
Functional Characteristics of the Nav1.1 p.Arg1596Cys Mutation Associated with Varying Severity of Epilepsy Phenotypes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
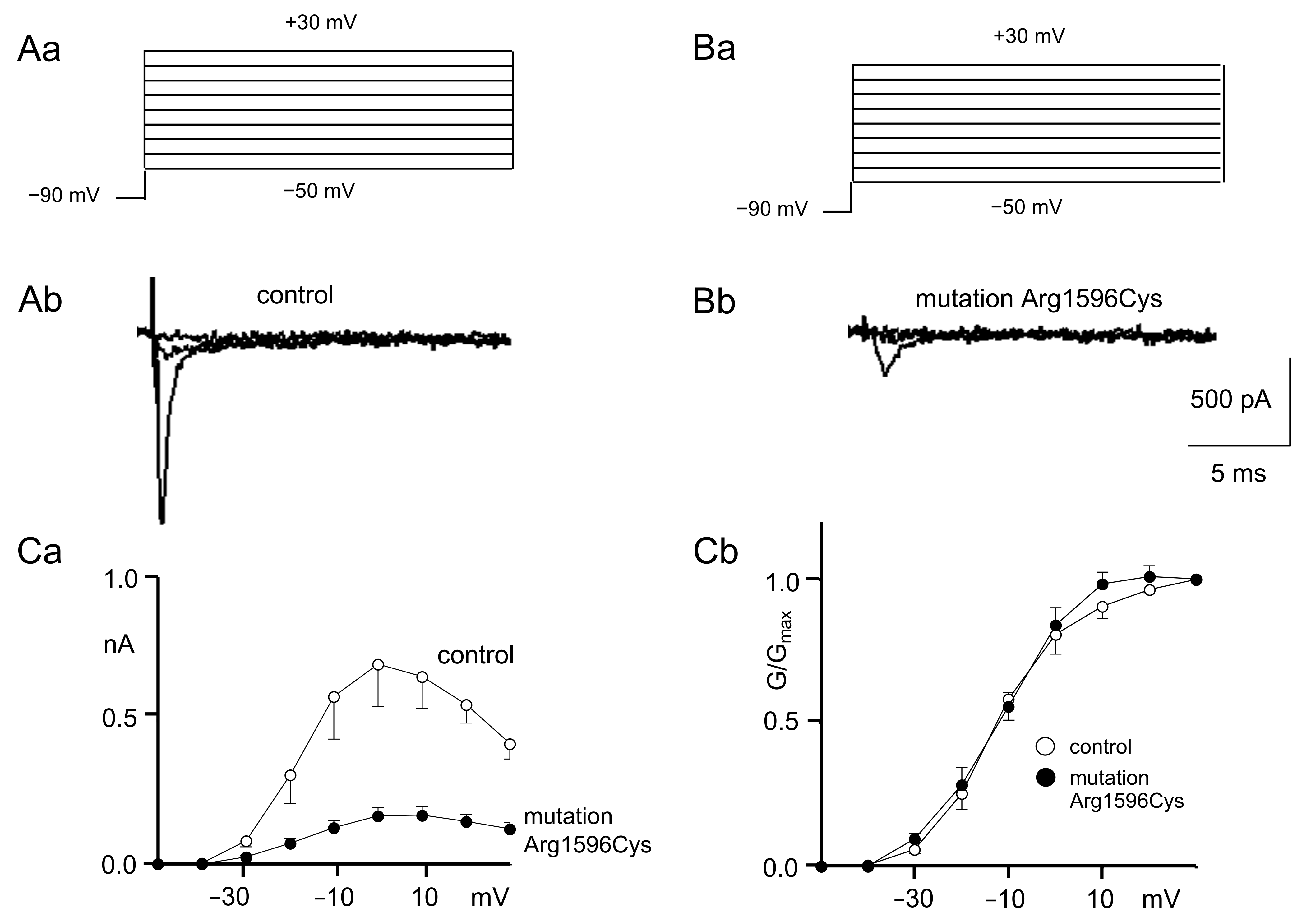
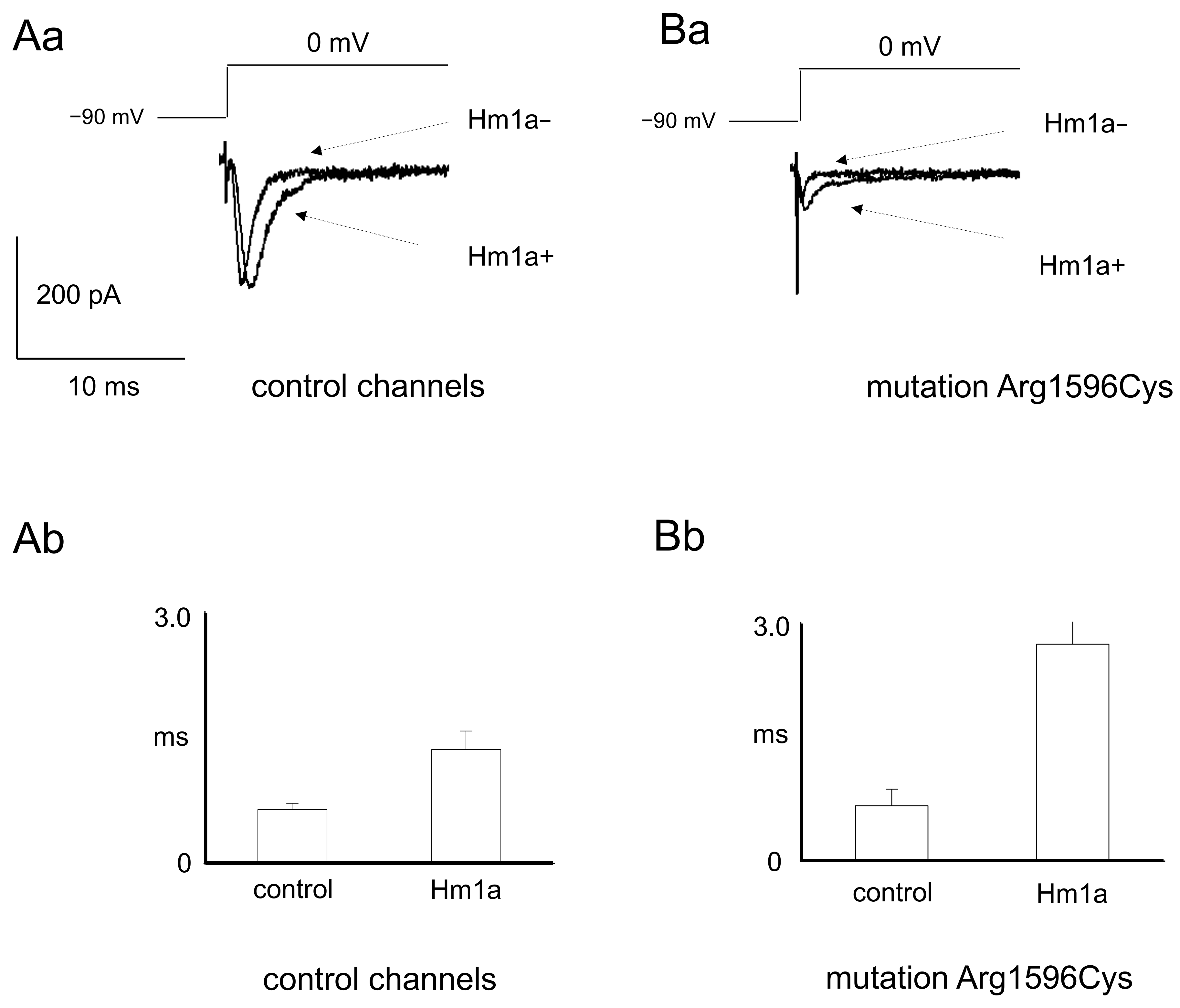
2.1. Patch Clamp Recordings
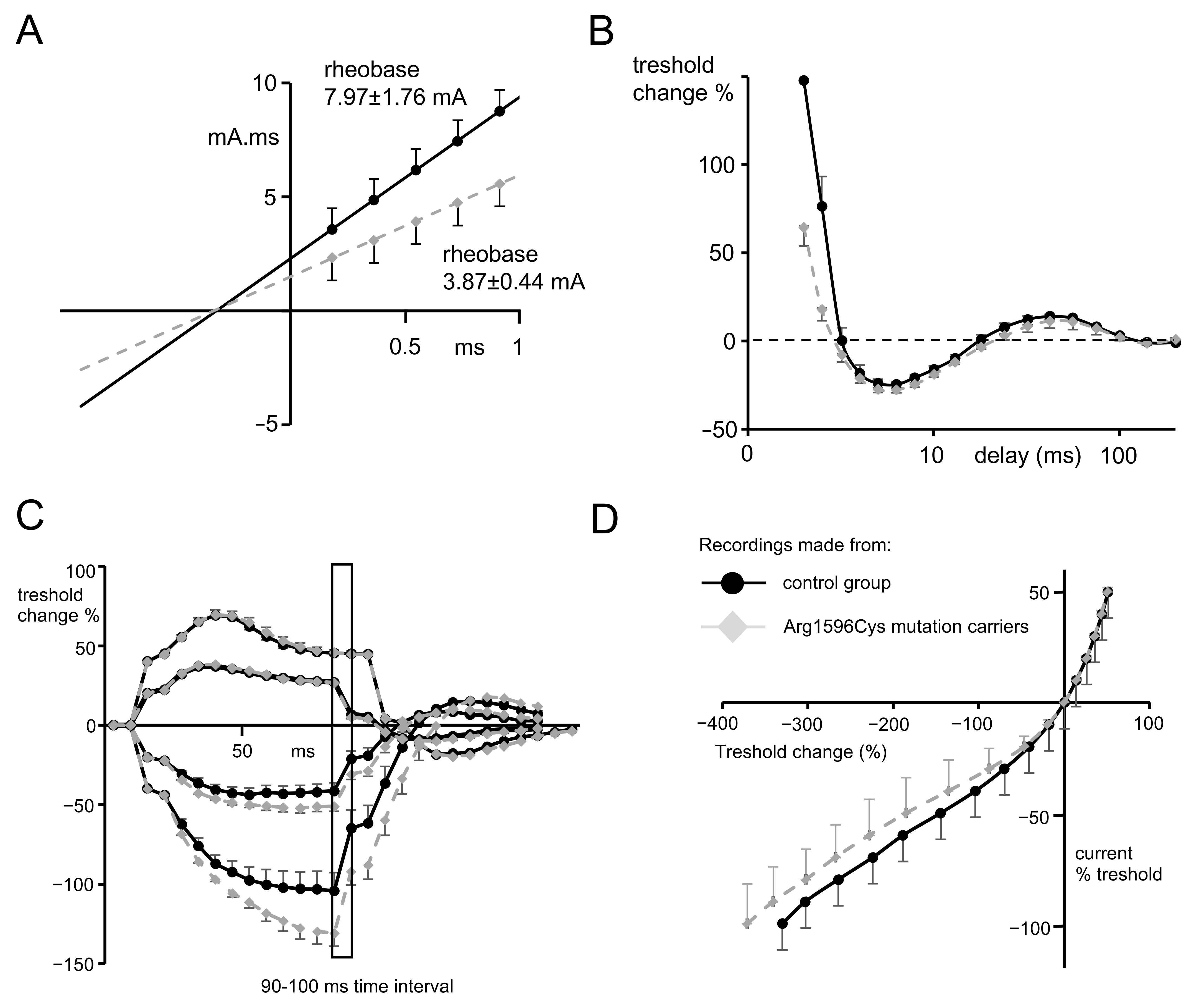
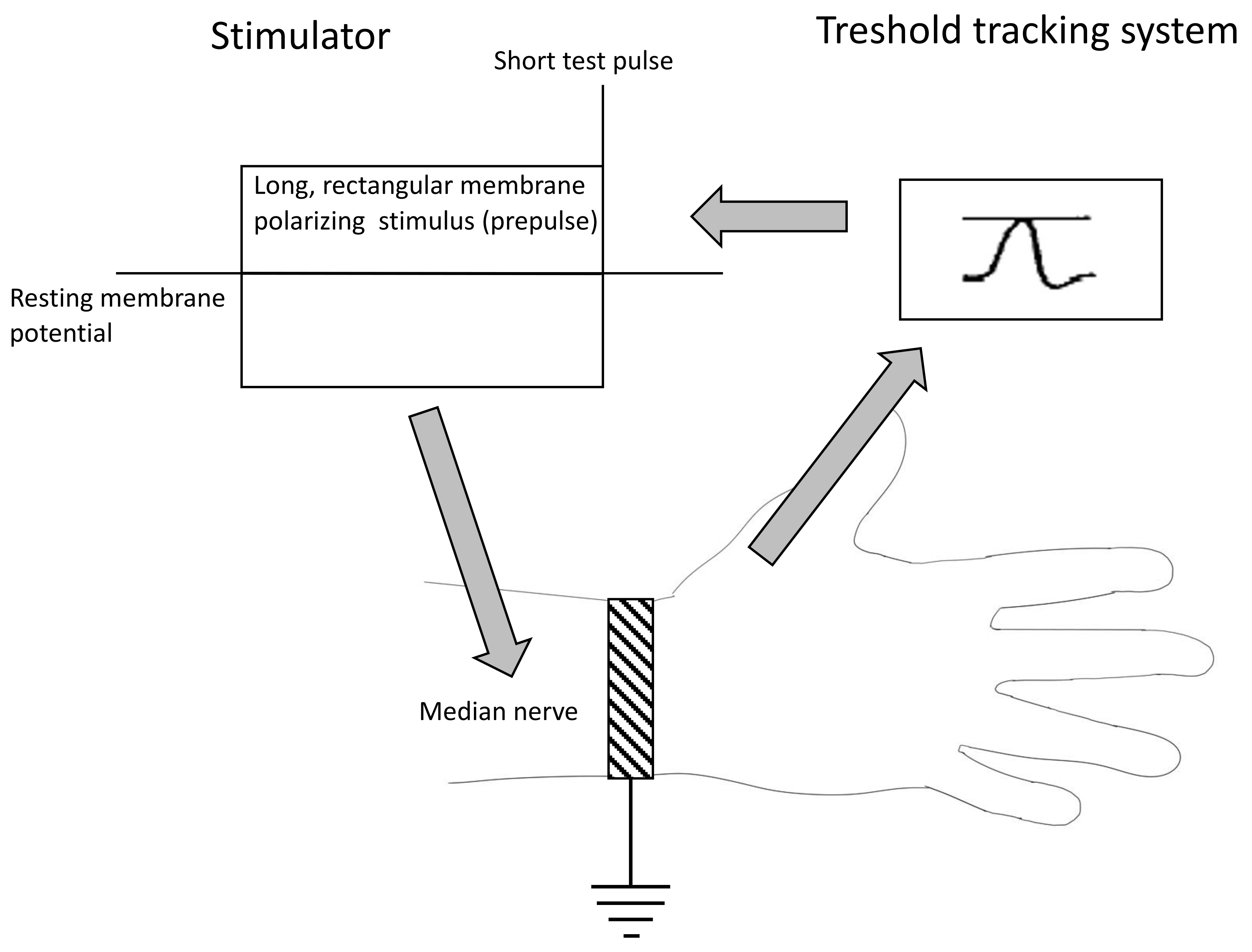
2.2. Nerve Excitability Study
- Preliminary nerve conduction study (NCS).
- Strength–duration time constants.
- Recovery cycle.
- Threshold electrotonus.
- Current–voltage relationship.
2.3. WES Analysis
3. Discussion
3.1. Patch Clamp Findings
3.2. NES Examination Findings
3.3. Why Does Reduced Nav1.1 Sodium Current Cause Epilepsy?
3.4. WES Findings
4. Materials and Methods
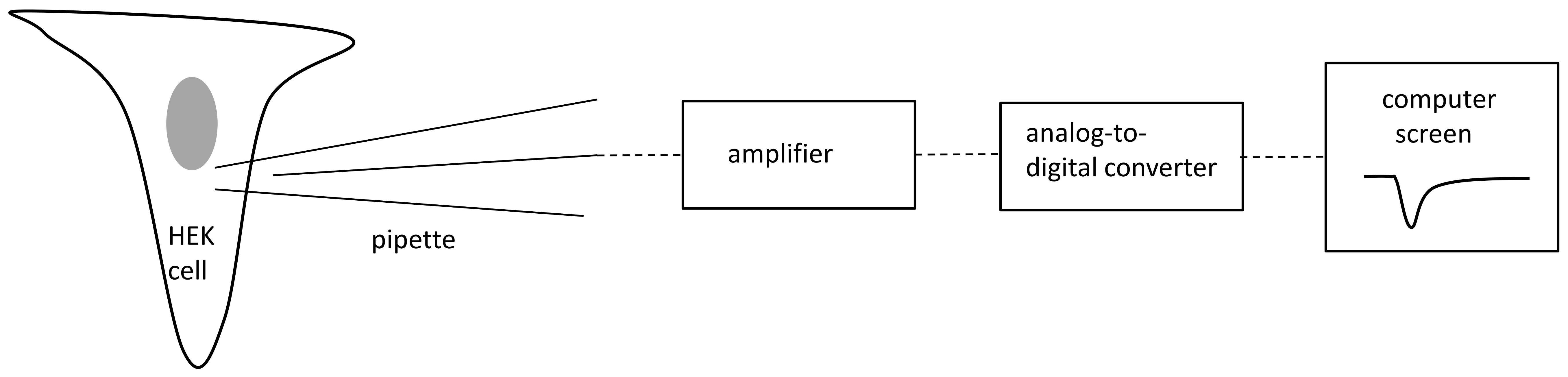
4.1. Patch Clamp
4.1.1. Cell Culture and Transfection
4.1.2. Patch Clamp Recordings
4.2. Nerve Excitability Study
4.2.1. Clinical Characteristics of the Participants
4.2.2. The Methodology of Nerve Excitability Study (NES)
- The strength–duration relationship between the duration and strength of pulse necessary to evoke a compound muscle-evoked potential (MEP) with a preset amplitude (40% of maximum) was calculated automatically with the use of the Weiss empirical law, Q=Ixt = Irh (t + SDTC), where Q is the stimulus charge, I is the stimulus current of duration t, SDTC is the strength–duration time constant, and Irh is the rheobasic current. Measurements revealed the nerve fiber rheobase (the lowest current with an infinite duration that induces a response, calculated as a slope of the straight line fitted to the points on the charge–stimulus duration plot), and chronaxie (equal to the strength–duration time constant), found as the x intercept of the above-mentioned straight line [40]. Rheobase is influenced mainly by the activity and properties of persistent sodium currents generated in the nodes [22].
- The subprotocol for the threshold electrotonus contained four series of subthreshold depolarizing and hyperpolarizing prepulses set to +/− 20 and +/− 40% of the unconditioned threshold current, lasting for different durations (between 1 and 200 ms). The test pulse generated after each prepulse was automatically adjusted to reach the preset amplitude of the fiber response. The threshold electrotonus curve represents membranes’ behavior during prolonged de- and hyperpolarizations and is shaped by the activity of voltage-dependent sodium and voltage-dependent slow potassium and inward rectifying channels [41].
- The recovery cycle measured membrane excitability changes after the generation of the compound action potential. The protocol tracks 3 phases of returning to normal excitability: the relative refractory period (RRP), the superexcitable period and the late subexcitability period. The RRP reflects the recovery of inactivated voltage-dependent sodium channels in the nodes [42], where the superexcitability is influenced by prolonged internodal depolarization and late subexcitability by the slow kinetics of voltage-dependent potassium channels from the afterhyperpolarization [43].
- The current–threshold relationship reveals the dependence between the different levels of fiber resting potential shaped by subthreshold depolarizing and hyperpolarizing pulses with a fixed length and the current necessary to evoke the CAP with a fixed amplitude. The current–threshold curve is sensitive to processes modifying membrane polarization; during hyperpolarizing pulses, the activity of inward rectifying potassium channels limits the decrease in membrane excitability [10].
4.2.3. Statistical Analyses
4.3. Exome Sequencing and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldin, A.L.; Snutch, T.; Lubbert, H.; Dowsett, A.; Marshall, J.; Auld, V.; Downey, W.; Fritz, L.C.; Lester, H.A.; Dunn, R. Messenger RNA Coding for Only the α Subunit of the Rat Brain Na Channel Is Sufficient for Expression of Functional Channels in Xenopus Oocytes. Proc. Natl. Acad. Sci. USA 1986, 83, 7503–7507. [Google Scholar] [CrossRef]
- Berecki, G.; Bryson, A.; Terhag, J.; Maljevic, S.; Gazina, E.V.; Hill, S.L.; Petrou, S. SCN1A Gain of Function in Early Infantile Encephalopathy. Ann. Neurol. 2019, 85, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Brünger, T.; Feng, T.; Fons, C.; Lehikoinen, A.; Panagiotakaki, E.; Vintan, M.A.; Symonds, J.; Andrew, J.; Arzimanoglou, A.; et al. The Gain of Function SCN1A Disorder Spectrum: Novel Epilepsy Phenotypes and Therapeutic Implications. Brain 2022, 145, 3816–3831. [Google Scholar] [CrossRef] [PubMed]
- Dlugos, D.J.; Ferraro, T.N.; Buono, R.J. Novel De Novo Mutation of a Conserved SCN1A Amino-Acid Residue (R1596). Pediatr. Neurol. 2007, 37, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Trouillard, O.; Saint-Martin, C.; Gourfinkel-An, I.; Bouteiller, D.; Carpentier, W.; Keren, B.; Abert, B.; Gautier, A.; Baulac, S.; et al. Spectrum of SCN1A Gene Mutations Associated with Dravet Syndrome: Analysis of 333 Patients. J. Med. Genet. 2009, 46, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.O.; Bellows, S.; Mcmahon, J.M.; Iona, X.; Damiano, J.; Dibbens, L.; Kelley, K.; Gill, D.; Cross, J.H.; Berkovic, S.F.; et al. Atypical Multifocal Dravet Syndrome Lacks Generalized Seizures and May Show Later Cognitive Decline. Dev. Med. Child Neurol. 2014, 56, 85–90. [Google Scholar] [CrossRef]
- Møller, R.S.; Larsen, L.H.G.; Johannesen, K.M.; Talvik, I.; Talvik, T.; Vaher, U.; Miranda, M.J.; Farooq, M.; Nielsen, J.E.K.; Lavard Svendsen, L.; et al. Gene Panel Testing in Epileptic Encephalopathies and Familial Epilepsies. Mol. Syndromol. 2016, 7, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Zacharska, D.; Szczepanik, E.; Terczynska, I.; Goszczanska-Ciuchta, A.; Zalewska-Miszkurka, Z.; Tataj, R.; Bal, J. From Focal Epilepsy to Dravet Syndrome –Heterogeneity of the Phenotype Due to SCN1A Mutations of the p.Arg1596 Amino Acid Residue in the Nav1.1 Subunit. Neurol. Neurochir. Pol. 2015, 49, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Huynh, W.; Kiernan, M.C. Peripheral Nerve Axonal Excitability Studies: Expanding the Neurophysiologist’s Armamentarium. Cerebellum Ataxias 2015, 2, 4. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Bostock, H.; Park, S.B.; Kaji, R.; Krarup, C.; Krishnan, A.V.; Kuwabara, S.; Lin, C.S.Y.; Misawa, S.; Moldovan, M.; et al. Measurement of Axonal Excitability: Consensus Guidelines. Clin. Neurophysiol. 2020, 131, 308–323. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium Channel Nav1.6 Is Localized at Nodes of Ranvier, Dendrites, and Synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar] [CrossRef]
- Tomlinson, S.E.; Tan, S.V.; Kullmann, D.M.; Griggs, R.C.; Burke, D.; Hanna, M.G.; Bostock, H. Nerve Excitability Studies Characterize KV1.1 Fast Potassium Channel Dysfunction in Patients with Episodic Ataxia Type 1. Brain 2010, 133, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.V.; Wraige, E.; Lascelles, K.; Bostock, H. Episodic Ataxia Type 1 without Episodic Ataxia: The Diagnostic Utility of Nerve Excitability Studies in Individuals with KCNA1 Mutations. Dev. Med. Child Neurol. 2013, 55, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Bostock, H.; Ip, J.; Hayes, M.; Watson, S.; Kiernan, M.C. Axonal Function in a Family with Episodic Ataxia Type 2 Due to a Novel Mutation. J. Neurol. 2008, 255, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Gawlak, M.; Szulczyk, B.; Berłowski, A.; Grzelka, K.; Stachurska, A.; Pełka, J.; Czarzasta, K.; Małecki, M.; Kurowski, P.; Nurowska, E.; et al. Age-Dependent Expression of Nav1.9 Channels in Medial Prefrontal Cortex Pyramidal Neurons in Rats. Dev. Neurobiol. 2017, 77, 1371–1384. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Ghosh, S.; Sharma, H.; Bhaskar, R.; Narayanan, K.B. A Comprehensive Review of Emerging Trends and Innovative Therapies in Epilepsy Management. Brain Sci. 2023, 13, 1305. [Google Scholar] [CrossRef] [PubMed]
- Kluckova, D.; Kolnikova, M.; Lacinova, L.; Jurkovicova-Tarabova, B.; Foltan, T.; Demko, V.; Kadasi, L.; Ficek, A.; Soltysova, A. A Study among the Genotype, Functional Alternations, and Phenotype of 9 SCN1A Mutations in Epilepsy Patients. Sci. Rep. 2020, 10, 10288. [Google Scholar] [CrossRef] [PubMed]
- Escayg, A.; Goldin, A.L. Sodium Channel SCN1A and Epilepsy: Mutations and Mechanisms. Epilepsia 2010, 51, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.L.; Milligan, C.J.; Richardson, R.J.; Jancovski, N.; Grunnet, M.; Jacobson, L.H.; Undheim, E.A.B.; Mobli, M.; Chow, C.Y.; Herzig, V.; et al. Selective NaV1.1 Activation Rescues Dravet Syndrome Mice from Seizures and Premature Death. Proc. Natl. Acad. Sci. USA 2018, 115, E8077–E8085. [Google Scholar] [CrossRef]
- McMoneagle, E.; Zhou, J.; Zhang, S.; Huang, W.; Josiah, S.S.; Ding, K.; Wang, Y.; Zhang, J. Neuronal K+-Cl- Cotransporter KCC2 as a Promising Drug Target for Epilepsy Treatment. Acta Pharmacol. Sin. 2023, 45, 1–22. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Khan, T.; Devaraju, K.S.; Singh, P.; Vaibhav, K.; Gaur, P. Pharmacological and Therapeutic Approaches in the Treatment of Epilepsy. Biomedicines 2021, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Nodera, H.; Kaji, R. Nerve Excitability Testing and Its Clinical Application to Neuromuscular Diseases. Clin. Neurophysiol. 2006, 117, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Duflocq, A.; Le Bras, B.; Bullier, E.; Couraud, F.; Davenne, M. Nav1.1 Is Predominantly Expressed in Nodes of Ranvier and Axon Initial Segments. Mol. Cell. Neurosci. 2008, 39, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, T.; Kobayashi, K.; Yamanaka, H.; Obata, K.; Dai, Y.; Noguchi, K. Comparative Study of the Distribution of the α-Subunits of Voltage-Gated Sodium Channels in Normal and Axotomized Rat Dorsal Root Ganglion Neurons. J. Comp. Neurol. 2008, 510, 188–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Atianjoh, F.; Gauda, E.B.; Yaster, M.; Li, Y.; Tao, Y.X. Increased Expression of Sodium Channel Subunit Nav1.1 in the Injured Dorsal Root Ganglion After Peripheral Nerve Injury. Anat. Rec. 2011, 294, 1406–1411. [Google Scholar] [CrossRef]
- Sibarov, D.A.; Tsytsarev, V.; Volnova, A.; Vaganova, A.N.; Alves, J.; Rojas, L.; Sanabria, P.; Ignashchenkova, A.; Savage, E.D.; Inyushin, M. Arc Protein, a Remnant of Ancient Retrovirus, Forms Virus-like Particles, Which Are Abundantly Generated by Neurons during Epileptic Seizures, and Affects Epileptic Susceptibility in Rodent Models. Front. Neurol. 2023, 14, 1201104. [Google Scholar] [CrossRef]
- Zanatta, G.; Sula, A.; Miles, A.J.; Ng, L.C.T.; Torella, R.; Pryde, D.C.; DeCaen, P.G.; Wallace, B.A. Valproic Acid Interactions with the NavMs Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. USA 2019, 116, 26549–26554. [Google Scholar] [CrossRef]
- Szulczyk, B.; Nurowska, E. Valproic Acid Inhibits TTX-Resistant Sodium Currents in Prefrontal Cortex Pyramidal Neurons. Biochem. Biophys. Res. Commun. 2017, 491, 291–295. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced Sodium Current in GABAergic Interneurons in a Mouse Model of Severe Myoclonic Epilepsy in Infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef]
- Tai, C.; Abe, Y.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Impaired Excitability of Somatostatin- and Parvalbumin-Expressing Cortical Interneurons in a Mouse Model of Dravet Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E3139–E3148. [Google Scholar] [CrossRef]
- Ogiwara, I.; Iwasato, T.; Miyamoto, H.; Iwata, R.; Yamagata, T.; Mazaki, E.; Yanagawa, Y.; Tamamaki, N.; Hensch, T.K.; Itohara, S.; et al. Nav1.1 Haploinsufficiency in Excitatory Neurons Ameliorates Seizure-Associated Sudden Death in a Mouse Model of Dravet Syndrome. Hum. Mol. Genet. 2013, 22, 4784–4804. [Google Scholar] [CrossRef]
- Ohmori, I.; Ouchida, M.; Kobayashi, K.; Jitsumori, Y.; Mori, A.; Michiue, H.; Nishiki, T.; Ohtsuka, Y.; Matsui, H. CACNA1A Variants May Modify the Epileptic Phenotype of Dravet Syndrome. Neurobiol. Dis. 2013, 50, 209–217. [Google Scholar] [CrossRef]
- Singh, N.A.; Pappas, C.; Dahle, E.J.; Claes, L.R.F.; Pruess, T.H.; De Jonghe, P.; Thompson, J.; Dixon, M.; Gurnett, C.; Peiffer, A.; et al. A Role of SCN9A in Human Epilepsies, as a Cause of Febrile Seizures and as a Potential Modifier of Dravet Syndrome. PLoS Genet. 2009, 5, e1000649. [Google Scholar] [CrossRef]
- Chourasia, N.; Ossó-Rivera, H.; Ghosh, A.; Von Allmen, G.; Koenig, M.K. Expanding the Phenotypic Spectrum of CACNA1H Mutations. Pediatr. Neurol. 2019, 93, 50–55. [Google Scholar] [CrossRef]
- Klassen, T.; Davis, C.; Goldman, A.; Burgess, D.; Chen, T.; Wheeler, D.; McPherson, J.; Bourquin, T.; Lewis, L.; Villasana, D.; et al. Exome Sequencing of Ion Channel Genes Reveals Complex Profiles Confounding Personal Risk Assessment in Epilepsy. Cell 2011, 145, 1036–1048. [Google Scholar] [CrossRef]
- Kalantari, S.; Carlston, C.; Alsaleh, N.; Abdel-Salam, G.M.H.; Alkuraya, F.; Kato, M.; Matsumoto, N.; Miyatake, S.; Yamamoto, T.; Fares-Taie, L.; et al. Expanding the KIF4A-Associated Phenotype. Am. J. Med. Genet. Part A 2021, 185, 3728–3739. [Google Scholar] [CrossRef]
- Castro, P.J.S.; Fernandez, C.N.; Subirana, P.Q.; Ortiz, M.P. Vohwinkel Syndrome Secondary to Missense Mutation D66H in GJB2 Gene (Connexin 26) Can Include Epileptic Manifestations. Seizure 2010, 19, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Dworakowska, B.; Nurowska, E.; Dołowy, K. Hydrocortisone Inhibition of Wild-Type and AD200Q Nicotinic Acetylcholine Receptors. Chem. Biol. Drug Des. 2018, 92, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Bostock, H.; Cikurel, K.; Burke, D. Threshold Tracking Techniques in the Study of Human Peripheral Nerve. Muscle Nerve 1998, 21, 137–158. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Burke, D.; Andersen, K.V.; Bostock, H. Multiple Measures of Axonal Excitability: A New Approach in Clinical Testing. Muscle Nerve 2000, 23, 399–409. [Google Scholar] [CrossRef]
- Burke, D.; Kiernan, M.C.; Bostock, H. Excitability of Human Axons. Clin. Neurophysiol. 2001, 112, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.R.; Reid, G.; Bostock, H. Action Potentials and Membrane Currents in the Human Node of Ranvier. Pflügers Arch. Eur. J. Physiol. 1995, 430, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Howells, J.; Czesnik, D.; Trevillion, L.; Burke, D. Excitability and the Safety Margin in Human Axons during Hyperthermia. J. Physiol. 2013, 591, 3063–3080. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group | SCN1A Mutation Carriers | ||||
|---|---|---|---|---|---|
| Mean | Range | Mean | Range | P (t-Test) | |
| Distal latency (ms) | 3.3 | 2.7–3.5 | 3.1 | 2.5–3.4 | 0.12 |
| Excitability parameters | |||||
| Stimulus (mA) for 50% max response | 8.85 | 7.11–12.94 | 5.64 | 4.17–7.1 | 0.09 |
| Strength–duration\time constant (ms) | 0.4 | 0.25–0.55 | 0.37 | 0.32–0.41 | 0.53 |
| Rheobase (mA) | 7.97 | 5.1–13 | 3.87 | 2.75–5.13 | 0.038 |
| Stimulus–response\slope | 6.43 | 5.43–8.64 | 5.14 | 3.84–5.85 | 0.13 |
| Peak response\(mV) | 0.53 | 0.31–0.67 | 0.63 | 0.27–0.78 | 0.5 |
| Resting I/V slope | 0.61 | 0.52–0.69 | 0.5 | 0.24–0.64 | 0.2 |
| RRP (ms) | 3.41 | 2.95–3.74 | 2.88 | 2.66–3.12 | 0.045 |
| Superexcitability (%) | −23.8 | −18.4–(−32.9) | −24.6 | −18–(−34.2) | 0.51 |
| Subexcitability (%) | 16.48 | 11.54–20.8 | 10.1 | 9.3–13.1 | 0.14 |
| Refractoriness at 2.5 ms (%) | 26.25 | 21.24–33.12 | 26.9 | 23.3–33.2 | 0.86 |
| TEd (10–20 ms) | 61.64 | 2.83 | 68.06 | 1.85 | 0.3 |
| TEd (40–60 ms) | 51.2 | 44–57 | 53.5 | 47–58 | 0.68 |
| TEd (90–100 ms) | 45.2 | 41.6–49.4 | 44.6 | 40.27–48.6 | 0.77 |
| TEh (10–20 ms) | −67.8 | −66.1-(−78.2) | −77.2 | −75.7-(−83.1) | 0.03 |
| TEh (20–40 ms) | −85.12 | −83.6-(−96.1) | −96.3 | −93.3-(−107.1) | 0.045 |
| TEh (slope 101–140 ms) | 1.79 | 1.69–2.28 | 2.26 | 2.07–2.8 | 0.046 |
| Gene | Reference Sequence | cDNA | Protein | Zygosity | Effect | gnomAD | CADD | Classification |
|---|---|---|---|---|---|---|---|---|
| CACNA1H | NM_021098.3 | c.4772G>A | p.(Ser1591Asn) | het | missense | 0.00006 | 8.171 | VUS |
| KIF4A | NM_012310.5 | c.2555A>T | p.(Gln852Leu) | het | missense | 0.00001 | 21.3 | VUS |
| GJB2 | NM_004004.6 | c.35del | p.(Gly12ValfsTer2) | het | frameshift | 0.00643 | - | Pathogenic |
| Subject No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Age at investigation | 53 y | 16 y | 51 y | 17 y | 15 y |
| Gender | M | M | M | M | F |
| Prenatal history | Uneventful | Uneventful | Uneventful | Uneventful | Uneventful |
| Psychomotor development | Normal | Normal | Normal | Normal | Normal |
| Age at seizure onset | 13 y | 14 m | 4 y | 3 y | 4 y |
| Triggering factors at onset | Fatigue | Fever | Physical effort, hyperthermia? | Fever | Fever |
| Age at last seizure | 53 y | 6 y | 39 y | 12 y/convulsive status epilepticus | 10 v |
| Types of seizures | GTCS | Myoclonic, GTCS, absence, focal impaired awareness, predominantly on the right side | GTCS | GTCS | GTCS |
| Clusters of seizures | No | Yes | No | No | None |
| Convulsive status epilepticus | No | Yes | No | Yes | None |
| Non-convulsive status epilepticus | No | No | No | No | None |
| Cognitive development | Normal | Normal | Normal | Normal | Normal |
| ASD | No | Asperger syndrome | No | No | No |
| Neurological examination | Normal | Ataxia | Normal | Slight ataxia, clumsiness | Normal |
| EEG (interictal) | Normal | Focal, lateralized and generalized spikes, multispikes, spikes–waves predominantly on the left side | Generalized spikes–waves | Normal | Normal |
| Neuroimaging | MRI-normal | MRI-normal | MRI-normal | MRI-normal | MRI-normal |
| AED previous | CBZ | VPA; VPA + OXCB; VPA + LTG; VPA + LEV | CBZ; CBZ + VPA; OXCB + VPA; VPA | VPA | VPA |
| AED current | VPA 1800 mg/d | VPA 600 mg/d | VPA 1000 mg/d | VPA 700 mg/d | VPA 600 mg/d |
| SCN1A-related epilepsy phenotype | Epilepsy with GTCS | Dravet syndrome—atypical | Epilepsy with GTCS | GEFS(+) | Epilepsy with GTCS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witkowski, G.; Szulczyk, B.; Nurowska, E.; Jurek, M.; Pasierski, M.; Lipiec, A.; Charzewska, A.; Dawidziuk, M.; Milewski, M.; Owsiak, S.; et al. Functional Characteristics of the Nav1.1 p.Arg1596Cys Mutation Associated with Varying Severity of Epilepsy Phenotypes. Int. J. Mol. Sci. 2024, 25, 1745. https://doi.org/10.3390/ijms25031745
Witkowski G, Szulczyk B, Nurowska E, Jurek M, Pasierski M, Lipiec A, Charzewska A, Dawidziuk M, Milewski M, Owsiak S, et al. Functional Characteristics of the Nav1.1 p.Arg1596Cys Mutation Associated with Varying Severity of Epilepsy Phenotypes. International Journal of Molecular Sciences. 2024; 25(3):1745. https://doi.org/10.3390/ijms25031745
Chicago/Turabian StyleWitkowski, Grzegorz, Bartlomiej Szulczyk, Ewa Nurowska, Marta Jurek, Michal Pasierski, Agata Lipiec, Agnieszka Charzewska, Mateusz Dawidziuk, Michal Milewski, Szymon Owsiak, and et al. 2024. "Functional Characteristics of the Nav1.1 p.Arg1596Cys Mutation Associated with Varying Severity of Epilepsy Phenotypes" International Journal of Molecular Sciences 25, no. 3: 1745. https://doi.org/10.3390/ijms25031745
APA StyleWitkowski, G., Szulczyk, B., Nurowska, E., Jurek, M., Pasierski, M., Lipiec, A., Charzewska, A., Dawidziuk, M., Milewski, M., Owsiak, S., Rola, R., Sienkiewicz Jarosz, H., & Hoffman-Zacharska, D. (2024). Functional Characteristics of the Nav1.1 p.Arg1596Cys Mutation Associated with Varying Severity of Epilepsy Phenotypes. International Journal of Molecular Sciences, 25(3), 1745. https://doi.org/10.3390/ijms25031745