, X.; Wei, P.; Wang, Y.; Li, P.; Shao, K. Regulation of pyroptosis in cardiovascular pathologies: Role of noncoding RNAs. Mol. Ther. Nucleic Acids 2021, 25, 220–236. [Google Scholar] [CrossRef]
- Tong, R.; Jia, T.; Shi, R.; Yan, F. Inhibition of microRNA-15 protects H9c2 cells against CVB3-induced myocardial injury by targeting NLRX1 to regulate the NLRP3 inflammasome. Cell. Mol. Biol. Lett. 2020, 25, 6. [Google Scholar] [CrossRef]
- Li, X.; Long, J.; Zong, L.; Zhang, C.; Yang, Z.; Guo, S. ZNF561-AS1 Regulates Cell Proliferation and Apoptosis in Myocardial Infarction Through miR-223-3p/NLRP3 Axis. Cell Transplant. 2022, 31, 9636897221077928. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xiang, D.K.; Li, S.N.; Yang, L.H.; Gao, L.F.; Feng, C. MicroRNA-495 Ameliorates Cardiac Microvascular Endothelial Cell Injury and Inflammatory Reaction by Suppressing the NLRP3 Inflammasome Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 798–815. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Tian, R.; Chen, Q.; Wang, L.; Gu, Q.; Zhao, H.; Huang, C.; Liu, Y.; Li, J.; Yang, X.; et al. miR-330-5p inhibits NLRP3 inflammasome-mediated myocardial ischaemia-reperfusion injury by targeting TIM3. Cardiovasc. Drugs Ther. 2021, 35, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 769. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 2207–2219. [Google Scholar] [CrossRef]
- Sothivelr, V.; Hasan, M.Y.; Mohd Saffian, S.; Zainalabidin, S.; Ugusman, A.; Mahadi, M.K. Revisiting miRNA-21 as a Therapeutic Strategy for Myocardial Infarction: A Systematic Review. J. Cardiovasc. Pharmacol. 2022, 80, 393–406. [Google Scholar] [CrossRef]
- Rayner, K.J.; Moore, K.J. MicroRNA control of high-density lipoprotein metabolism and function. Circ. Res. 2014, 114, 183–192. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmo, H.R.P.; Bonilha, I.; Barreto, J.; Tognolini, M.; Zanotti, I.; Sposito, A.C. High-Density Lipoproteins at the Interface between the NLRP3 Inflammasome and Myocardial Infarction. Int. J. Mol. Sci. 2024, 25, 1290. https://doi.org/10.3390/ijms25021290
Carmo HRP, Bonilha I, Barreto J, Tognolini M, Zanotti I, Sposito AC. High-Density Lipoproteins at the Interface between the NLRP3 Inflammasome and Myocardial Infarction. International Journal of Molecular Sciences. 2024; 25(2):1290. https://doi.org/10.3390/ijms25021290
Chicago/Turabian StyleCarmo, Helison R. P., Isabella Bonilha, Joaquim Barreto, Massimiliano Tognolini, Ilaria Zanotti, and Andrei C. Sposito. 2024. "High-Density Lipoproteins at the Interface between the NLRP3 Inflammasome and Myocardial Infarction" International Journal of Molecular Sciences 25, no. 2: 1290. https://doi.org/10.3390/ijms25021290
APA StyleCarmo, H. R. P., Bonilha, I., Barreto, J., Tognolini, M., Zanotti, I., & Sposito, A. C. (2024). High-Density Lipoproteins at the Interface between the NLRP3 Inflammasome and Myocardial Infarction. International Journal of Molecular Sciences, 25(2), 1290. https://doi.org/10.3390/ijms25021290