
Ubiquitin Carboxyl-Terminal Hydrolase L1 and Its Role in Parkinson’s Disease

Abstract
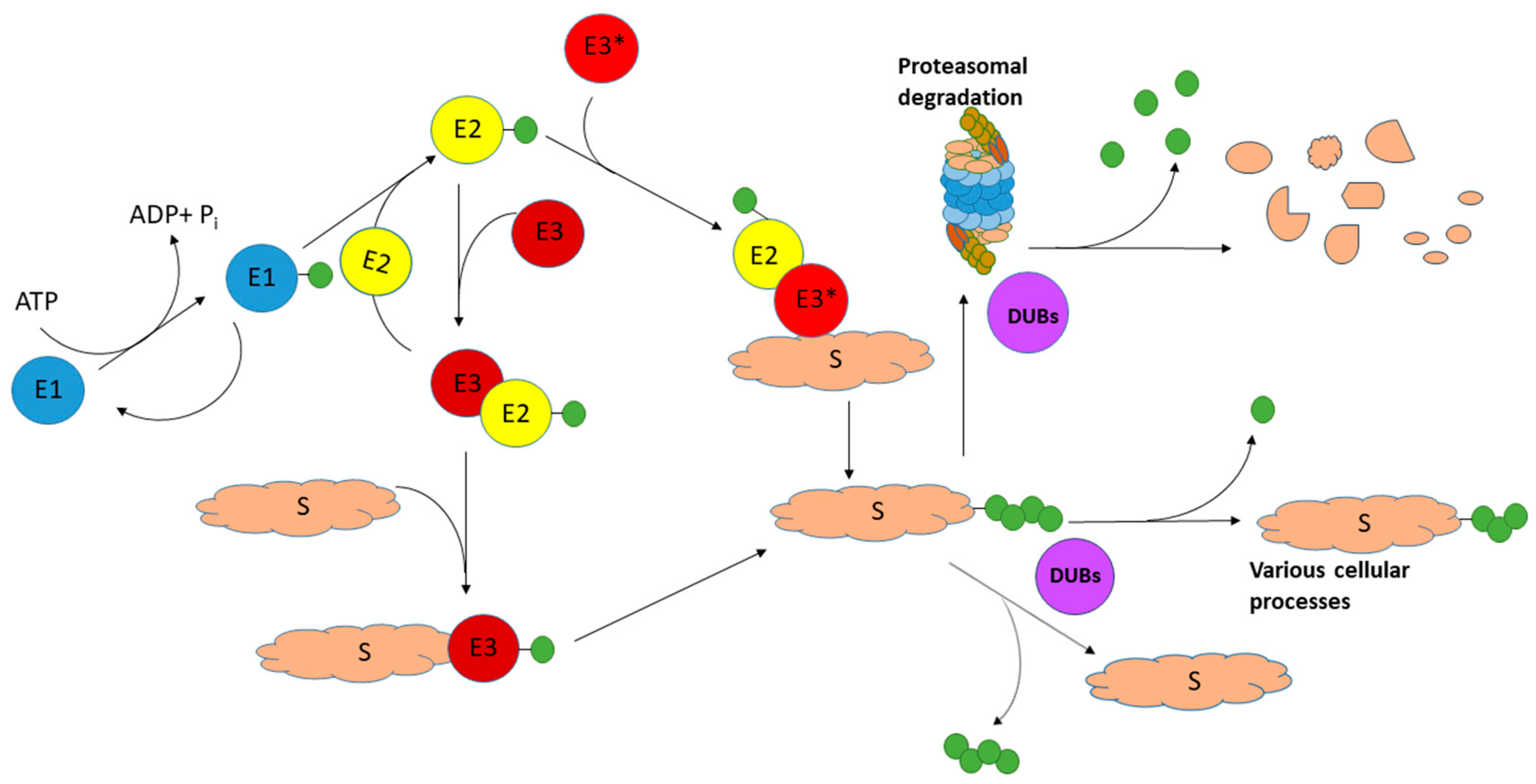
:1. Introduction
2. UCHL1 Is a Unique Member in the Family of UCHL Enzymes
3. UCHL1 Is a Highly Expressed Protein That Interacts with Many Proteins in Brain Cells
4. Mutations and Posttranslational Modification of UCHL1 Have a Significant Impact on the Catalytic Activity and Aggregability
5. Experimental Models of UCHL1 Deficiency and Postmortem Brain Studies Demonstrate a Role of This Protein in PD Pathogenesis
6. Mechanisms Underlying the Contribution of UCHL1 to the Pathogenesis of PD
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Obeso, J.A.; Jon Stoessl, A.; Stamelou, M. Editors’ Note: The 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson’s Disease: A Multisystem Disorder. Neurosci. Bull. 2023, 39, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. 2023, 8, 95–121. [Google Scholar] [CrossRef]
- Domingo, A.; Klein, C. Genetics of Parkinson disease. Handb. Clin. Neurol. 2018, 147, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, S.; Arena, G.; Valente, E. Genetics and Molecular Biology of Parkinson Disease. In Movement Disorders: Genetics and Molecular Biology of Parkinson Disease, 2nd ed.; LeDoux, M.S., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 243–257. [Google Scholar] [CrossRef]
- Lill, C.M.; Klein, C. What would Dr. James Parkinson think today? The role of genetics in Parkinson’s disease. Mov. Disord. 2017, 32, 1115–1116. [Google Scholar] [CrossRef] [PubMed]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef]
- Gialluisi, A.; Reccia, M.G.; Modugno, N.; Nutile, T.; Lombardi, A.; Di Giovannantonio, L.G.; Pietracupa, S.; Ruggiero, D.; Scala, S.; Gambardella, S.; et al. Identification of sixteen novel candidate genes for late onset Parkinson’s disease. Mol. Neurodegener. 2021, 16, 35. [Google Scholar] [CrossRef]
- Schulte, C.; Gasser, T. Genetic basis of Parkinson’s disease: Inheritance, penetrance, and expression. Appl. Clin. Genet. 2011, 4, 67–80. [Google Scholar] [CrossRef]
- Buneeva, O.; Medvedev, A. Atypical Ubiquitination and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 3705. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Schaffer, A.A.; et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinsons disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Bentivoglio, A.R.; Cassetta, E.; Dixon, P.H.; Davis, M.B.; Ferraris, A.; Ialongo, T.; Frontali, M.; Wood, N.W.; Albanese, A. Identification of a novel primary torsion dystonia locus (DYT13) on chromosome 1p36 in an Italian family with cranial-cervical or upper limb onset. Neurol. Sci. 2001, 22, 95–96. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Lockhart, P.J.; Lincoln, S.; Hulihan, M.; Kachergus, J.; Wilkes, K.; Bisceglio, G.; Mash, D.C.; Farrer, M.J. DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function. J. Med. Genet. 2004, 41, e22. [Google Scholar] [CrossRef]
- Ibáñez, P.; De Michele, G.; Bonifati, V.; Lohmann, E.; Thobois, S.; Pollak, P.; Agid, Y.; Heutink, P.; Dürr, A.; Brice, A. French Parkinson’s Disease Genetics Study Group. Screening for DJ-1 mutations in early onset autosomal recessive parkinsonism. Neurology 2003, 61, 1429–1431. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 2002, 51, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Hicks, A.A.; Pétursson, H.; Jónsson, T.; Stefánsson, H.; Jóhannsdóttir, H.S.; Sainz, J.; Frigge, M.L.; Kong, A.; Gulcher, J.R.; Stefánsson, K.; et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann. Neurol. 2002, 52, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.A.; Li, Y.J.; Noureddine, M.A.; Zuchner, S.; Qin, X.; Pericak-Vance, M.A.; Vance, J.M. Identification of risk and age-at-onset genes on chromosome 1p in Parkinson disease. Am. J. Hum. Genet. 2005, 77, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, N.; Nichols, W.C.; Uniacke, S.K.; Halter, C.; Rudolph, A.; Shults, C.; Conneally, P.M.; Foroud, T. Parkinson Study Group. Significant linkage of Parkinson disease to chromosome 2q36-37. Am. J. Hum. Genet. 2003, 72, 1053–1057. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Q.Y.; Yu, R.H.; Guo, J.F.; Tang, B.S.; Yan, X.X. The contribution of GIGYF2 to Parkinson’s disease: A meta-analysis. Neurol. Sci. 2015, 36, 2073–2079. [Google Scholar] [CrossRef]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. Rev. Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef]
- Bello, A.I.; Goswami, R.; Brown, S.L.; Costanzo, K.; Shores, T.; Allan, S.; Odah, R.; Mohan, R.D. Deubiquitinases in Neurodegeneration. Cells 2022, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Amer-Sarsour, F.; Kordonsky, A.; Berdichevsky, Y.; Prag, G.; Ashkenazi, A. Deubiquitylating enzymes in neuronal health and disease. Cell Death Dis. 2021, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Joo, J.Y.; Baek, K.H. The potential roles of deubiquitinating enzymes in brain diseases. Ageing Res Rev. 2020, 61, 101088. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.Y.Ø.; Okarmus, J.; Meyer, M. Role of Deubiquitinases in Parkinson’s Disease-Therapeutic Perspectives. Cells 2023, 12, 651. [Google Scholar] [CrossRef]
- Shrestha, R.K.; Ronau, J.A.; Davies, C.W.; Guenette, R.G.; Strieter, E.R.; Paul, L.N.; Das, C. Insights into the mechanism of deubiquitination by JAMM deubiquitinases from cocrystal structures of the enzyme with the substrate and product. Biochemistry 2014, 53, 3199–3217. [Google Scholar] [CrossRef]
- Cruz, L.; Soares, P.; Correia, M. Ubiquitin-Specific Proteases: Players in Cancer Cellular Processes. Pharmaceuticals 2021, 14, 848. [Google Scholar] [CrossRef]
- Kitamura, H. Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders. Int. J. Mol. Sci. 2023, 24, 3219. [Google Scholar] [CrossRef]
- Schlüter, D.; Schulze-Niemand, E.; Stein, M.; Naumann, M. Ovarian tumor domain proteases in pathogen infection. Trends Microbiol. 2022, 30, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Zhou, R.; Wan, S.; Su, D.; Wang, S.L.; Hess, J. Ubiquitin Carboxyl-Terminal Hydrolases and Human Malignancies: The Novel Prognostic and Therapeutic Implications for Head and Neck Cancer. Front. Oncol. 2021, 10, 592501. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Liu, H.; Tobar-Tosse, F.; Chand Dakal, T.; Ludwig, M.; Holz, F.G.; Loeffler, K.U.; Wüllner, U.; Herwig-Carl, M.C. Ubiquitin Carboxyl-Terminal Hydrolases (UCHs): Potential Mediators for Cancer and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3910. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhao, C.; Ge, F.; Li, Y.; Cao, J.; Ying, M.; Lu, J.; He, Q.; Yang, B.; Dai, X.; et al. Machado-Joseph Deubiquitinases: From Cellular Functions to Potential Therapy Targets. Front. Pharmacol. 2020, 11, 1311. [Google Scholar] [CrossRef] [PubMed]
- Lill, J.R.; Wertz, I.E. Toward understanding ubiquitin-modifying enzymes: From pharmacological targeting to proteomics. Trends Pharmacol. Sci. 2014, 35, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Wei- dlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef]
- Coleman, K.E.; Huang, T.T. In a class of its own: A new family of deubiquitinases promotes genome stability. Mol. Cell 2018, 70, 1–3. [Google Scholar] [CrossRef]
- Kwasna, D.; Rehman, S.A.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164. [Google Scholar] [CrossRef]
- Haahr, P.; Borgermann, N.; Guo, X.; Typas, D.; Achuthankutty, D.; Hoffmann, S.; Shearer, R.; Sixma, T.K.; Mailand, N. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol. Cell 2018, 70, 165–174.e6. [Google Scholar] [CrossRef]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Thompson, R.J. The demonstration of new human brain-specific proteins by high-resolution two-dimensional polyacrylamide gel electrophoresis. J. Neurol. Sci. 1981, 49, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D.; Lee, K.M.; Deshpande, S.; Duerksen-Hughes, P.; Boss, J.M.; Pohl, J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 1989, 246, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Carmine Belin, A.; Westerlund, M.; Bergman, O.; Nissbrandt, H.; Lind, C.; Sydow, O.; Galter, D. S18Y in ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) associated with decreased risk of Parkinson’s disease in Sweden. Parkinsonism Relat. Disord. 2007, 13, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Wintermeyer, P.; Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Berg, D.; Becker, G.; Leroy, E.; Polymeropoulos, M.; Berger, K.; et al. Mutation analysis and association studies of the UCHL1 gene in German Parkinson’s disease patients. Neuroreport 2000, 11, 2079–2082. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Puong, K.Y.; Fook-Chong, S.; Chua, E.; Shen, H.; Yuen, Y.; Pavanni, R.; Wong, M.C.; Puvan, K.; Zhao, Y. Case-control study of UCHL1 S18Y variant in Parkinson’s disease. Mov. Disord. 2006, 21, 1765–1768. [Google Scholar] [CrossRef]
- Satoh, J.; Kuroda, Y. A polymorphic variation of serine to tyrosine at codon 18 in the ubiquitin C-terminal hydrolase-L1 gene is associated with a reduced risk of sporadic Parkinson’s disease in a Japanese population. J. Neurol. Sci. 2001, 189, 113–117. [Google Scholar] [CrossRef]
- Hutter, C.M.; Samii, A.; Factor, S.A.; Nutt, J.G.; Higgins, D.S.; Bird, T.D.; Griffith, A.; Roberts, J.W.; Leis, B.C.; Montimurro, J.S.; et al. Lack of evidence for an association between UCHL1 S18Y and Parkinson’s disease. Eur. J. Neurol. 2008, 15, 134–139. [Google Scholar] [CrossRef]
- Momose, Y.; Murata, M.; Kobayashi, K.; Tachikawa, M.; Nakabayashi, Y.; Kanazawa, I.; Toda, T. Association studies of multiple candidate genes for Parkinson’s disease using single nucleotide polymorphisms. Ann. Neurol. 2002, 51, 133–136. [Google Scholar] [CrossRef]
- Savettieri, G.; De Marco, E.V.; Civitelli, D.; Salemi, G.; Nicoletti, G.; Annesi, G.; Ciro Candiano, I.C.; Quattrone, A. Lack of association between ubiquitin carboxy-terminal hydrolase L1 gene polymorphism and PD. Neurology 2001, 57, 560–561. [Google Scholar] [CrossRef] [PubMed]
- Mellick, G.D.; Silburn, P.A. The ubiquitin carboxy-terminal hydrolase-L1 gene S18Y polymorphism does not confer protection against idiopathic Parkinson’s disease. Neurosci. Lett. 2000, 293, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Burgunder, J.M.; An, X.K.; Wu, Y.; Chen, W.J.; Zhang, J.H.; Wang, Y.C.; Xu, Y.M.; Gou, Y.R.; Yuan, G.G.; et al. Lack of evidence for association of a UCH-L1 S18Y polymorphism with Parkinson’s disease in a Han-Chinese population. Neurosci. Lett. 2008, 442, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Lu, C.S.; Peng, R.; Teo, Y.Y.; Wu-Chou, Y.H.; Chen, R.S.; Weng, Y.H.; Chen, C.M.; Fung, H.C.; Tan, L.C.; et al. Analysis of the UCHL1 genetic variant in Parkinson’s disease among chinese. Neurobiol. Aging 2010, 31, 2194–2196. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Sasaki, S.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Shimada, H.; Kawamura, N.; et al. UCHL1 S18Y variant is a risk factor for Parkinson’s disease in Japan. BMC Neurol. 2012, 12, 62. [Google Scholar] [CrossRef]
- Lowe, J.; McDermott, H.; Landon, M.; Mayer, R.J.; Wilkinson, K.D. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol. 1990, 161, 153–160. [Google Scholar] [CrossRef]
- Barrachina, M.; Castaño, E.; Dalfó, E.; Maes, T.; Buesa, C.; Ferrer, I. Reduced ubiquitin C-terminal hydrolase-1 expression levels in dementia with Lewy bodies. Neurobiol. Dis. 2006, 22, 265–273. [Google Scholar] [CrossRef]
- Dang, L.C.; Melandri, F.D.; Stein, R.L. Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry 1998, 37, 1868–1879. [Google Scholar] [CrossRef]
- Larsen, C.N.; Price, J.S.; Wilkinson, K.D. Substrate binding and catalysis by ubiquitin C-terminal hydrolases: Identification of two active site residues. Biochemistry 1996, 35, 6735–6744. [Google Scholar] [CrossRef]
- Zhou, Z.R.; Zhang, Y.H.; Liu, S.; Song, A.X.; Hu, H.Y. Length of the active-site crossover loop defines the substrate specificity of ubiquitin C-terminal hydrolases for ubiquitin chains. Biochem. J. 2012, 441, 143–149. [Google Scholar] [CrossRef]
- Luchansky, S.J.; Lansbury, P.T., Jr.; Stein, R.L. Substrate recognition and catalysis by UCH-L1. Biochemistry 2006, 45, 14717–14725. [Google Scholar] [CrossRef] [PubMed]
- Setsuie, R.; Sakurai, M.; Sakaguchi, Y.; Wada, K. Ubiquitin dimers control the hydrolase activity of UCH-L3. Neurochem. Int. 2009, 54, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Grou, C.P.; Pinto, M.P.; Mendes, A.V.; Domingues, P.; Azevedo, J.E. The de novo synthesis of ubiquitin: Identification of deubiquitinases acting on ubiquitin precursors. Sci. Rep. 2015, 5, 12836. [Google Scholar] [CrossRef] [PubMed]
- Panyain, N.; Godinat, A.; Thawani, A.R.; Lachiondo-Ortega, S.; Mason, K.; Elkhalifa, S.; Smith, L.M.; Harrigan, J.A.; Tate, E.W. Activity-based protein profiling reveals deubiquitinase and aldehyde dehydrogenase targets of a cyanopyrrolidine probe. RSC Med. Chem. 2021, 12, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Bheda, A.; Gullapalli, A.; Caplow, M.; Pagano, J.S.; Shackelford, J. Ubiquitin editing enzyme UCH L1 and microtubule dynamics: Implication in mitosis. Cell Cycle 2010, 9, 980–994. [Google Scholar] [CrossRef]
- Das, C.; Hoang, Q.Q.; Kreinbring, C.A.; Luchansky, S.J.; Meray, R.K.; Ray, S.S.; Lansbury, P.T.; Ringe, D.; Petsko, G.A. Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. USA 2006, 103, 4675–4680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fallon, L.; Lashuel, H.A.; Liu, Z.; Lansbury, P.T., Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002, 111, 209–218. [Google Scholar] [CrossRef]
- Bilguvar, K.; Tyagi, N.K.; Ozkara, C.; Tuysuz, B.; Bakircioglu, M.; Choi, M.; Delil, S.; Caglayan, A.O.; Baranoski, J.F.; Erturk, O.; et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 3489–3494. [Google Scholar] [CrossRef]
- Wen, Y.; Wu, Q.; Shi, Q.; Xie, Y.; Dan, W.; Chen, Y.; Ma, L. UCH-L1 inhibition aggravates mossy fiber sprouting in the pentylenetetrazole kindling model. Biochem. Biophys. Res. Commun. 2018, 503, 2312–2318. [Google Scholar] [CrossRef]
- Matuszczak, E.; Tylicka, M.; Komarowska, M.D.; Debek, W.; Hermanowicz, A. Ubiquitin carboxy-terminal hydrolase L1—Physiology and pathology. Cell Biochem. Funct. 2020, 38, 533–540. [Google Scholar] [CrossRef]
- Ham, S.J.; Lee, D.; Xu, W.J.; Cho, E.; Choi, S.; Min, S.; Park, S.; Chung, J. Loss of UCHL1 rescues the defects related to Parkinson’s disease by suppressing glycolysis. Sci. Adv. 2021, 7, eabg4574. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, N.; Li, M.; Hong, T.; Meng, W.; Ouyang, T. Ubiquitin C terminal hydrolase L1: A new cancer marker and therapeutic target with dual effects (Review). Oncol. Lett. 2023, 25, 123. [Google Scholar] [CrossRef]
- Cartier, A.E.; Djakovic, S.N.; Salehi, A.; Wilson, S.M.; Masliah, E.; Patrick, G.N. Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci. 2009, 29, 7857–7868. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Abiko, Y.; Katayama, Y.; Kaji, T.; Kumagai, Y. S-Mercuration of ubiquitin carboxyl-terminal hydrolase L1 through Cys152 by methylmercury causes inhibition of its catalytic activity and reduction of monoubiquitin levels in SH-SY5Y cells. J. Toxicol. Sci. 2015, 40, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Grethe, C.; Schmidt, M.; Kipka, G.-M.; O’Dea, R.; Gallant, K.; Janning, P.; Gersch, M. Structural basis for specific inhibition of the deubiquitinase UCHL1. Nat. Commun. 2022, 13, 5950. [Google Scholar] [CrossRef] [PubMed]
- Osaka, H.; Wang, Y.L.; Takada, K.; Takizawa, S.; Setsuie, R.; Li, H.; Sato, Y.; Nishikawa, K.; Sun, Y.J.; Sakurai, M.; et al. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum. Mol. Genet. 2003, 12, 1945–1958. [Google Scholar] [CrossRef]
- Mi, Z.; Graham, S.H. Role of UCHL1 in the pathogenesis of neurodegenerative diseases and brain injury. Ageing Res. Rev. 2023, 86, 101856. [Google Scholar] [CrossRef]
- Butterfield, D.A. Ubiquitin carboxyl-terminal hydrolase L-1 in brain: Focus on its oxidative/nitrosative modification and role in brains of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2021, 177, 278–286. [Google Scholar] [CrossRef]
- Doran, J.F.; Jackson, P.; Kynoch, P.A.; Thompson, R.J. Isolation of PGP 9.5, a new human neurone-specific protein detected by high-resolution two-dimensional electrophoresis. J. Neurochem. 1983, 40, 1542–1547. [Google Scholar] [CrossRef]
- Pukaß, K.; Richter-Landsberg, C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents α-synuclein aggregate formation by activating the autophagic pathway: Implications for multiple system atrophy. Front. Cell Neurosci. 2015, 9, 163. [Google Scholar] [CrossRef]
- Sjölin, K.; Kultima, K.; Larsson, A.; Freyhult, E.; Zjukovskaja, C.; Alkass, K.; Burman, J. Distribution of five clinically important neuroglial proteins in the human brain. Mol. Brain 2022, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.J.; Doran, J.F.; Jackson, P.; Dhillon, A.P.; Rode, J. PGP 9.5—A new marker for vertebrate neurons and neuroendocrine cells. Brain Res. 1983, 278, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, L.M.; Alm, P.; Wharton, J.; Polak, J.M. Protein gene product 9.5 (PGP 9.5). A new neuronal marker visualizing the whole uterine innervation and pregnancy-induced and developmental changes in the guinea pig. Histochemistry 1988, 90, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.; Rubin, P.; Thomson, A.R.; Rocca, D.; Henley, J.M. The ubiquitin C-terminal hydrolase L1 (UCH-L1) C terminus plays a key role in protein stability, but its farnesylation is not required for membrane association in primary neurons. J. Biol. Chem. 2014, 289, 36140–36149. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Meray, R.K.; Grammatopoulos, T.N.; Fredenburg, R.A.; Cookson, M.R.; Liu, Y.; Logan, T.; Lansbury, P.T., Jr. Membrane-associated farnesylated UCH-L1 promotes alpha-synuclein neurotoxicity and is a therapeutic target for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, R.Y.; Turko, I.V. Mass spectrometry assessment of ubiquitin carboxyl-terminal hydrolase L1 partitioning between soluble and particulate brain homogenate fractions. Anal. Chem. 2013, 85, 6011–6017. [Google Scholar] [CrossRef]
- Buneeva, O.; Kopylov, A.; Kaloshina, S.; Zgoda, V.; Medvedev, A. 20S and 26S proteasome-binding proteins of the rabbit brain: A proteomic dataset. Data Brief 2021, 38, 107276. [Google Scholar] [CrossRef]
- Buneeva, O.A.; Kopylov, A.T.; Zgoda, V.G.; Gnedenko, O.V.; Kaloshina, S.A.; Medvedeva, M.V.; Ivanov, A.S.; Medvedev, A.E. Comparative analysis of proteins associated with 26S and 20S proteasomes isolated from rabbit brain and liver. Biomed. Khim. 2022, 68, 18–31. [Google Scholar] [CrossRef]
- Sahu, I.; Mali, S.M.; Sulkshane, P.; Xu, C.; Rozenberg, A.; Morag, R.; Sahoo, M.P.; Singh, S.K.; Ding, Z.; Wang, Y.; et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021, 12, 6173. [Google Scholar] [CrossRef]
- Wilson, P.O.; Barber, P.C.; Hamid, Q.A.; Power, B.F.; Dhillon, A.P.; Rode, J.; Day, I.N.; Thompson, R.J.; Polak, J.M. The immunolocalization of protein gene product 9.5 using rabbit polyclonal and mouse monoclonal antibodies. Br. J. Exp. Pathol. 1988, 69, 91–104. [Google Scholar]
- Buneeva, O.; Kopylov, A.; Kapitsa, I.; Ivanova, E.; Zgoda, V.; Medvedev, A. The Effect of Neurotoxin MPTP and Neuroprotector Isatin on the Profile of Ubiquitinated Brain Mitochondrial Proteins. Cells 2018, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Jamin, A.; Berthelot, L.; Couderc, A.; Chemouny, J.M.; Boedec, E.; Dehoux, L.; Abbad, L.; Dossier, C.; Daugas, E.; Monteiro, R.C.; et al. Autoantibodies against podocytic UCHL1 are associated with idiopathic nephrotic syndrome relapses and induce proteinuria in mice. J. Autoimmun. 2018, 89, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.W.; Wang, C.; Zhou, Y.; Hou, M.M.; Wang, X.; Tang, H.D.; Wu, Y.W.; Ma, J.F.; Chen, S.D. Plasma epidermal growth factor decreased in the early stage of Parkinson’s disease. Aging Dis. 2015, 6, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hüll, M.; Lichtner, P.; et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Molecular chaperones in Parkinson’s disease—Present and future. J. Parkinsons Dis. 2011, 1, 299–320. [Google Scholar] [CrossRef] [PubMed]
- Dimant, H.; Ebrahimi-Fakhari, D.; McLean, P.J. Molecular chaperones and co-chaperones in Parkinson disease. Neuroscientist 2012, 18, 589–601. [Google Scholar] [CrossRef]
- Uryu, K.; Richter-Landsberg, C.; Welch, W.; Sun, E.; Goldbaum, O.; Norris, E.H.; Pham, C.T.; Yazawa, I.; Hilburger, K.; Micsenyi, M.; et al. Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am. J. Pathol. 2006, 168, 947–961. [Google Scholar] [CrossRef]
- Kabuta, T.; Furuta, A.; Aoki, S.; Furuta, K.; Wada, K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 2008, 283, 23731–23738. [Google Scholar] [CrossRef]
- Andersson, F.I.; Werrell, E.F.; McMorran, L.; Crone, W.J.; Das, C.; Hsu, S.T.; Jackson, S.E. The effect of Parkinson’s-disease-associated mutations on the deubiquitinating enzyme UCH-L1. J. Mol. Biol. 2011, 407, 261–272. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.P.; Park, C.M.; Kim, K.S.; Kim, E.; Jeong, M.; Shin, J.Y.; Yun, C.H.; Kim, K.; Chock, P.B.; Chae, H.Z. Structural and biochemical analyses reveal ubiquitin C-terminal hydrolase-L1 as a specific client of the peroxiredoxin II chaperone. Arch. Biochem. Biophys. 2018, 640, 61–74. [Google Scholar] [CrossRef] [PubMed]
- McKeon, J.E.; Sha, D.; Li, L.; Chin, L.S. Parkin-mediated K63-polyubiquitination targets ubiquitin C-terminal hydrolase L1 for degradation by the autophagy-lysosome system. Cell Mol. Life Sci. 2015, 72, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- McNaught, K.S.; Shashidharan, P.; Perl, D.P.; Jenner, P.; Olanow, C.W. Aggresome-related biogenesis of Lewy bodies. Eur. J. Neurosci. 2002, 16, 2136–2148. [Google Scholar] [CrossRef]
- Leverenz, J.B.; Umar, I.; Wang, Q.; Montine, T.J.; McMillan, P.J.; Tsuang, D.W.; Jin, J.; Pan, C.; Shin, J.; Zhu, D.; et al. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 2007, 17, 139–145. [Google Scholar] [CrossRef]
- Buneeva, O.; Kopylov, A.; Gnedenko, O.; Medvedeva, M.; Veselovsky, A.; Ivanov, A.; Zgoda, V.; Medvedev, A. Proteomic Profiling of Mouse Brain Pyruvate Kinase Binding Proteins: A Hint for Moonlighting Functions of PKM1? Int. J. Mol. Sci. 2023, 24, 7634. [Google Scholar] [CrossRef] [PubMed]
- Bouron, A.; Aubry, L.; Loreth, D.; Fauvarque, M.-O.; Meyer-Schwesinger, C. Role of the deubiquitinating enzyme UCH-L1 in mitochondrial function. Front. Cell. Neurosci. 2023, 17, 1149954. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.J.; Jeong, J.E.; Baek, J.Y.; Jeong, J.; Kim, S.; Kim, Y.M.; Kim, Y.; Nam, J.H.; Huh, S.H.; et al. N-terminal truncated UCH-L1 prevents Parkinson’s disease associated damage. PLoS ONE 2014, 9, e99654. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Li, H.; Kawamura, R.; Osaka, H.; Wang, Y.L.; Hara, Y.; Hirokawa, T.; Manago, Y.; Amano, T.; Noda, M.; et al. Alterations of structure and hydrolase activity of parkinsonism-associated human ubiquitin carboxyl-terminal hydrolase L1 variants. Biochem. Biophys. Res. Commun. 2003, 304, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.N.; Krantz, B.A.; Wilkinson, K.D. Substrate Specificity of Deubiquitinating Enzymes: Ubiquitin C-Terminal Hydrolases. Biochemistry 1998, 37, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.Y.; Liu, H.; Yao, C.J.; Zhu, X.X.; Chen, D.J.; Yang, J.; Lu, Y.J.; Cao, J.Y. Association between ubiquitin carboxy-terminal hydrolase-L1 S18Y variant and risk of Parkinson’s disease: The impact of ethnicity and onset age. Neurol. Sci. 2015, 36, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Rydning, S.L.; Backe, P.H.; Sousa, M.M.L.; Iqbal, Z.; Øye, A.M.; Sheng, Y.; Yang, M.; Lin, X.; Slupphaug, G.; Nordenmark, T.H.; et al. Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum. Mol. Genet. 2017, 26, 1031–1040. [Google Scholar] [CrossRef]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef]
- Contu, V.R.; Kotake, Y.; Toyama, T.; Okuda, K.; Miyara, M.; Sakamoto, S.; Samizo, S.; Sanoh, S.; Kumagai, Y.; Ohta, S. Endogenous neurotoxic dopamine derivative covalently binds to Parkinson’s disease-associated ubiquitin C-terminal hydrolase L1 and alters its structure and function. J. Neurochem. 2014, 130, 826–838. [Google Scholar] [CrossRef]
- Guingab-Cagmat, J.D.; Stevens, S.M., Jr.; Ratliff, M.V.; Zhang, Z.; Gold, M.S.; Anagli, J.; Wang, K.K.; Kobeissy, F.H. Identification of tyrosine nitration in UCH-L1 and GAPDH. Electrophoresis 2011, 32, 1692–1705. [Google Scholar] [CrossRef]
- Nakamura, T.; Oh, C.K.; Liao, L.; Zhang, X.; Lopez, K.M.; Gibbs, D.; Deal, A.K.; Scott, H.R.; Spencer, B.; Masliah, E.; et al. Noncanonical transnitrosylation network contributes to synapse loss in Alzheimer’s disease. Science 2021, 371, eaaw0843. [Google Scholar] [CrossRef] [PubMed]
- Koharudin, L.M.; Liu, H.; Di Maio, R.; Kodali, R.B.; Graham, S.H.; Gronenborn, A.M. Cyclopentenone prostaglandin-induced unfolding and aggregation of the Parkinson disease-associated UCH-L1. Proc. Natl. Acad. Sci. USA 2010, 107, 6835–6840. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Wakasugi, N.; Tomita, T.; Kikuchi, T.; Mukoyama, M.; Ando, K. Gracile axonal dystrophy (GAD), a new neurological mutant in the mouse. Proc. Soc. Exp. Biol. Med. 1988, 187, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood, N.W. Molecular pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2749–2755, Erratum in Hum Mol Genet 2005, 14, 2749. [Google Scholar] [CrossRef] [PubMed]
- Saigoh, K.; Wang, Y.L.; Suh, J.G.; Yamanishi, T.; Sakai, Y.; Kiyosawa, H.; Harada, T.; Ichihara, N.; Wakana, S.; Kikuchi, T.; et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999, 23, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Kyratzi, E.; Pitychoutis, P.M.; Papadopoulou-Daifoti, Z.; Perier, C.; Vila, M.; Maniati, M.; Ulusoy, A.; Kirik, D.; Park, D.S.; et al. Selective neuroprotective effects of the S18Y polymorphic variant of UCH-L1 in the dopaminergic system. Hum. Mol. Genet. 2012, 21, 874–889. [Google Scholar] [CrossRef] [PubMed]
- Kyratzi, E.; Pavlaki, M.; Stefanis, L. The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Hum. Mol. Genet. 2008, 17, 2160–2171. [Google Scholar] [CrossRef]
- Walters, B.J.; Campbell, S.L.; Chen, P.C.; Taylor, A.P.; Schroeder, D.G.; Dobrunz, L.E.; Artavanis-Tsakonas, K.; Ploegh, H.L.; Wilson, J.A.; Cox, G.A.; et al. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol. Cell Neurosci. 2008, 39, 539–548. [Google Scholar] [CrossRef]
- Chen, F.; Sugiura, Y.; Myers, K.G.; Liu, Y.; Lin, W. Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc. Natl. Acad. Sci. USA 2010, 107, 1636–1641. [Google Scholar] [CrossRef]
- Setsuie, R.; Wang, Y.L.; Mochizuki, H.; Osaka, H.; Hayakawa, H.; Ichihara, N.; Li, H.; Furuta, A.; Sano, Y.; Sun, Y.J.; et al. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem. Int. 2007, 50, 119–129. [Google Scholar] [CrossRef]
- Auburger, G.; Kessler, K.; Kang, J.S.; Gispert, S.; Stoltenburg, G.; Braak, H. Is the PARK5 I93M mutation a cause of Parkinson’s disease with cognitive deficits and cortical Lewy Pathology? In Abstracts 16th International Congress on Parkinson’s Disease and Related Disorders, Berlin, June 2005. Parkinson. Rel. Disord. 2005, 11 (Suppl. S2), 252. [Google Scholar]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Nihira, T.; Ren, Y.R.; Cao, X.Q.; Wada, K.; Setsule, R.; Kabuta, T.; Wada, K.; Hattori, N.; Mizuno, Y.; et al. Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease. J. Neurochem. 2009, 108, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, H.; Li, L.; Liu, W.; Ma, W.; Fang, J.; Tang, L.; et al. Pilot study: Molecular risk factors for diagnosing sporadic Parkinson’s disease based on gene expression in blood in MPTP-induced rhesus monkeys. Oncotarget 2017, 8, 105606–105614. [Google Scholar] [CrossRef] [PubMed]
- Benskey, M.; Behrouz, B.; Sunryd, J.; Pappas, S.S.; Baek, S.H.; Huebner, M.; Lookingland, K.J.; Goudreau, J.L. Recovery of hypothalamic tuberoinfundibular dopamine neurons from acute toxicant exposure is dependent upon protein synthesis and associated with an increase in parkin and ubiquitin carboxy-terminal hydrolase-L1 expression. Neurotoxicology 2012, 33, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W. The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): A promising biomarker and potential therapeutic target of human diseases. Genes Dis. 2020, 8, 640–654. [Google Scholar] [CrossRef]
- Park, G.H.; Park, J.H.; Chung, K.C. Precise control of mitophagy through ubiquitin proteasome system and deubiquitin proteases and their dysfunction in Parkinson’s disease. BMB Rep. 2021, 54, 592–600. [Google Scholar] [CrossRef]
- Buneeva, O.A.; Medvedev, A.E. DJ-1 Protein and Its Role in the Development of Parkinson’s Disease: Studies on Experimental Models. Biochemistry 2021, 86, 627–640. [Google Scholar] [CrossRef]
- Jara, J.H.; Genç, B.; Cox, G.A.; Bohn, M.C.; Roos, R.P.; Macklis, J.D.; Ulupınar, E.; Özdinler, P.H. Corticospinal Motor Neurons Are Susceptible to Increased ER Stress and Display Profound Degeneration in the Absence of UCHL1 Function. Cereb. Cortex 2015, 25, 259–272. [Google Scholar] [CrossRef]
- Meray, R.K.; Lansbury, P.T., Jr. Reversible monoubiquitination regulates the Parkinson disease-associated ubiquitin hydrolase UCH-L1. J Biol Chem. 2007, 282, 10567–10575. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, F.M.; von Stockum, S.; Giacomello, M.; Goliand, I.; Kakimoto, P.; Marchesan, E.; De Stefani, D.; Kowaltowski, A.J.; Ziviani, E.; Shirihai, O.S. A new target for an old DUB: UCH-L1 regulates mitofusin-2 levels, altering mitochondrial morphology, function and calcium uptake. Redox Biol. 2020, 37, 101676. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Symbol | Gene | Protein Product | Relation to UPS | Type of PD | Inheritance | References |
|---|---|---|---|---|---|---|
| PARK1 | SNCA | Alpha-synuclein | Ubiquitination substrate | Classical and early-onset PD | AD * | [14,15] |
| PARK2 | Parkin | Parkin | E3 ubiquitin ligase | Early-onset PD | AR ** | [16,17] |
| PARK5 | UCHL1 | Ubiquitin C-terminal hydrolase L1 | Deubiquitinase | Classical PD | AD | [18] |
| PARK6 | PINK1 | PTEN-induced kinase 1 | Phosphorylates ubiquitination substrate E3 ubiquitin ligase | Early-onset PD | AR | [19,20] |
| PARK7 | DJ-1 | DJ-1 | Ubiquitination substrate | Early-onset PD | AR | [21,22,23] |
| PARK8 | LRRK2 | Leucine-rich repeat kinase 2 | Ubiquitination substrate | Classical PD | AD | [24,25,26] |
| PARK10 | USP24 | Ubiquitin-specific peptidase 24 | Deubiquitinase | Late-onset PD | Risk factor | [27,28] |
| PARK11 | GIGYF2 | Grb10-interacting GYF protein-2 | Could promote ligand-induced ubiquitination of IGF1R | Classical PD | AD | [29,30] |
| PARK13 | HTRA2 | Htra2 serine peptidase 2 | Cell stress response | Susceptibility locus for PD | AD | [31] |
| PARK15 | FBXO7 | Fbxo7 | E3 ubiquitin ligase | Early-onset PD | AR | [32] |
| # | Accession Number | Gene Name | Protein Name | Function | Localization | References |
|---|---|---|---|---|---|---|
| 1 | P00533 | EGFR | Epidermal growth factor receptor | 3 | PM, EPR, M | [105] |
| 2 | P05067 | APP | Amyloid-beta precursor protein | 3 | PM, EPR, M | [106] |
| 3 | Q14568 | HSP90 * | Heat Shock Protein 90-alpha A2 | 4 | C, N, Mch, PM | [107,108,109,110,111,112] |
| 4 | P37840 | SNCA * | Alpha-synuclein | 3 | C, M, N | [79,92,113] |
| 5 | P32119 | PRDX2 | Peroxiredoxin-2 | 4 | C | [114,115] |
| 6 | P62987 | UBA52 * | Monoubiquitin | 5 | C, N, PM, Mch | [88] |
| 7 | O60260 | PARK2 * | E3 ubiquitin-protein ligase parkin | 5 | C, N, EPR, Mch | [116,117] |
| 8 | P34931 | HS71L * | Heat shock 70 kDa protein 1-like | 4 | C, N | [111,112] |
| 9 | P14618 | KPYM | Pyruvate kinase PKM | 1 | C | [83] |
| 10 | - | - | Tubulins a | 2 | C | [77,92] |
| Mutation | Hydrolase Activity Versus WT UCHL1, % | Reference |
|---|---|---|
| N-terminal deletion (residues 1–11) | 0 | [125] |
| E7A | 50% | [80] |
| S18Y | 113% | [126] |
| D30K | 0 | [88] |
| Q73R | 97% | [88,127] |
| C90S | <<1% or 0 | [88,127] |
| I93M | 50% | [18] |
| C152A | ~85% | [128] |
| H97Q, H97N | 85–87% | [127] |
| H161D | <1% | [127] |
| H161K, H161N, H161Q, H161Y | <<1% | [127] |
| D176N | 2.5% | [127] |
| R178Q | 400% | [129] |
| A216N | Not measurable | [129] |
| C-terminal deletion (residues 220–223) | Not determined | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buneeva, O.; Medvedev, A. Ubiquitin Carboxyl-Terminal Hydrolase L1 and Its Role in Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 1303. https://doi.org/10.3390/ijms25021303
Buneeva O, Medvedev A. Ubiquitin Carboxyl-Terminal Hydrolase L1 and Its Role in Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(2):1303. https://doi.org/10.3390/ijms25021303
Chicago/Turabian StyleBuneeva, Olga, and Alexei Medvedev. 2024. "Ubiquitin Carboxyl-Terminal Hydrolase L1 and Its Role in Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 2: 1303. https://doi.org/10.3390/ijms25021303
APA StyleBuneeva, O., & Medvedev, A. (2024). Ubiquitin Carboxyl-Terminal Hydrolase L1 and Its Role in Parkinson’s Disease. International Journal of Molecular Sciences, 25(2), 1303. https://doi.org/10.3390/ijms25021303