
Therapeutic Delivery of Soluble Fractalkine Ameliorates Vascular Dysfunction in the Diabetic Retina

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
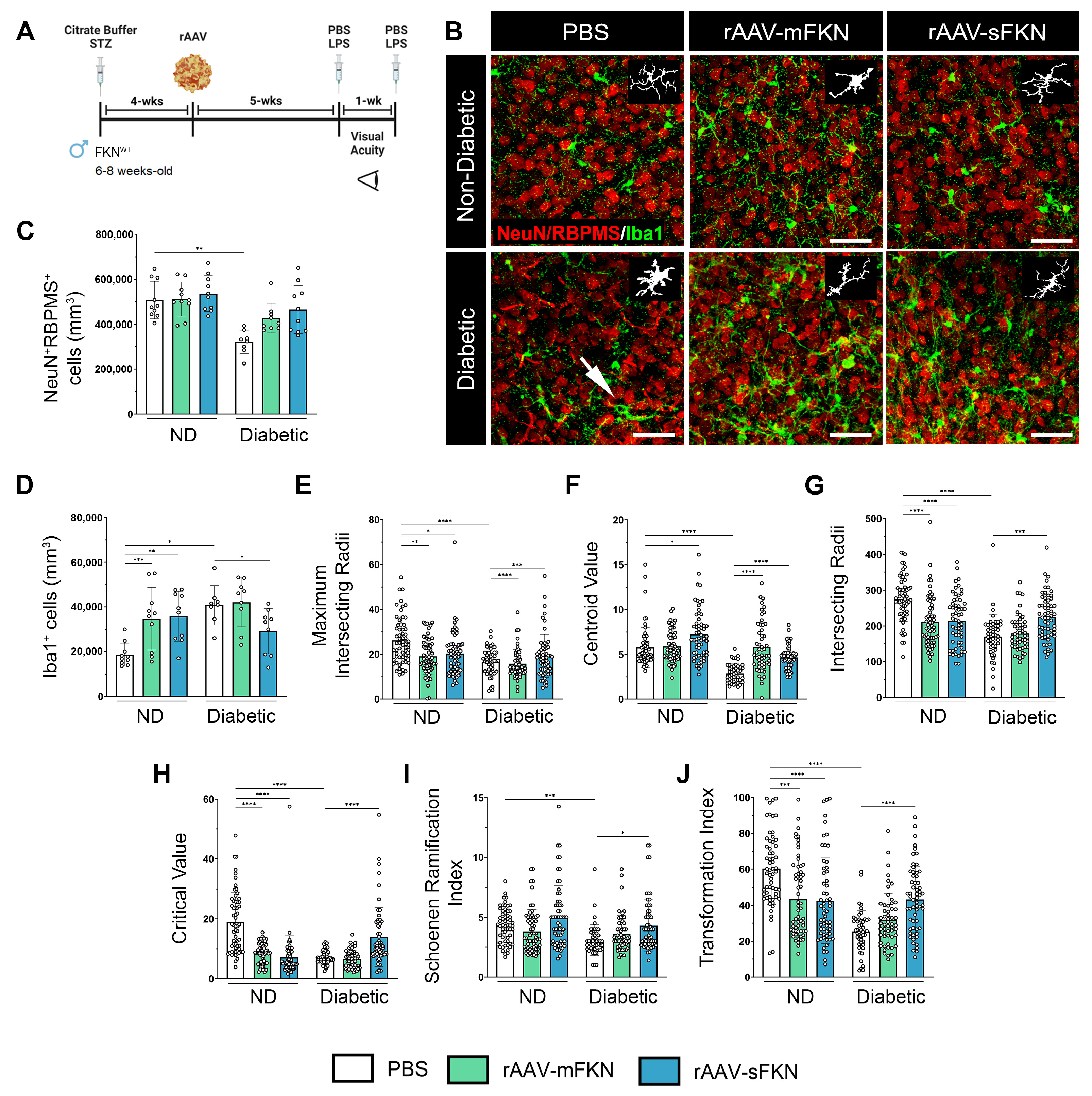
2.1. Neuronal Densities in Retinas Treated with sFKN or mFKN Are Comparable to Non-Diabetic Groups
2.2. rAAV-sFKN Administration Minimizes Microglial Hyperreactivity during Diabetes
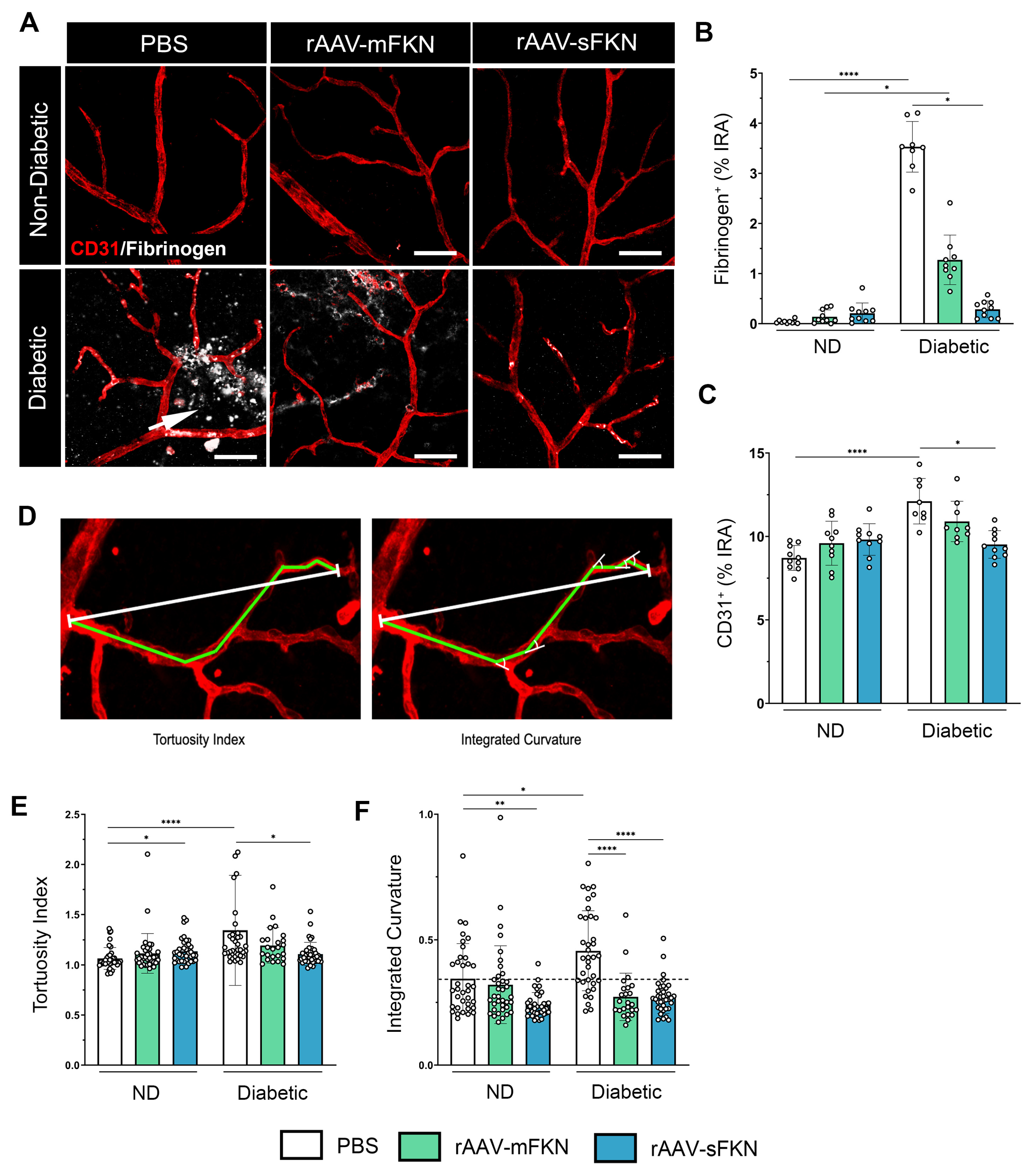
2.3. sFKN Alleviates Aberrant Vascular Permeability in the Diabetic Retina
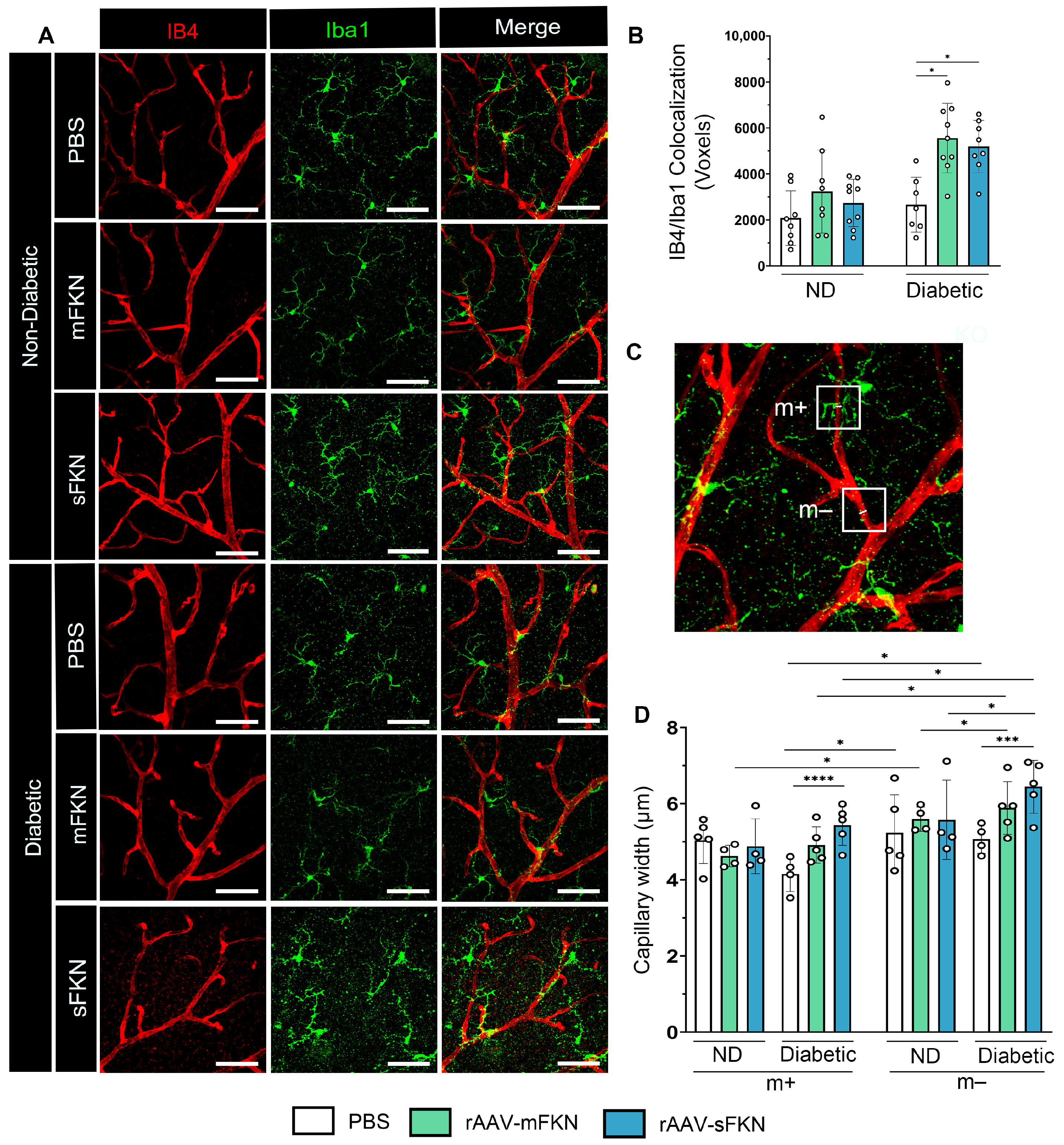
2.4. Administration of rAAVs Expressing mFKN or sFKN Increases Microglia–Vascular Interactions Influencing Capillary Constriction
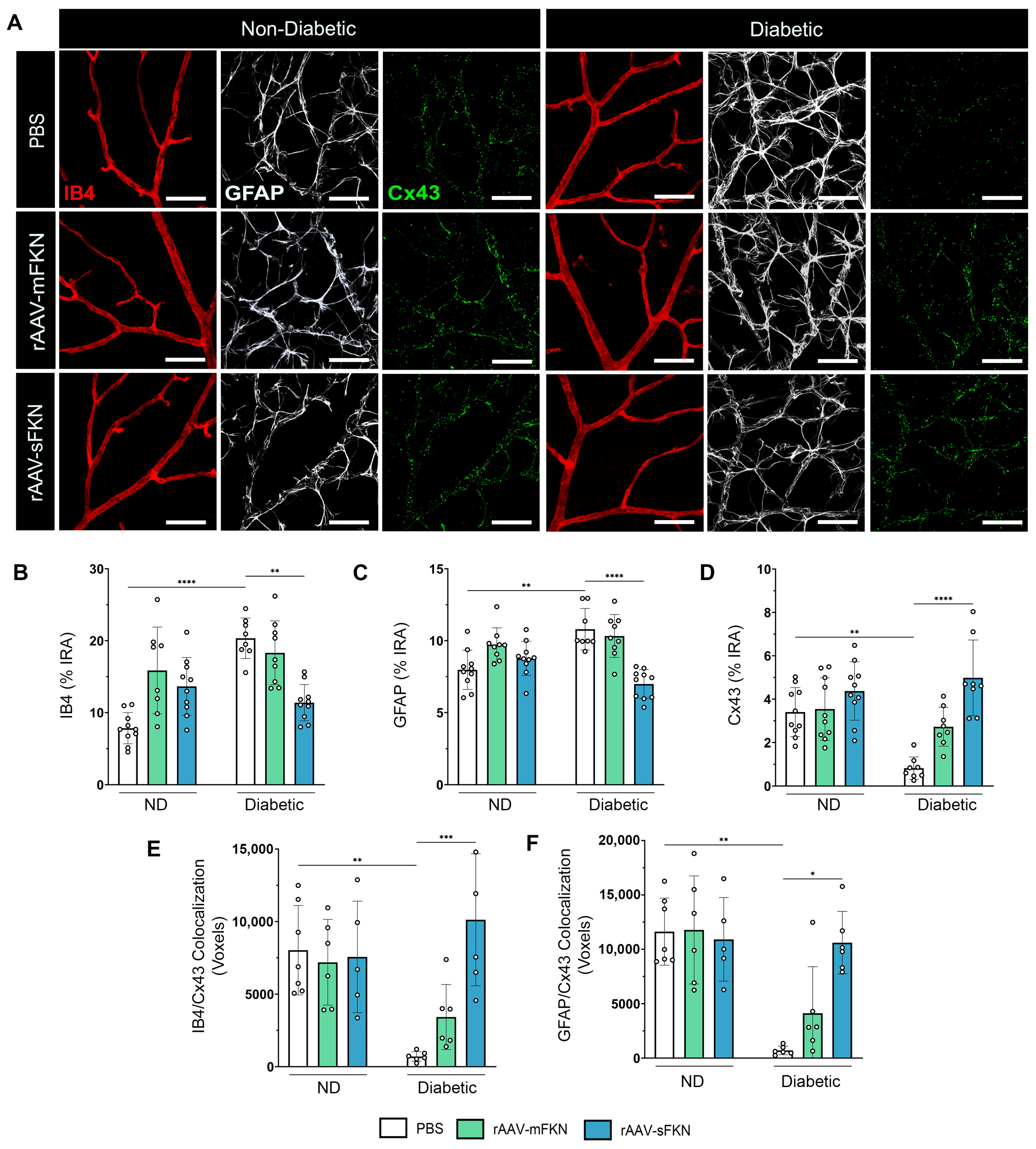
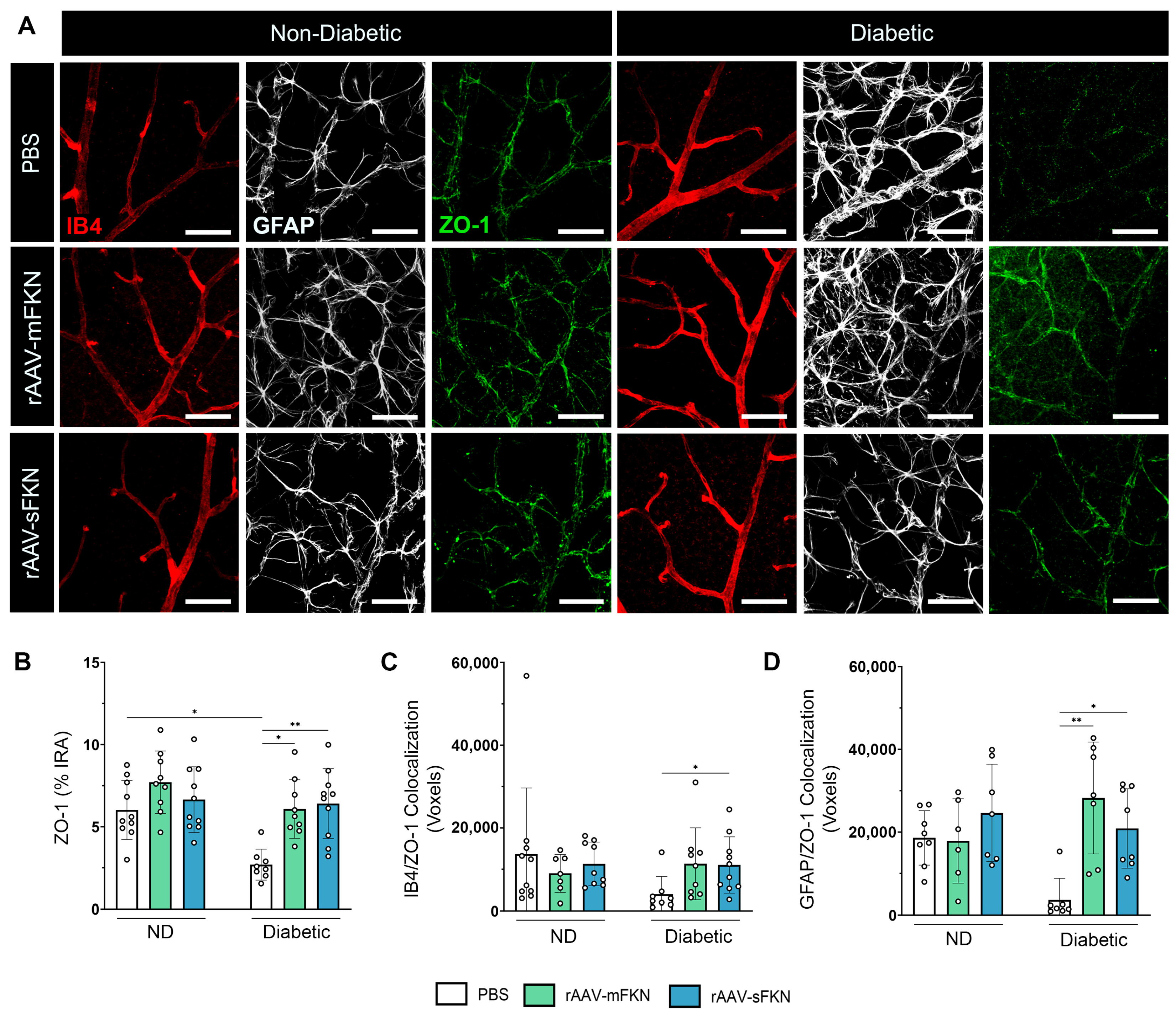
2.5. sFKN Minimizes Reactive Astrocytes and Reduces Tight- and Gap-Junction Expression during Diabetes
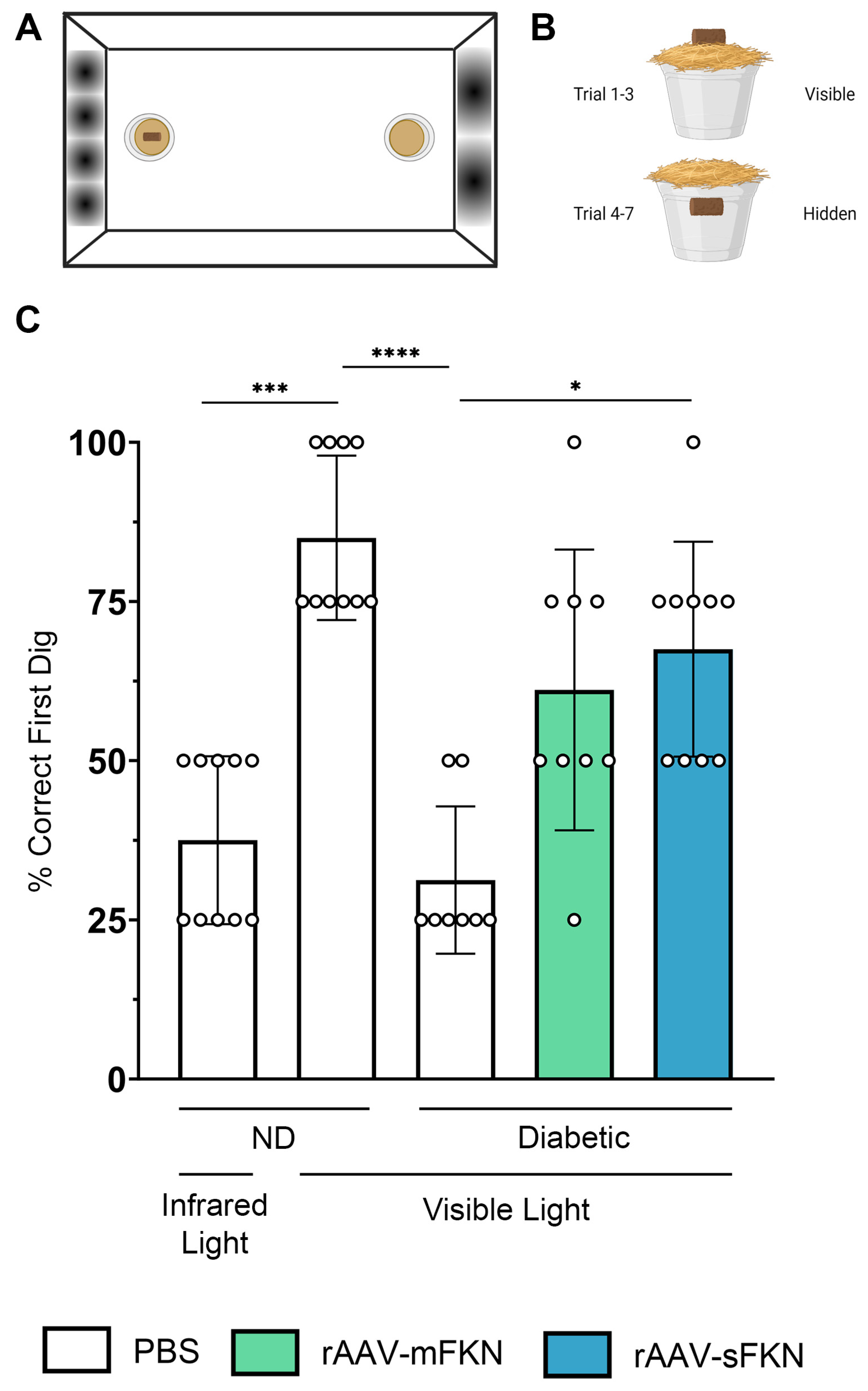
2.6. Visual Acuity Is Enhanced with rAAV-sFKN Treatment
3. Discussion
3.1. Understanding the Role of FKN Isoforms in Microglia and Vascular Function Using rAAV
3.2. FKN Overexpression Alleviates Vascular Damage in the Diabetic Retina
3.3. FKN Gene Therapy Reverses Tight- and Gap-Junction Loss
4. Materials and Methods
4.1. Mice
4.2. Recombinant Adeno-Associated Viral (rAAV) Vector Production
4.3. Two-Hit Inflammatory Streptozotocin (STZ)-Induced Model
4.4. Intra-Vitreal Injection with rAAVs
4.5. Tissue Collection
4.6. Immunofluorescence Staining
4.7. Confocal Microscopy
4.8. Sholl Analysis and Schoenen Ramification Index Analysis
4.9. Transformation Index Analysis
4.10. Retinal Tortuosity Analysis
4.11. Microglial and Vessel Interaction Analysis
4.12. Flow Cytometry of Blood and CNS Tissues
4.13. Visual Acuity Test
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lundeen, E.A.; Burke-Conte, Z.; Rein, D.B.; Wittenborn, J.S.; Saaddine, J.; Lee, A.Y.; Flaxman, A.D. Prevalence of Diabetic Retinopathy in the US in 2021. JAMA Ophthalmol. 2023, 141, 747–754. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat. Rev. Endocrinol. 2021, 17, 195–206. [Google Scholar] [CrossRef]
- Mills, S.A.; Jobling, A.I.; Dixon, M.A.; Bui, B.V.; Vessey, K.A.; Phipps, J.A.; Greferath, U.; Venables, G.; Wong, V.H.Y.; Wong, C.H.Y.; et al. Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic retinopathy. Proc. Natl. Acad. Sci. USA 2021, 118, e2112561118. [Google Scholar] [CrossRef]
- Ivanova, E.; Corona, C.; Eleftheriou, C.G.; Stout, R.F., Jr.; Korbelin, J.; Sagdullaev, B.T. AAV-BR1 targets endothelial cells in the retina to reveal their morphological diversity and to deliver Cx43. J. Comp. Neurol. 2022, 530, 1302–1317. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Chiba, H.; Fujita, H.; Kojima, T.; Saito, T.; Endo, T.; Sawada, N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell Physiol. 2006, 208, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.E.; Gourdie, R.G. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules 2020, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xin, Y.; He, Z.; Hu, W. Function of Connexins in the Interaction between Glial and Vascular Cells in the Central Nervous System and Related Neurological Diseases. Neural Plast. 2018, 2018, 6323901. [Google Scholar] [CrossRef]
- Cibelli, A.; Stout, R.; Timmermann, A.; de Menezes, L.; Guo, P.; Maass, K.; Seifert, G.; Steinhauser, C.; Spray, D.C.; Scemes, E. Cx43 carboxyl terminal domain determines AQP4 and Cx30 endfoot organization and blood brain barrier permeability. Sci. Rep. 2021, 11, 24334. [Google Scholar] [CrossRef]
- Hu, X.; Zhao, G.L.; Xu, M.X.; Zhou, H.; Li, F.; Miao, Y.; Lei, B.; Yang, X.L.; Wang, Z. Interplay between Muller cells and microglia aggravates retinal inflammatory response in experimental glaucoma. J. Neuroinflamm. 2021, 18, 303. [Google Scholar] [CrossRef]
- Tworig, J.M.; Coate, C.J.; Feller, M.B. Excitatory neurotransmission activates compartmentalized calcium transients in Muller glia without affecting lateral process motility. eLife 2021, 10, e73202. [Google Scholar] [CrossRef]
- Sarker, B.; Cardona, S.M.; Church, K.A.; Vanegas, D.; Velazquez, P.; Rorex, C.; Rodriguez, D.; Mendiola, A.S.; Kern, T.S.; Domingo, N.D.; et al. Defibrinogenation Ameliorates Retinal Microgliosis and Inflammation in A CX3CR1-Independent Manner. ASN Neuro 2022, 14, 17590914221131446. [Google Scholar] [CrossRef]
- Church, K.A.; Rodriguez, D.; Vanegas, D.; Gutierrez, I.L.; Cardona, S.M.; Madrigal, J.L.M.; Kaur, T.; Cardona, A.E. Models of microglia depletion and replenishment elicit protective effects to alleviate vascular and neuronal damage in the diabetic murine retina. J. Neuroinflamm. 2022, 19, 300. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Garza, R.; Cardona, S.M.; Mythen, S.A.; Lira, S.A.; Akassoglou, K.; Cardona, A.E. Fractalkine Signaling Attenuates Perivascular Clustering of Microglia and Fibrinogen Leakage during Systemic Inflammation in Mouse Models of Diabetic Retinopathy. Front. Cell Neurosci. 2016, 10, 303. [Google Scholar] [CrossRef]
- Rodriguez, D.; Church, K.A.; Pietramale, A.N.; Cardona, S.M.; Vanegas, D.; Rorex, C.; Leary, M.C.; Muzzio, I.A.; Nash, K.R.; Cardona, A.E. Fractalkine isoforms differentially regualte microglia-mediated inflammation and enhance visual function in the diabetic retina. J. Neuroinflamm. 2024, in press. [Google Scholar]
- Cardona, S.M.; Mendiola, A.S.; Yang, Y.C.; Adkins, S.L.; Torres, V.; Cardona, A.E. Disruption of Fractalkine Signaling Leads to Microglial Activation and Neuronal Damage in the Diabetic Retina. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hussain, R.M.; Weng, C.Y. Voretigene Neparvovec in Retinal Diseases: A Review of the Current Clinical Evidence. Clin. Ophthalmol. 2020, 14, 3855–3869. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.N.; Subbarayan, M.S.; Grimmig, B.; Weesner, J.A.; Moss, L.; Peters, M.; Weeber, E.; Nash, K.; Bickford, P.C. Two forms of CX3CL1 display differential activity and rescue cognitive deficits in CX3CL1 knockout mice. J. Neuroinflamm. 2020, 17, 157. [Google Scholar] [CrossRef]
- Daily, J.L.; Nash, K.; Jinwal, U.; Golde, T.; Rogers, J.; Peters, M.M.; Burdine, R.D.; Dickey, C.; Banko, J.L.; Weeber, E.J. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS ONE 2011, 6, e27221. [Google Scholar] [CrossRef]
- Nash, K.R.; Lee, D.C.; Hunt, J.B., Jr.; Morganti, J.M.; Selenica, M.L.; Moran, P.; Reid, P.; Brownlow, M.; Guang-Yu Yang, C.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Gayen, M.; Singh, N.; Gao, F.; He, W.; Hu, X.; Tsai, L.H.; Yan, R. The intracellular domain of CX3CL1 regulates adult neurogenesis and Alzheimer’s amyloid pathology. J. Exp. Med. 2019, 216, 1891–1903. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Church, K.A.; Cardona, S.M.; Vanegas, D.; Garcia, S.A.; Macklin, W.; Lira, S.A.; Ransohoff, R.M.; Kokovay, E.; Lin, C.A.; et al. Defective fractalkine-CX3CR1 signaling aggravates neuroinflammation and affects recovery from cuprizone-induced demyelination. J. Neurochem. 2022, 162, 430–443. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, G.; Liu, W.; Ni, Y.; Zhou, W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS ONE 2012, 7, e35446. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xie, H.; Zhang, C.; Wang, T.; Tian, H.; Lu, L.; Xu, J.Y.; Xu, G.T.; Liu, L.; Zhang, J. Enhancing fractalkine/CX3CR1 signalling pathway can reduce neuroinflammation by attenuating microglia activation in experimental diabetic retinopathy. J. Cell Mol. Med. 2022, 26, 1229–1244. [Google Scholar] [CrossRef]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Meng, Z.; Luo, J. Connexin 43 (Cx43) regulates high-glucose-induced retinal endothelial cell angiogenesis and retinal neovascularization. Front. Endocrinol. 2022, 13, 909207. [Google Scholar] [CrossRef]
- Howarth, A.G.; Hughes, M.R.; Stevenson, B.R. Detection of the tight junction-associated protein ZO-1 in astrocytes and other nonepithelial cell types. Am. J. Physiol. 1992, 262, C461–C469. [Google Scholar] [CrossRef]
- Puller, C.; de Sevilla Muller, L.P.; Janssen-Bienhold, U.; Haverkamp, S. ZO-1 and the spatial organization of gap junctions and glutamate receptors in the outer plexiform layer of the mammalian retina. J. Neurosci. 2009, 29, 6266–6275. [Google Scholar] [CrossRef] [PubMed]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef]
- Tien, T.; Barrette, K.F.; Chronopoulos, A.; Roy, S. Effects of high glucose-induced Cx43 downregulation on occludin and ZO-1 expression and tight junction barrier function in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6518–6525. [Google Scholar] [CrossRef] [PubMed]
- Bobbie, M.W.; Roy, S.; Trudeau, K.; Munger, S.J.; Simon, A.M.; Roy, S. Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.; Kovacs-Oller, T.; Sagdullaev, B.T. Vascular Pericyte Impairment and Connexin43 Gap Junction Deficit Contribute to Vasomotor Decline in Diabetic Retinopathy. J. Neurosci. 2017, 37, 7580–7594. [Google Scholar] [CrossRef]
- Desjardins, D.M.; Yates, P.W.; Dahrouj, M.; Liu, Y.; Crosson, C.E.; Ablonczy, Z. Progressive Early Breakdown of Retinal Pigment Epithelium Function in Hyperglycemic Rats. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.Z.; Le, Y.Z. Significance of outer blood-retina barrier breakdown in diabetes and ischemia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Le May, C.; Chu, K.; Hu, M.; Ortega, C.S.; Simpson, E.R.; Korach, K.S.; Tsai, M.J.; Mauvais-Jarvis, F. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9232–9237. [Google Scholar] [CrossRef]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single dose streptozotocin-induced diabetes: Considerations for study design in islet transplantation models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P. Animal models of sepsis. Virulence 2014, 5, 143–153. [Google Scholar] [CrossRef]
- Chen, Z.; Jalabi, W.; Shpargel, K.B.; Farabaugh, K.T.; Dutta, R.; Yin, X.; Kidd, G.J.; Bergmann, C.C.; Stohlman, S.A.; Trapp, B.D. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J. Neurosci. 2012, 32, 11706–11715. [Google Scholar] [CrossRef]
- Raetzsch, C.F.; Brooks, N.L.; Alderman, J.M.; Moore, K.S.; Hosick, P.A.; Klebanov, S.; Akira, S.; Bear, J.E.; Baldwin, A.S.; Mackman, N.; et al. Lipopolysaccharide inhibition of glucose production through the Toll-like receptor-4, myeloid differentiation factor 88, and nuclear factor kappa b pathway. Hepatology 2009, 50, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.J.; Xie, H.; Zhang, C.Y.; Qin, H.F.; Zeng, X.W.; Lou, H.; Zhang, L.; Xu, G.T.; Zhang, J.F.; Xu, G.X. Is Iba-1 protein expression a sensitive marker for microglia activation in experimental diabetic retinopathy? Int. J. Ophthalmol. 2021, 14, 200–208. [Google Scholar] [CrossRef]
- Ferreira, T.A.; Blackman, A.V.; Oyrer, J.; Jayabal, S.; Chung, A.J.; Watt, A.J.; Sjostrom, P.J.; van Meyel, D.J. Neuronal morphometry directly from bitmap images. Nat. Methods 2014, 11, 982–984. [Google Scholar] [CrossRef]
- Arshadi, C.; Gunther, U.; Eddison, M.; Harrington, K.I.S.; Ferreira, T.A. SNT: A unifying toolbox for quantification of neuronal anatomy. Nat. Methods 2021, 18, 374–377. [Google Scholar] [CrossRef]
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4. [Google Scholar] [CrossRef]
- Reeves, A.M.; Shigetomi, E.; Khakh, B.S. Bulk loading of calcium indicator dyes to study astrocyte physiology: Key limitations and improvements using morphological maps. J. Neurosci. 2011, 31, 9353–9358. [Google Scholar] [CrossRef]
- Fujita, H.; Tanaka, J.; Toku, K.; Tateishi, N.; Suzuki, Y.; Matsuda, S.; Sakanaka, M.; Maeda, N. Effects of GM-CSF and ordinary supplements on the ramification of microglia in culture: A morphometrical study. Glia 1996, 18, 269–281. [Google Scholar] [CrossRef]
- Scott, A.; Powner, M.B.; Fruttiger, M. Quantification of vascular tortuosity as an early outcome measure in oxygen induced retinopathy (OIR). Exp. Eye Res. 2014, 120, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Dholakia, K.Y.; Guevara-Torres, A.; Feng, G.; Power, D.; Schallek, J. In Vivo Capillary Structure and Blood Cell Flux in the Normal and Diabetic Mouse Eye. Investig. Ophthalmol. Vis. Sci. 2022, 63, 18. [Google Scholar] [CrossRef] [PubMed]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 2010, 5, e13693. [Google Scholar] [CrossRef]
- Cardona, S.M.; Kim, S.V.; Church, K.A.; Torres, V.O.; Cleary, I.A.; Mendiola, A.S.; Saville, S.P.; Watowich, S.S.; Parker-Thornburg, J.; Soto-Ospina, A.; et al. Role of the Fractalkine Receptor in CNS Autoimmune Inflammation: New Approach Utilizing a Mouse Model Expressing the Human CX3CR1(I249/M280) Variant. Front. Cell Neurosci. 2018, 12, 365. [Google Scholar] [CrossRef] [PubMed]
- Normandin, M.E.; Garza, M.C.; Ramos-Alvarez, M.M.; Julian, J.B.; Eresanara, T.; Punjaala, N.; Vasquez, J.H.; Lopez, M.R.; Muzzio, I.A. Navigable Space and Traversable Edges Differentially Influence Reorientation in Sighted and Blind Mice. Psychol. Sci. 2022, 33, 925–947. [Google Scholar] [CrossRef] [PubMed]
- Keinath, A.T.; Julian, J.B.; Epstein, R.A.; Muzzio, I.A. Environmental Geometry Aligns the Hippocampal Map during Spatial Reorientation. Curr. Biol. 2017, 27, 309–317. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, D.; Church, K.A.; Smith, C.T.; Vanegas, D.; Cardona, S.M.; Muzzio, I.A.; Nash, K.R.; Cardona, A.E. Therapeutic Delivery of Soluble Fractalkine Ameliorates Vascular Dysfunction in the Diabetic Retina. Int. J. Mol. Sci. 2024, 25, 1727. https://doi.org/10.3390/ijms25031727
Rodriguez D, Church KA, Smith CT, Vanegas D, Cardona SM, Muzzio IA, Nash KR, Cardona AE. Therapeutic Delivery of Soluble Fractalkine Ameliorates Vascular Dysfunction in the Diabetic Retina. International Journal of Molecular Sciences. 2024; 25(3):1727. https://doi.org/10.3390/ijms25031727
Chicago/Turabian StyleRodriguez, Derek, Kaira A. Church, Chelsea T. Smith, Difernando Vanegas, Sandra M. Cardona, Isabel A. Muzzio, Kevin R. Nash, and Astrid E. Cardona. 2024. "Therapeutic Delivery of Soluble Fractalkine Ameliorates Vascular Dysfunction in the Diabetic Retina" International Journal of Molecular Sciences 25, no. 3: 1727. https://doi.org/10.3390/ijms25031727
APA StyleRodriguez, D., Church, K. A., Smith, C. T., Vanegas, D., Cardona, S. M., Muzzio, I. A., Nash, K. R., & Cardona, A. E. (2024). Therapeutic Delivery of Soluble Fractalkine Ameliorates Vascular Dysfunction in the Diabetic Retina. International Journal of Molecular Sciences, 25(3), 1727. https://doi.org/10.3390/ijms25031727