
GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine

Abstract
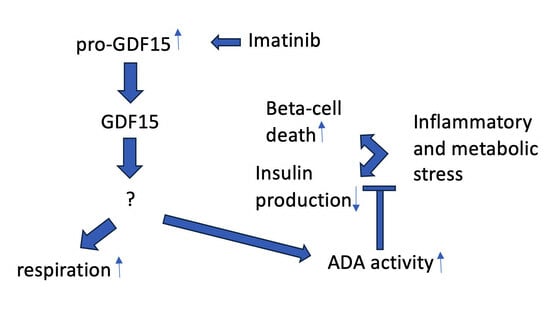
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. GDF15 Protein Expression in Human Islet and EndoC-betaH1 Cells
2.2. Imatinib Increases GDF15 Protein Expression in EndoC-betaH1 Cells
2.3. The Proliferation of EndoC-betaH1 Cells Is Unaffected by GDF15
2.4. ATP-Coupled Oxygen Consumption Is Stimulated by GDF15 under Control Conditions but Not in Cytokine-Treated Human Islets
2.5. GDF15 Increases Insulin Release and Content in EndoC-betaH1 Cells Treated with Cytokines or Palmitate + High Glucose
2.6. GDF15 Partially Protects EndoC-betaH1 and Human Islet Cells from Palmitate + High-Glucose- and Cytokine-Induced Cell Death
2.7. GDF15 Stimulates Adenosine Deminase (ADA)-Mediated Conversion of Adenosine to Inosine
2.8. ADA Downregulation Counteracts GDF15-Induced Protection against EndoC-betaH1 Cell Death
3. Discussion
4. Materials and Methods
4.1. Human Pancreatic Islet Culture
4.2. Human EndoC-betaH1 Cell Culture
4.3. Immunocytochemistry (ICC)
4.4. Immunoblot Analysis
4.5. Evaluation of Cell Viability
4.6. Cell Proliferation
4.7. Glucose-Stimulated Insulin Secretion (GSIS)
4.8. Oxygen Consumption and Extracellular Acidification Rates
4.9. siRNA-Mediated Silencing of Adenosine Deaminase (ADA)
4.10. Inosine Detection
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | Adenosine deaminase |
| AMPK | AMP-dependent kinase |
| BSA | Bovine serum albumin |
| ECAR | Extracellular acidification rate |
| DAPI | 4’,6-Diamidino-2-phenylindole |
| FCCP | Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| FPKM | Fragments per kilobase of exon model per million reads mapped |
| GDF15 | Growth differentiation factor 15 |
| GFRAL | Glial cell-derived neurotrophic factor family receptor alpha-like |
| IFN-γ | Interferon-γ |
| IL-1β | Interleukin 1β |
| KRBH | Krebs-Ringer bicarbonate buffer |
| OCR | Oxygen consumption rate |
| PI | Propidium iodide |
| RET | Rearranged during transfection |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TXNIP | Thioredoxin-interacting protein |
References
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Jin, X.; Hsiao, E.C.; McGrath, S.A.; Esquela, A.F.; Koniaris, L.G. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 2005, 23, 543–548. [Google Scholar] [PubMed]
- Jena, J.; García-Peña, L.M.; Pereira, R.O. The roles of FGF21 and GDF15 in mediating the mitochondrial integrated stress response. Front. Endocrinol. 2023, 14, 1264530. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Day, E.A.; Townsend, L.K.; Djordjevic, D.; Jørgensen, S.B.; Steinberg, G.R. GDF15: Emerging biology and therapeutic applications for obesity and cardiometabolic disease. Nat. Rev. Endocrinol. 2021, 17, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Fu, J. GDF15 as a key disease target and biomarker: Linking chronic lung diseases and ageing. Mol. Cell Biochem. 2023, 24, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lodi, R.S.; Yu, B.; Xia, L.; Liu, F. Roles and Regulation of Growth differentiation factor-15 in the Immune and tumor microenvironment. Hum. Immunol. 2021, 82, 937–944. [Google Scholar] [CrossRef]
- Xue, X.H.; Tao, L.L.; Su, D.Q.; Guo, C.J.; Liu, H. Diagnostic utility of GDF15 in neurodegenerative diseases: A systematic review and meta-analysis. Brain Behav. 2022, 12, e2502. [Google Scholar] [CrossRef]
- Asrih, M.; Wei, S.; Nguyen, T.T.; Yi, H.S.; Ryu, D.; Gariani, K. Overview of growth differentiation factor 15 in metabolic syndrome. J. Cell Mol. Med. 2023, 27, 1157–1167. [Google Scholar] [CrossRef]
- Yang, L.; Chang, C.C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjær, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P.; Silvestre, R.A.; Díez, J.J. Growth differentiation factor 15 (GDF-15) in endocrinology. Endocrine 2023, 81, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Pare, G.; Hess, S.; Ford, R.J.; Sjaarda, J.; Raman, K.; McQueen, M.; Lee, S.; Haenel, H.; Steinberg, G.R.; et al. Growth Differentiation Factor 15 as a Novel Biomarker for Metformin. Diabetes Care 2017, 40, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Recarte, D.; Barroso, E.; Palomer, X.; Wahli, W.; Vázquez-Carrera, M. Knocking on GDF15’s door for the treatment of type 2 diabetes mellitus. Trends Endocrinol. Metab. 2022, 33, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Recarte, D.; Barroso, E.; Zhang, M.; Rada, P.; Pizarro-Delgado, J.; Peña, L.; Palomer, X.; Valverde, Á.M.; Wahli, W.; Vázquez-Carrera, M. A positive feedback loop between AMPK and GDF15 promotes metformin antidiabetic effects. Pharmacol. Res. 2023, 187, 106578. [Google Scholar] [CrossRef] [PubMed]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 2020, 578, E24. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Smith, B.K.; Mohammadi-Shemirani, P.; Morrow, M.R.; Gutgesell, R.M.; Lu, R.; Raphenya, A.R.; Kabiri, M.; McArthur, A.G.; et al. Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat. Metab. 2019, 1, 1202–1208. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Sang, T.; Chen, C.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. NAG-1/GDF15 protects against streptozotocin-induced type 1 diabetes by inhibiting apoptosis, preserving beta-cell function, and suppressing inflammation in pancreatic islets. Mol. Cell Endocrinol. 2022, 549, 111643. [Google Scholar] [CrossRef]
- Zhang, H.; Mulya, A.; Nieuwoudt, S.; Vandanmagsar, B.; McDowell, R.; Heintz, E.C.; Zunica, E.R.M.; Collier, J.J.; Bozadjieva-Kramer, N.; Seeley, R.J.; et al. GDF15 Mediates the Effect of Skeletal Muscle Contraction on Glucose-Stimulated Insulin Secretion. Diabetes 2023, 72, 1070–1082. [Google Scholar] [CrossRef]
- Mohammad, M.G.; Saeed, R.; Mohammed, A.K.; Khalique, A.; Hamad, M.; El-Huneidi, W.; Hamad, M.; Taneera, J. GDF15 plays a critical role in insulin secretion in INS-1 cells and human pancreatic islets. Exp. Biol. Med. 2023, 248, 339–349. [Google Scholar] [CrossRef]
- Lertpatipanpong, P.; Lee, J.; Kim, I.; Eling, T.; Oh, S.Y.; Seong, J.K.; Baek, S.J. The anti-diabetic effects of NAG-1/GDF15 on HFD/STZ-induced mice. Sci. Rep. 2021, 11, 15027. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Dusaulcy, R.; Gosmain, Y.; Philippe, J.; Somm, E.; Jornayvaz, F.R.; Kang, B.E.; Jo, Y.; Choi, M.J.; Yi, H.S.; et al. Growth differentiation factor-15 prevents glucotoxicity and connexin-36 downregulation in pancreatic beta-cells. Mol. Cell Endocrinol. 2022, 541, 111503. [Google Scholar] [CrossRef] [PubMed]
- Veneri, D.; Franchini, M.; Bonora, E. Imatinib and regression of type 2 diabetes. N. Engl. J. Med. 2005, 352, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Gitelman, S.E.; Bundy, B.N.; Ferrannini, E.; Lim, N.; Blanchfield, J.L.; DiMeglio, L.A.; Felner, E.I.; Gaglia, J.L.; Gottlieb, P.A.; Long, S.A.; et al. Imatinib therapy for patients with recent-onset type 1 diabetes: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2021, 9, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Hägerkvist, R.; Sandler, S.; Mokhtari, D.; Welsh, N. Amelioration of diabetes by imatinib mesylate (Gleevec): Role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J. 2007, 21, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Elksnis, A.; Schiffer, T.A.; Palm, F.; Wang, Y.; Cen, J.; Turpaev, K.; Ngamjariyawat, A.; Younis, S.; Huang, S.; Shen, Y.; et al. Imatinib protects against human beta-cell death via inhibition of mitochondrial respiration and activation of AMPK. Clin. Sci. 2021, 135, 2243–2263. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.J.; Eling, T. Growth differentiation factor 15 (GDF15): A survival protein with therapeutic potential in metabolic diseases. Pharmacol. Ther. 2019, 198, 46–58. [Google Scholar] [CrossRef]
- Fred, R.G.; Kappe, C.; Ameur, A.; Cen, J.; Bergsten, P.; Ravassard, P.; Scharfmann, R.; Welsh, N. Role of the AMP kinase in cytokine-induced human EndoC-βH1 cell death. Mol. Cell Endocrinol. 2015, 414, 53–63. [Google Scholar] [CrossRef]
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 β-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis. 2011, 2, e134. [Google Scholar] [CrossRef]
- Piro, S.; Rabuazzo, A.M.; Renis, M.; Purrello, F. Effects of metformin on oxidative stress, adenine nucleotides balance, and glucose-induced insulin release impaired by chronic free fatty acids exposure in rat pancreatic islets. J. Endocrinol. Investig. 2012, 35, 504–510. [Google Scholar] [CrossRef]
- Liu, H.; Liu, J.; Si, L.; Guo, C.; Liu, W.; Liu, Y. GDF-15 promotes mitochondrial function and proliferation in neuronal HT22 cells. J. Cell Biochem. 2019, 120, 10530–10547. [Google Scholar] [CrossRef]
- Xu, G.; Chen, J.; Jo, S.; Grayson, T.B.; Ramanadham, S.; Koizumi, A.; Germain-Lee, E.L.; Lee, S.J.; Shalev, A. Deletion of Gdf15 Reduces ER Stress-induced Beta-cell Apoptosis and Diabetes. Endocrinology 2022, 163, bqac030. [Google Scholar] [CrossRef] [PubMed]
- Ngamjariyawat, A.; Cen, J.; Said, R.; Incedal, C.; Idevall-Hagren, O.; Welsh, N. Metabolic stress-induced human beta-cell death is mediated by increased intracellular levels of adenosine. Front. Endocrinol. 2023, 14, 1060675. [Google Scholar] [CrossRef]
- Fred, R.G.; Boddeti, S.K.; Lundberg, M.; Welsh, N. Imatinib mesylate stimulates low-density lipoprotein receptor-related protein 1-mediated ERK phosphorylation in insulin-producing cells. Clin. Sci. 2015, 128, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, D.; Al-Amin, A.; Turpaev, K.; Li, T.; Idevall-Hagren, O.; Li, J.; Wuttke, A.; Fred, R.G.; Ravassard, P.; Scharfmann, R.; et al. Imatinib mesilate-induced phosphatidylinositol 3-kinase signalling and improved survival in insulin-producing cells: Role of Src homology 2-containing inositol 5’-phosphatase interaction with c-Abl. Diabetologia 2013, 56, 1327–1338. [Google Scholar] [CrossRef]
- Mahmood, D.F.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef]
- Mendsaikhan, A.; Takeuchi, S.; Walker, D.G.; Tooyama, I. Differences in Gene Expression Profiles and Phenotypes of Differentiated SH-SY5Y Neurons Stably Overexpressing Mitochondrial Ferritin. Front. Mol. Neurosci. 2019, 11, 470. [Google Scholar] [CrossRef]
- Schulten, H.J.; Bakhashab, S. Meta-Analysis of Microarray Expression Studies on Metformin in Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3173. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Csóka, B.; Pacher, P.; Haskó, G. Adenosine signalling in diabetes mellitus—Pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 2015, 11, 228–241. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanet J. Rare Dis. 2018, 13, 65. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Sakhatskyy, P.; Newton, J.; Shamirian, P.; Hsiao, V.; Curren, S.; Gabino Miranda, G.A.; Pedroza, M.; Blackburn, M.R.; Rounds, S. Sustained adenosine exposure causes lung endothelial apoptosis: A possible contributor to cigarette smoke-induced endothelial apoptosis and lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L361–L370. [Google Scholar] [CrossRef]
- Yaguchi, T.; Saito, M.; Yasuda, Y.; Nishizaki, T. Caspase-4 activation in association with decreased adenosine deaminase activity may be a factor for gastric ulcer. Digestion 2010, 81, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Kanno, T.; Nagaya, T.; Kuribayashi, K.; Nakano, T.; Gotoh, A.; Nishizaki, T. Adenosine deaminase inhibitor EHNA exhibits a potent anticancer effect against malignant pleural mesothelioma. Cell Physiol. Biochem. 2015, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Mello Pde, A.; Filippi-Chiela, E.C.; Nascimento, J.; Beckenkamp, A.; Santana, D.B.; Kipper, F.; Casali, E.A.; Nejar Bruno, A.; Paccez, J.D.; Zerbini, L.F.; et al. Adenosine uptake is the major effector of extracellular ATP toxicity in human cervical cancer cells. Mol. Biol. Cell 2014, 25, 2905–2918. [Google Scholar] [CrossRef]
- Conte, M.; Giuliani, C.; Chiariello, A.; Iannuzzi, V.; Franceschi, C.; Salvioli, S. GDF15, an emerging key player in human aging. Ageing Res. Rev. 2022, 75, 101569. [Google Scholar] [CrossRef]
- Krizhanovskii, C.; Kristinsson, H.; Elksnis, A.; Wang, X.; Gavali, H.; Bergsten, P.; Scharfman, R.; Welsh, N. EndoC-bH1 cells display increased sensitivity to sodium palmitate when cultured in DMEM/F12 medium. Islets 2017, 9, e1296995. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngamjariyawat, A.; Cen, J.; Wang, X.; Welsh, N. GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine. Int. J. Mol. Sci. 2024, 25, 801. https://doi.org/10.3390/ijms25020801
Ngamjariyawat A, Cen J, Wang X, Welsh N. GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine. International Journal of Molecular Sciences. 2024; 25(2):801. https://doi.org/10.3390/ijms25020801
Chicago/Turabian StyleNgamjariyawat, Anongnad, Jing Cen, Xuan Wang, and Nils Welsh. 2024. "GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine" International Journal of Molecular Sciences 25, no. 2: 801. https://doi.org/10.3390/ijms25020801
APA StyleNgamjariyawat, A., Cen, J., Wang, X., & Welsh, N. (2024). GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine. International Journal of Molecular Sciences, 25(2), 801. https://doi.org/10.3390/ijms25020801