
Human Fallopian Tube-Derived Organoids with TP53 and RAD51D Mutations Recapitulate an Early Stage High-Grade Serous Ovarian Cancer Phenotype In Vitro

 ,
,
Abstract
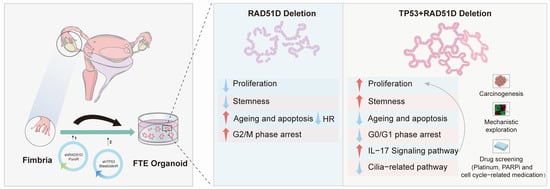
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Establishment of Human Fallopian Tube-Derived Organoids and the Stable Knockdown of TP53 and RAD51D
2.2. Effects of TP53 and RAD51D Knockdown on Organoid Proliferation and Ciliated Cell Differentiation
2.3. Impact of TP53 ± RAD51D Knockdown on Homologous Recombination, Cell Senescence, and Apoptosis in Human Fallopian Tube Organoids
2.4. Cell Cycle in TP53 ± RAD51D Knockdown Organoids
2.5. Transcriptomic Profiles of Fallopian Tube Organoids of Varied Genotypes
2.6. TP53 + RAD51D Knockdown Upregulating the IL-17 Signaling Pathway
2.7. Sensitivity of TP53 ± RAD51D Knockdown Organoids to Platinum-Based PARP Inhibitors and Cell cycle-related Medication
3. Discussion
4. Materials and Methods
4.1. Epithelial Progenitor Isolation from Fallopian Tube Samples
4.2. Organoid Culture
4.3. Cloning of shRNAs and Virus Production
4.4. Western Blotting
4.5. Organoid Growth Assay
4.6. Immunofluorescent and IHC
4.7. Flow Cytometry and Cell Cycle Analysis
4.8. Cell Senescence and β-galactosidase
4.9. Apoptosis Caspase-Glo3/7 3D Assay
4.10. Drug Sensitivity Assays
4.11. RNA Extraction and qRT-PCR
4.12. RNA Sequencing and Analysis
4.13. ELISA
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Henderson, J.T.; Webber, E.M.; Sawaya, G.F. Screening for Ovarian Cancer. JAMA 2018, 319, 595. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih Ie, M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanova, A.; Vang, R.; Kshirsagar, M.; Lu, D.; Marks, M.A.; Shih Ie, M.; Kurman, R.J. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: An immunohistochemical and nucleotide sequencing analysis. Mod. Pathol. 2011, 24, 1248–1253. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Levanon, K.; Ng, V.; Piao, H.Y.; Zhang, Y.; Chang, M.C.; Roh, M.H.; Kindelberger, D.W.; Hirsch, M.S.; Crum, C.P.; Marto, J.A.; et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 2010, 29, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; Li, J.; Jia, S.; Meng, Y.; Zhang, C.; Wang, L.; Di, L.J. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin. Cancer Res. 2017, 23, 7400–7411. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.T.; Baggerly, K.A.; Patterson, A.P.; Liu, J.; Broaddus, R.; Frumovitz, M.; Atkinson, E.N.; Smith, D.I.; Hartmann, L.; Fishman, D.; et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin. Cancer Res. 2005, 11, 6116–6126. [Google Scholar] [CrossRef]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Bernstein, K.A. RAD-ical New Insights into RAD51 Regulation. Genes 2018, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Xu, J.; Dai, Y.; Gao, Y.; Chai, R.; Lu, C.; Yu, B.; Kang, Y.; Xu, C. RAD51D Secondary Mutation-Mediated Resistance to PARP-Inhibitor-Based Therapy in HGSOC. Int. J. Mol. Sci. 2023, 24, 14476. [Google Scholar] [CrossRef]
- Smiraldo, P.G.; Gruver, A.M.; Osborn, J.C.; Pittman, D.L. Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res. 2005, 65, 2089–2096. [Google Scholar] [CrossRef]
- Dai, Y.L.; Wei, J.S.; Xu, J.; Li, K.; Wang, J.; Cha, R.R.; Kang, Y. Progress in the Application of Ovarian and Fallopian Tube Organoid. Reprod. Dev. Med. 2021, 5, 174. [Google Scholar] [CrossRef]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019, 10, 5367. [Google Scholar] [CrossRef]
- Hoffmann, K.; Berger, H.; Kulbe, H.; Thillainadarasan, S.; Mollenkopf, H.J.; Zemojtel, T.; Taube, E.; Darb-Esfahani, S.; Mangler, M.; Sehouli, J.; et al. Stable expansion of high-grade serous ovarian cancer organoids requires a low-Wnt environment. EMBO J. 2020, 39, e104013. [Google Scholar] [CrossRef] [PubMed]
- Lõhmussaar, K.; Kopper, O.; Korving, J.; Begthel, H.; Vreuls, C.P.H.; van Es, J.H.; Clevers, H. Assessing the origin of high-grade serous ovarian cancer using CRISPR-modification of mouse organoids. Nat. Commun. 2020, 11, 2660. [Google Scholar] [CrossRef]
- Zhang, S.; Iyer, S.; Ran, H.; Dolgalev, I.; Gu, S.; Wei, W.; Foster, C.J.R.; Loomis, C.A.; Olvera, N.; Dao, F.; et al. Genetically Defined, Syngeneic Organoid Platform for Developing Combination Therapies for Ovarian Cancer. Cancer Discov. 2021, 11, 362–383. [Google Scholar] [CrossRef]
- Iyer, S.; Zhang, S.; Yucel, S.; Horn, H.; Smith, S.G.; Reinhardt, F.; Hoefsmit, E.; Assatova, B.; Casado, J.; Meinsohn, M.C.; et al. Genetically defined syngeneic mouse models of ovarian cancer as tools for the discovery of combination immunotherapy. Cancer Discov. 2020, 11, 384–407. [Google Scholar] [CrossRef] [PubMed]
- Yucer, N.; Ahdoot, R.; Workman, M.J.; Laperle, A.H.; Recouvreux, M.S.; Kurowski, K.; Naboulsi, D.J.; Liang, V.; Qu, Y.; Plummer, J.T.; et al. Human iPSC-derived fallopian tube organoids with BRCA1 mutation recapitulate early-stage carcinogenesis. Cell Rep. 2021, 37, 110146. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Hoffmann, K.; Fritsche, K.; Brinkmann, V.; Mollenkopf, H.J.; Thieck, O.; Teixeira da Costa, A.R.; Braicu, E.I.; Sehouli, J.; Mangler, M.; et al. Chronic Chlamydia infection in human organoids increases stemness and promotes age-dependent CpG methylation. Nat. Commun. 2019, 10, 1194. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, L.; Li, Y.; Ma, X.; Dai, W.; Gao, X.; Rao, X.; Fu, G.; Wang, R.; Pan, M.; et al. Organoid modelling identifies that DACH1 functions as a tumour promoter in colorectal cancer by modulating BMP signalling. eBioMedicine 2020, 56, 102800. [Google Scholar] [CrossRef]
- Laury, A.R.; Hornick, J.L.; Perets, R.; Krane, J.F.; Corson, J.; Drapkin, R.; Hirsch, M.S. PAX8 reliably distinguishes ovarian serous tumors from malignant mesothelioma. Am. J. Surg. Pathol. 2010, 34, 627–635. [Google Scholar] [CrossRef]
- Paik, D.Y.; Janzen, D.M.; Schafenacker, A.M.; Velasco, V.S.; Shung, M.S.; Cheng, D.; Huang, J.; Witte, O.N.; Memarzadeh, S. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: A site for injury and serous cancer initiation. Stem Cells 2012, 30, 2487–2497. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, J.; G Manjón, A.; Kielbassa, K.; Feringa, F.M.; Freire, R.; Medema, R.H. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 2018, 46, 10132–10144. [Google Scholar] [CrossRef]
- Rodrigue, A.; Coulombe, Y.; Jacquet, K.; Gagné, J.P.; Roques, C.; Gobeil, S.; Poirier, G.; Masson, J.Y. The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. J. Cell Sci. 2013, 126 Pt 1, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef]
- Shpak, M.; Goldberg, M.M.; Cowperthwaite, M.C. Cilia gene expression patterns in cancer. Cancer Genom. Proteom. 2014, 11, 13–24. [Google Scholar]
- Huangfu, L.; Li, R.; Huang, Y.; Wang, S. The IL-17 family in diseases: From bench to bedside. Signal Transduct. Target. Ther. 2023, 8, 402. [Google Scholar] [CrossRef]
- Majumder, S.; McGeachy, M.J. IL-17 in the Pathogenesis of Disease: Good Intentions Gone Awry. Annu. Rev. Immunol. 2021, 39, 537–556. [Google Scholar] [CrossRef]
- Song, Y.; Yang, J.M. Role of interleukin (IL)-17 and T-helper (Th)17 cells in cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Zhang, J. Interleukin-17 promotes the development of ovarian cancer through upregulation of MTA1 expression. Am. J. Cancer Res. 2022, 12, 5646–5656. [Google Scholar]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2015, 34, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Reynolds, J.M.; Pappu, B.P.; Chen, G.; Martinez, G.J.; Dong, C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity 2011, 35, 611–621. [Google Scholar] [CrossRef]
- Nies, J.F.; Panzer, U. IL-17C/IL-17RE: Emergence of a Unique Axis in T(H)17 Biology. Front. Immunol. 2020, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Jungnickel, C.; Schmidt, L.H.; Bittigkoffer, L.; Wolf, L.; Wolf, A.; Ritzmann, F.; Kamyschnikow, A.; Herr, C.; Menger, M.D.; Spieker, T.; et al. IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene 2017, 36, 4182–4190. [Google Scholar] [CrossRef]
- Hatchi, E.; Livingston, D.M. Opening a Door to PARP Inhibitor-Induced Lethality in HR-Proficient Human Tumor Cells. Cancer Cell 2020, 37, 139–140. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmana, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Waterman, D.P.; Haber, J.E.; Smolka, M.B. Checkpoint Responses to DNA Double-Strand Breaks. Annu. Rev. Biochem. 2020, 89, 103–133. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef]
- Feng, W.; Jasin, M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 2017, 8, 525. [Google Scholar] [CrossRef]
- Dillon, M.T.; Good, J.S.; Harrington, K.J. Selective targeting of the G2/M cell cycle checkpoint to improve the therapeutic index of radiotherapy. Clin. Oncol. R. Coll. Radiol. 2014, 26, 257–265. [Google Scholar] [CrossRef]
- Larsen, B.D.; Benada, J.; Yung, P.Y.K.; Bell, R.A.V.; Pappas, G.; Urban, V.; Ahlskog, J.K.; Kuo, T.T.; Janscak, P.; Megeney, L.A.; et al. Cancer cells use self-inflicted DNA breaks to evade growth limits imposed by genotoxic stress. Science 2022, 376, 476–483. [Google Scholar] [CrossRef]
- Galhenage, P.; Zhou, Y.; Perry, E.; Loc, B.; Fietz, K.; Iyer, S.; Reinhardt, F.; Da Silva, T.; Botchkarev, V.; Chen, J.; et al. Replication stress and defective checkpoints make fallopian tube epithelial cells putative drivers of high-grade serous ovarian cancer. Cell Rep. 2023, 42, 113144. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, N.; Wang, S.; Wang, Y.; Yang, X.; Fan, J.; Chen, Y. Effects of Follicular Fluid on Physiological Characteristics and Differentiation of Fallopian Tube Epithelial Cells Implicating for Ovarian Cancer Pathogenesis. Int. J. Mol. Sci. 2023, 24, 10154. [Google Scholar] [CrossRef]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Lim, J.S.; Turner, N.C.; Yap, T.A. CDK4/6 Inhibitors: Promising Opportunities beyond Breast Cancer. Cancer Discov. 2016, 6, 697–699. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin. Cancer Res. 2013, 19, 5960–5971. [Google Scholar] [CrossRef] [PubMed]
- Au-Yeung, G.; Lang, F.; Azar, W.J.; Mitchell, C.; Jarman, K.E.; Lackovic, K.; Aziz, D.; Cullinane, C.; Pearson, R.B.; Mileshkin, L.; et al. Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin-Dependent Kinase 2 and AKT Inhibition. Clin. Cancer Res. 2017, 23, 1862–1874. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Chen, J.; Bae, E.; Zhang, L.; Hughes, K.; Parmigiani, G.; Braun, D.; Rebbeck, T.R. Penetrance of Breast and Ovarian Cancer in Women Who Carry a BRCA1/2 Mutation and Do Not Use Risk-Reducing Salpingo-Oophorectomy: An Updated Meta-Analysis. JNCI Cancer Spectr. 2020, 4, pkaa029. [Google Scholar] [CrossRef]
- Armes, J.E.; Trute, L.; White, D.; Southey, M.C.; Hammet, F.; Tesoriero, A.; Hutchins, A.M.; Dite, G.S.; McCredie, M.R.; Giles, G.G.; et al. Distinct molecular pathogeneses of early-onset breast cancers in BRCA1 and BRCA2 mutation carriers: A population-based study. Cancer Res. 1999, 59, 2011–2017. [Google Scholar] [PubMed]
- Li, S.; Silvestri, V.; Leslie, G.; Rebbeck, T.R.; Neuhausen, S.L.; Hopper, J.L.; Nielsen, H.R.; Lee, A.; Yang, X.; McGuffog, L.; et al. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J. Clin. Oncol. 2022, 40, 1529–1541. [Google Scholar] [CrossRef]
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.X.; He, H.W.; Cai, X.Y.; Zhou, J.; Cheng, Y.F.; Fan, J.; et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Liang, J.; Wang, L.; Li, W.; Jiang, S.; Xu, S.; Tang, L.; Du, Q.; Liu, G.; Meng, H.; et al. IL-17A functions and the therapeutic use of IL-17A and IL-17RA targeted antibodies for cancer treatment. Int. Immunopharmacol. 2023, 123, 110757. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Yang, L.; Shen, R.; Kong, B.; Chen, W.; Liang, J.; Tang, G.; Zhang, B. Th17 cells regulate the production of CXCL1 in breast cancer. Int. Immunopharmacol. 2018, 56, 320–329. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Huang, X.; Hao, J.; Tan, Y.Q.; Zhu, T.; Pandey, V.; Lobie, P.E. CXC Chemokine Signaling in Progression of Epithelial Ovarian Cancer: Theranostic Perspectives. Int. J. Mol. Sci. 2022, 23, 2642. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.L.; Kabir, S.M.; Lee, E.S.; Son, D.S. CXCR2-driven ovarian cancer progression involves upregulation of proinflammatory chemokines by potentiating NF-κB activation via EGFR-transactivated Akt signaling. PLoS ONE 2013, 8, e83789. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Liu, G.; Yang, F.; Guo, X.; Xiao, X.; Xue, F.; Mercado-Uribe, I.; Huang, J.; Lin, S.H.; et al. CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin. Cancer Res. 2010, 16, 3875–3886. [Google Scholar] [CrossRef]
- Peng, Y.; Kajiyama, H.; Yuan, H.; Nakamura, K.; Yoshihara, M.; Yokoi, A.; Fujikake, K.; Yasui, H.; Yoshikawa, N.; Suzuki, S.; et al. PAI-1 secreted from metastatic ovarian cancer cells triggers the tumor-promoting role of the mesothelium in a feedback loop to accelerate peritoneal dissemination. Cancer Lett. 2019, 442, 181–192. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, R.C.; Zhang, X.L.; Niu, X.L.; Qu, Y.; Li, L.Z.; Meng, X.Y. Interleukin-8 secretion by ovarian cancer cells increases anchorage-independent growth, proliferation, angiogenic potential, adhesion and invasion. Cytokine 2012, 59, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, W.; Zhang, N.; Di, W. The Role of CCL20-CCR6 Axis in Ovarian Cancer Metastasis. OncoTargets Ther. 2020, 13, 12739–12750. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Fritz, Y.; Dawes, S.M.; Diaconu, D.; Al-Attar, P.M.; Guzman, A.M.; Chen, C.S.; Fu, W.; Gudjonsson, J.E.; McCormick, T.S.; et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J. Immunol. 2013, 190, 2252–2262. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Y.; Xu, J.; Gong, X.; Wei, J.; Gao, Y.; Chai, R.; Lu, C.; Zhao, B.; Kang, Y. Human Fallopian Tube-Derived Organoids with TP53 and RAD51D Mutations Recapitulate an Early Stage High-Grade Serous Ovarian Cancer Phenotype In Vitro. Int. J. Mol. Sci. 2024, 25, 886. https://doi.org/10.3390/ijms25020886
Dai Y, Xu J, Gong X, Wei J, Gao Y, Chai R, Lu C, Zhao B, Kang Y. Human Fallopian Tube-Derived Organoids with TP53 and RAD51D Mutations Recapitulate an Early Stage High-Grade Serous Ovarian Cancer Phenotype In Vitro. International Journal of Molecular Sciences. 2024; 25(2):886. https://doi.org/10.3390/ijms25020886
Chicago/Turabian StyleDai, Yilin, Jing Xu, Xiaofeng Gong, Jinsong Wei, Yi Gao, Ranran Chai, Chong Lu, Bing Zhao, and Yu Kang. 2024. "Human Fallopian Tube-Derived Organoids with TP53 and RAD51D Mutations Recapitulate an Early Stage High-Grade Serous Ovarian Cancer Phenotype In Vitro" International Journal of Molecular Sciences 25, no. 2: 886. https://doi.org/10.3390/ijms25020886
APA StyleDai, Y., Xu, J., Gong, X., Wei, J., Gao, Y., Chai, R., Lu, C., Zhao, B., & Kang, Y. (2024). Human Fallopian Tube-Derived Organoids with TP53 and RAD51D Mutations Recapitulate an Early Stage High-Grade Serous Ovarian Cancer Phenotype In Vitro. International Journal of Molecular Sciences, 25(2), 886. https://doi.org/10.3390/ijms25020886