
New Obolenskvirus Phages Brutus and Scipio: Biology, Evolution, and Phage-Host Interaction

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
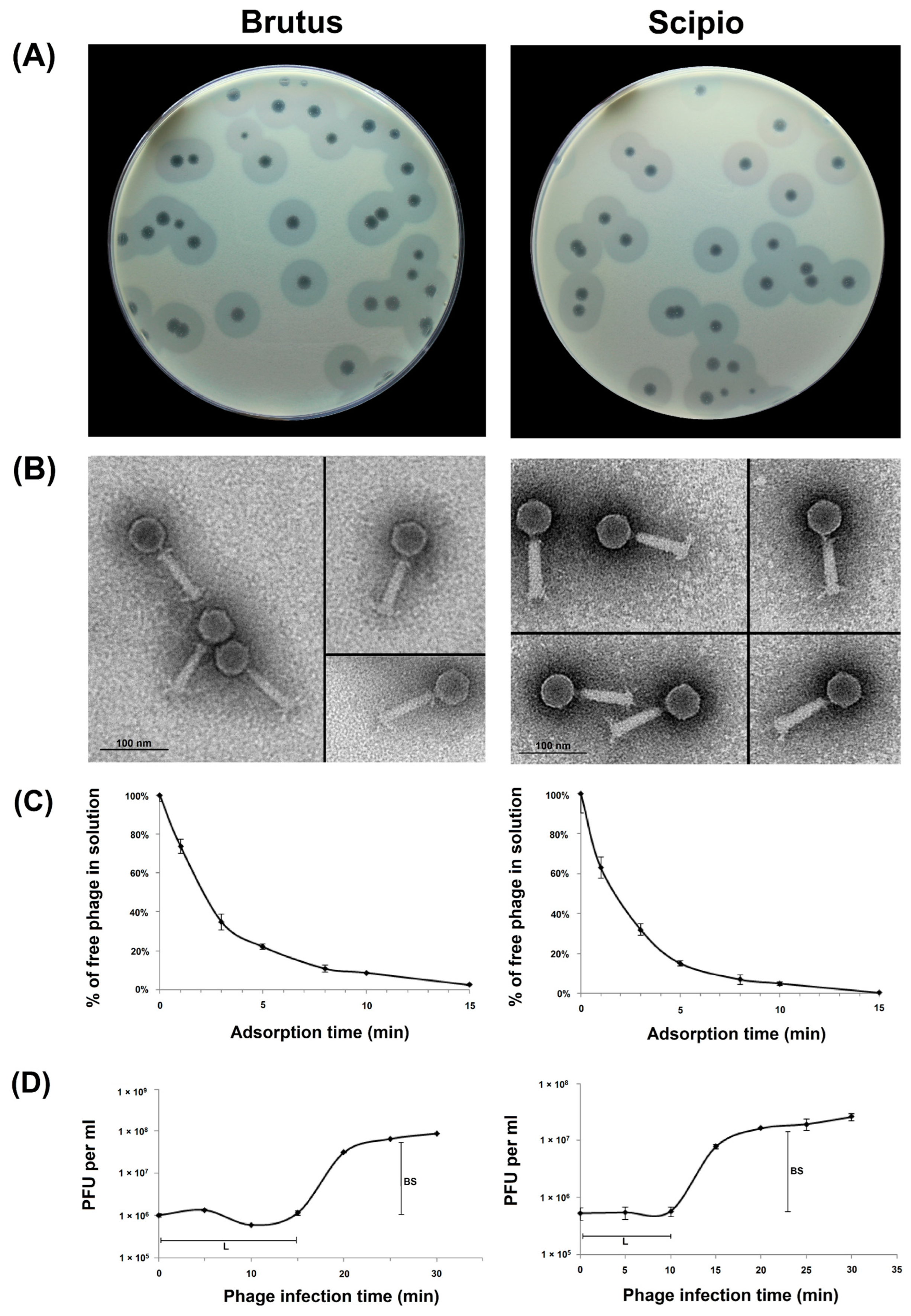
2.1. Phage Morphological Characteristics and Infection Parameters
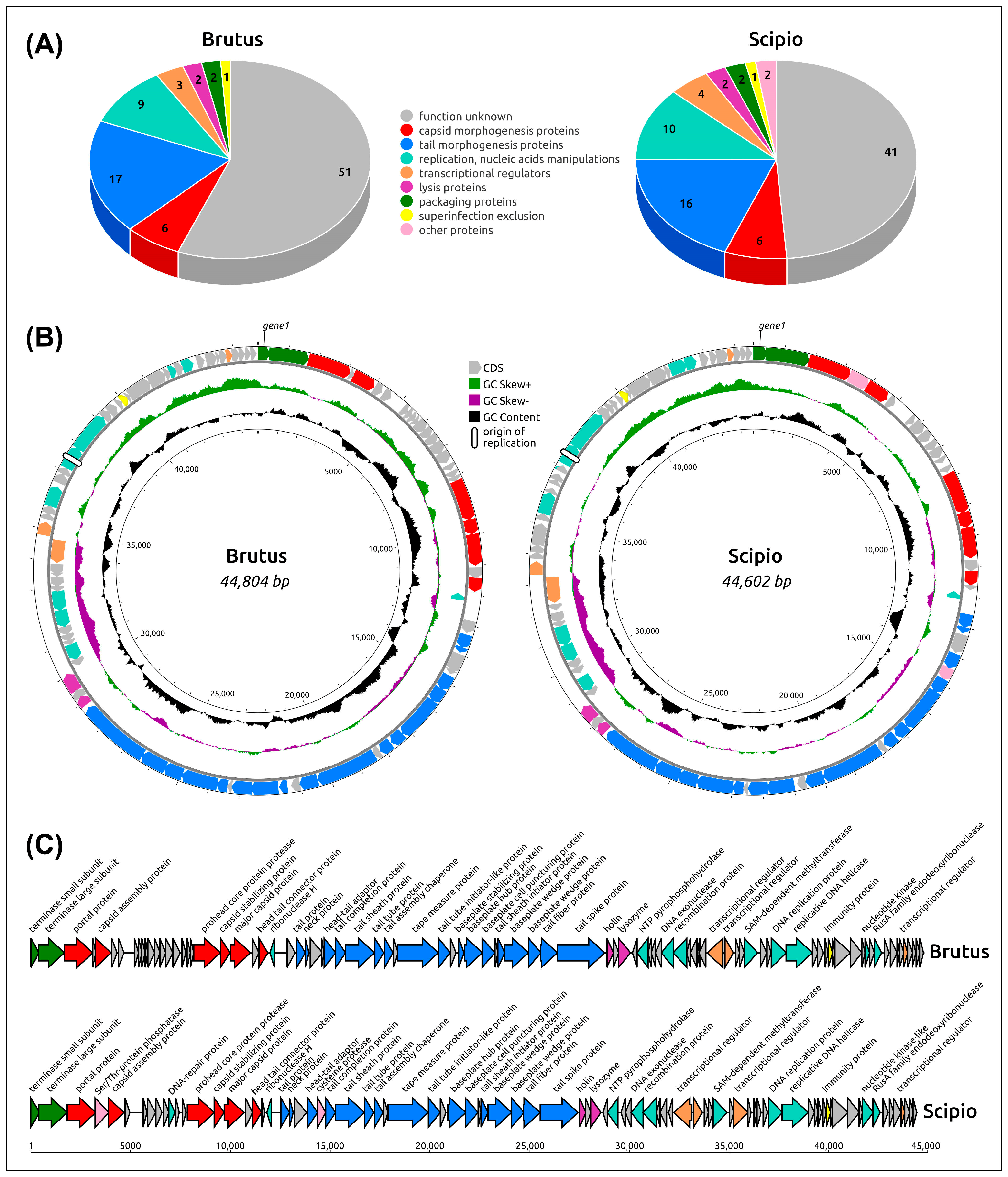
2.2. General Genome Features
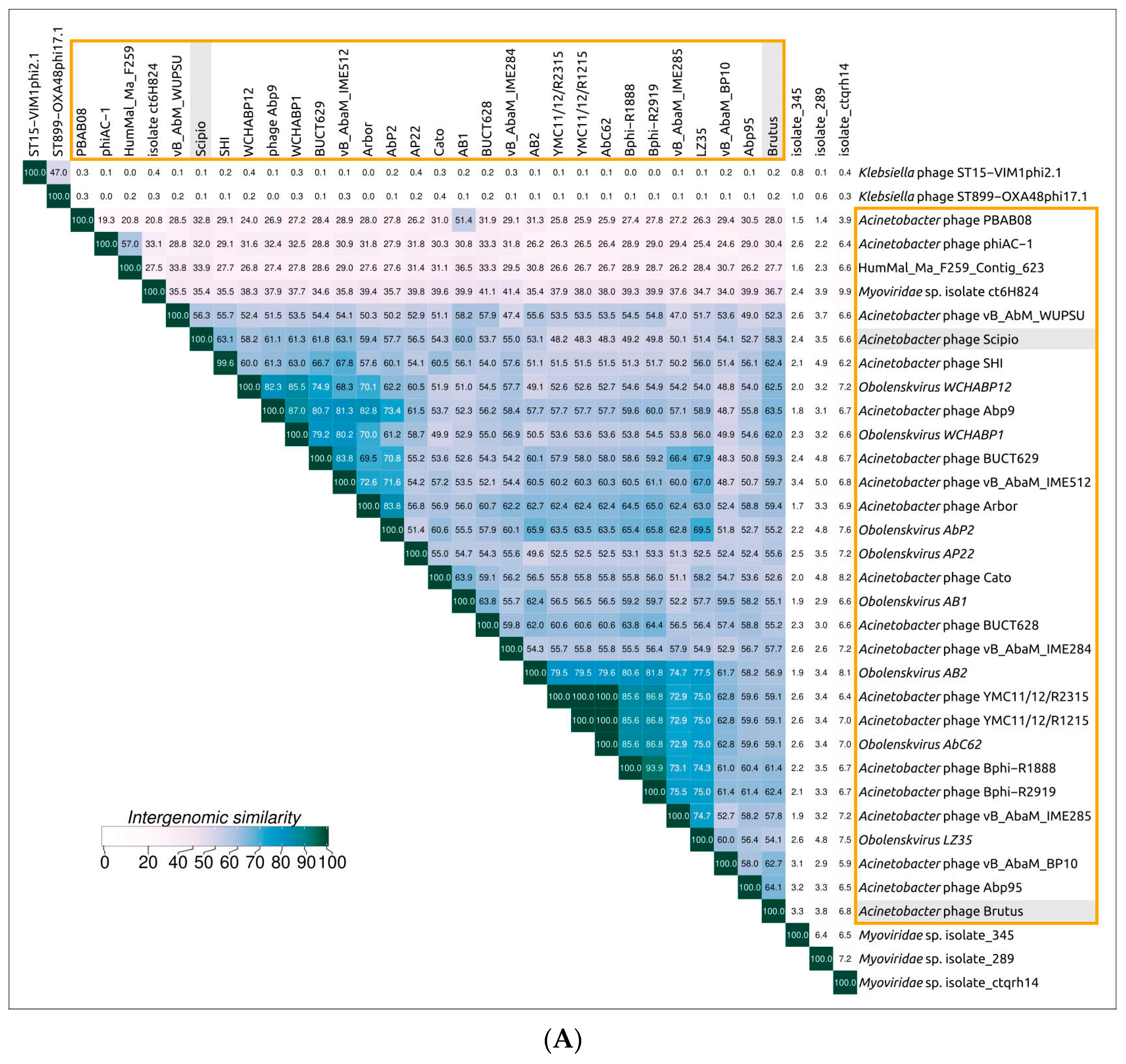
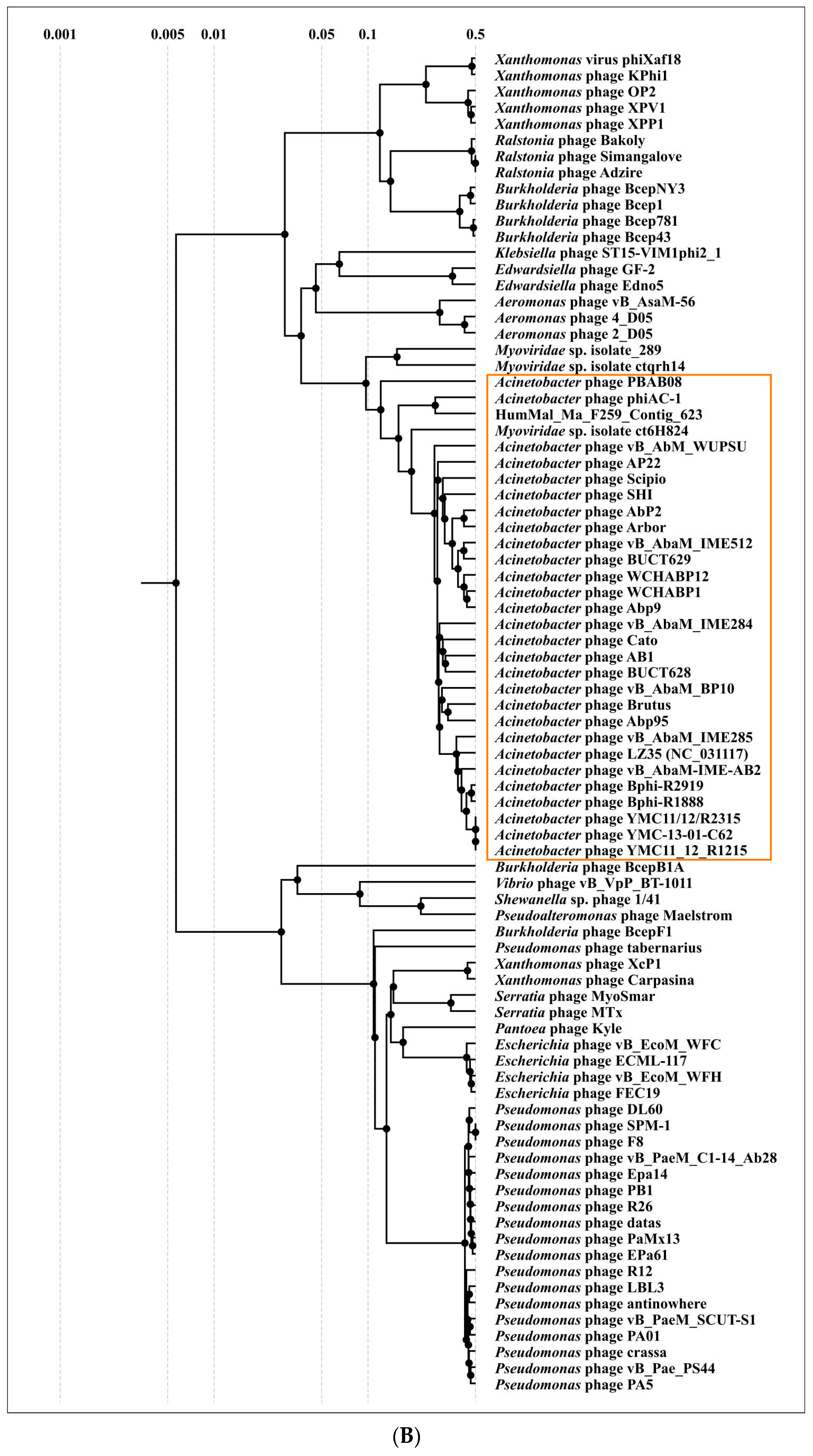
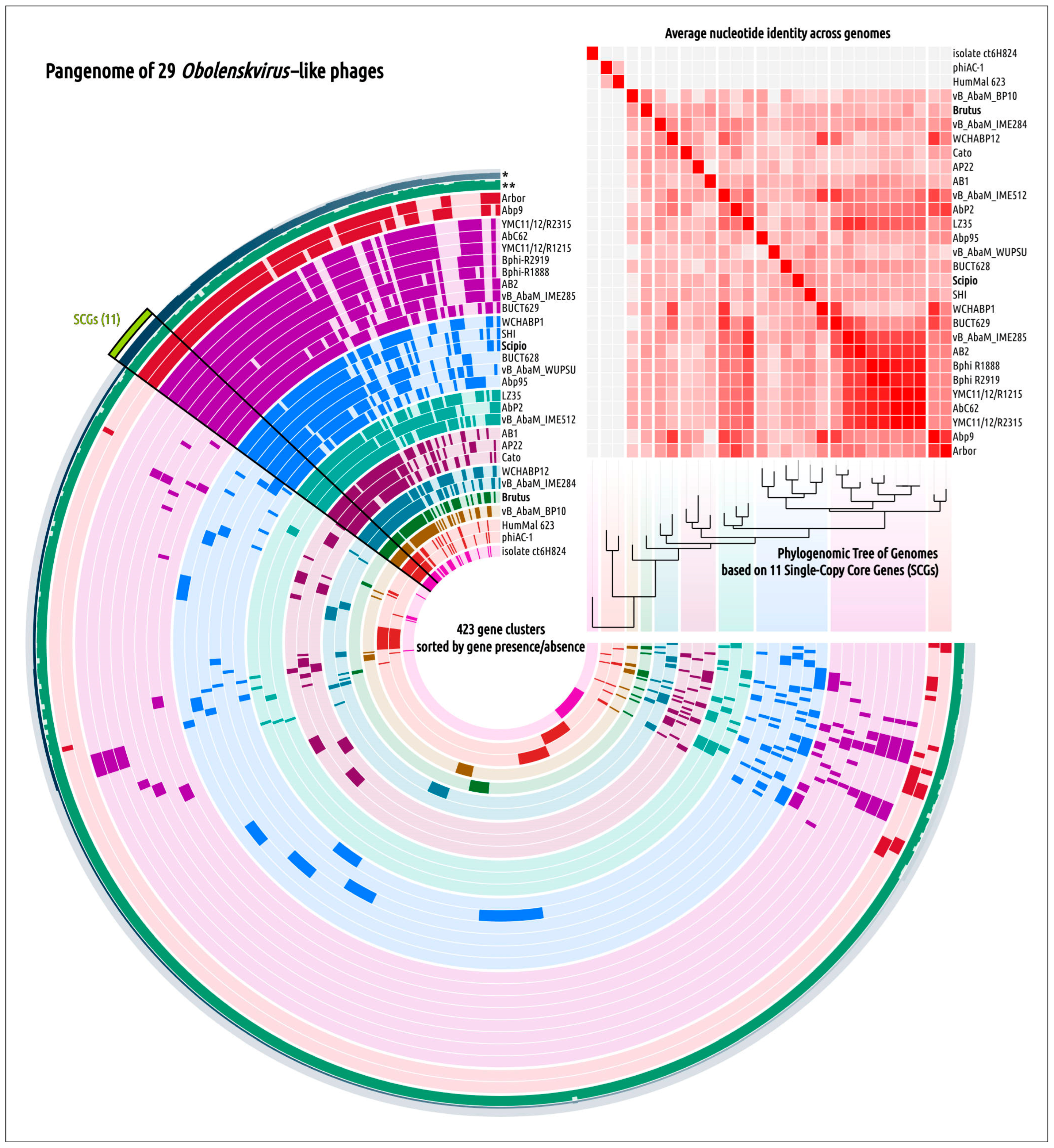
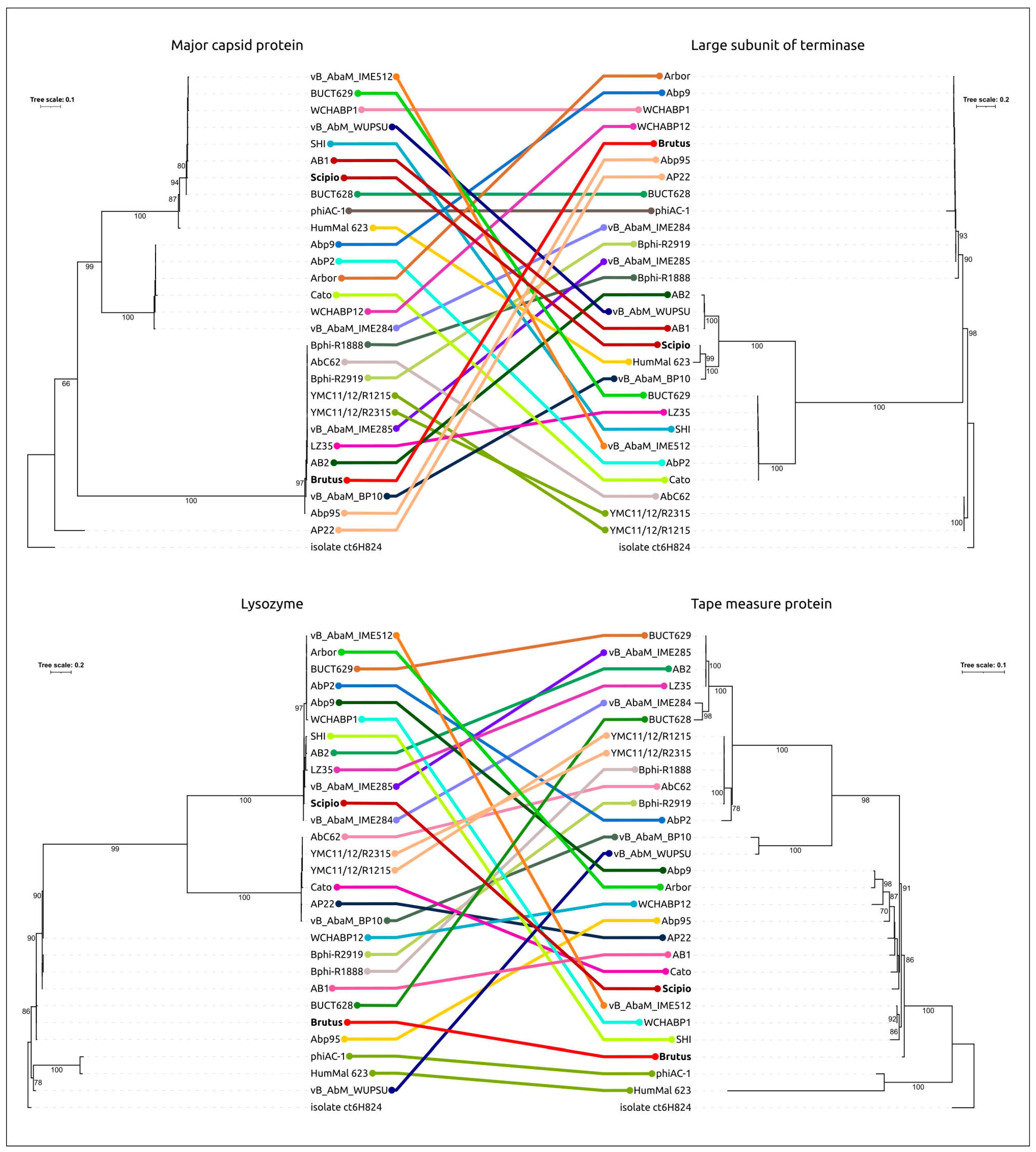
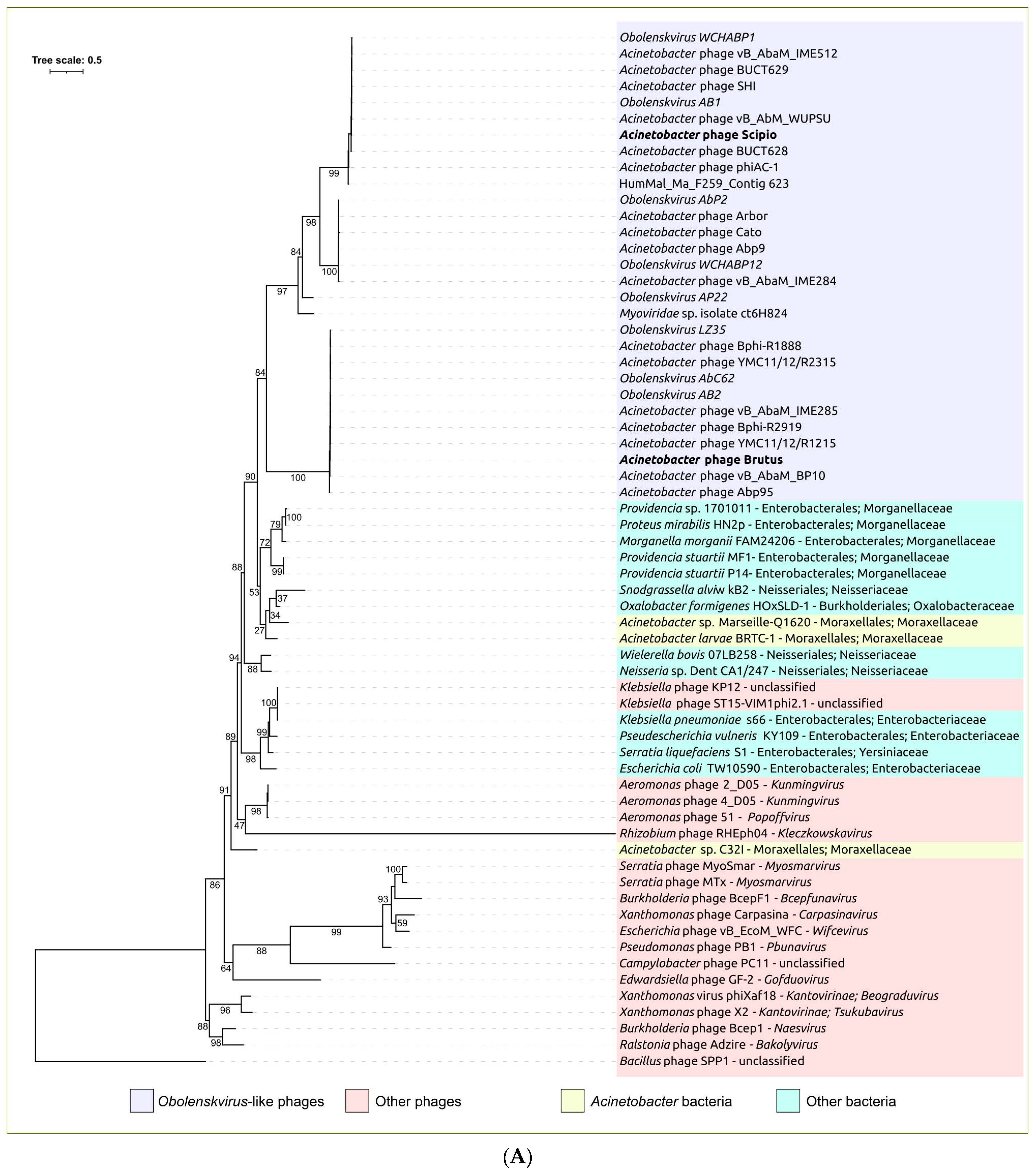
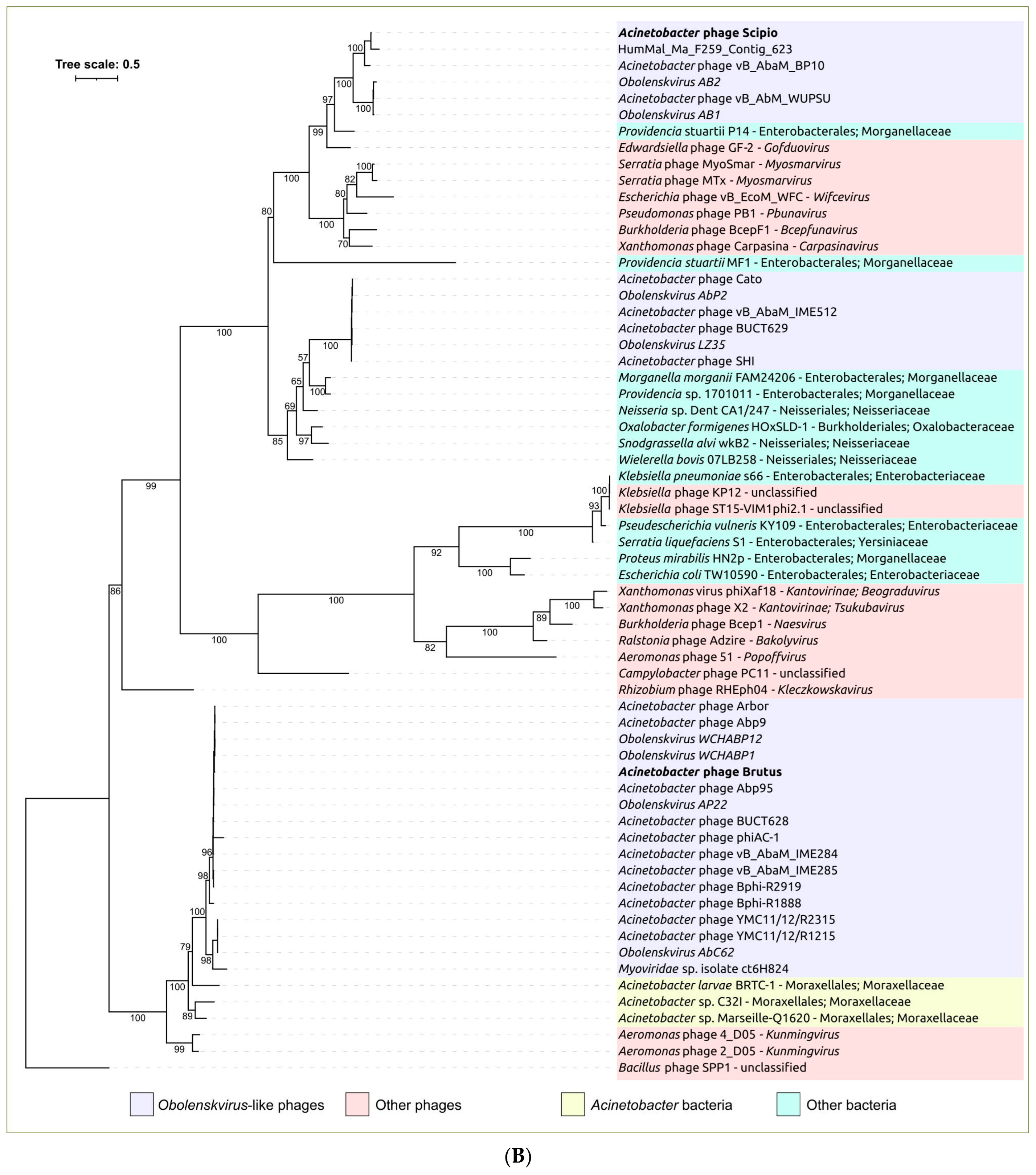
2.3. Search for Related Phages
2.4. Functional Genome Content
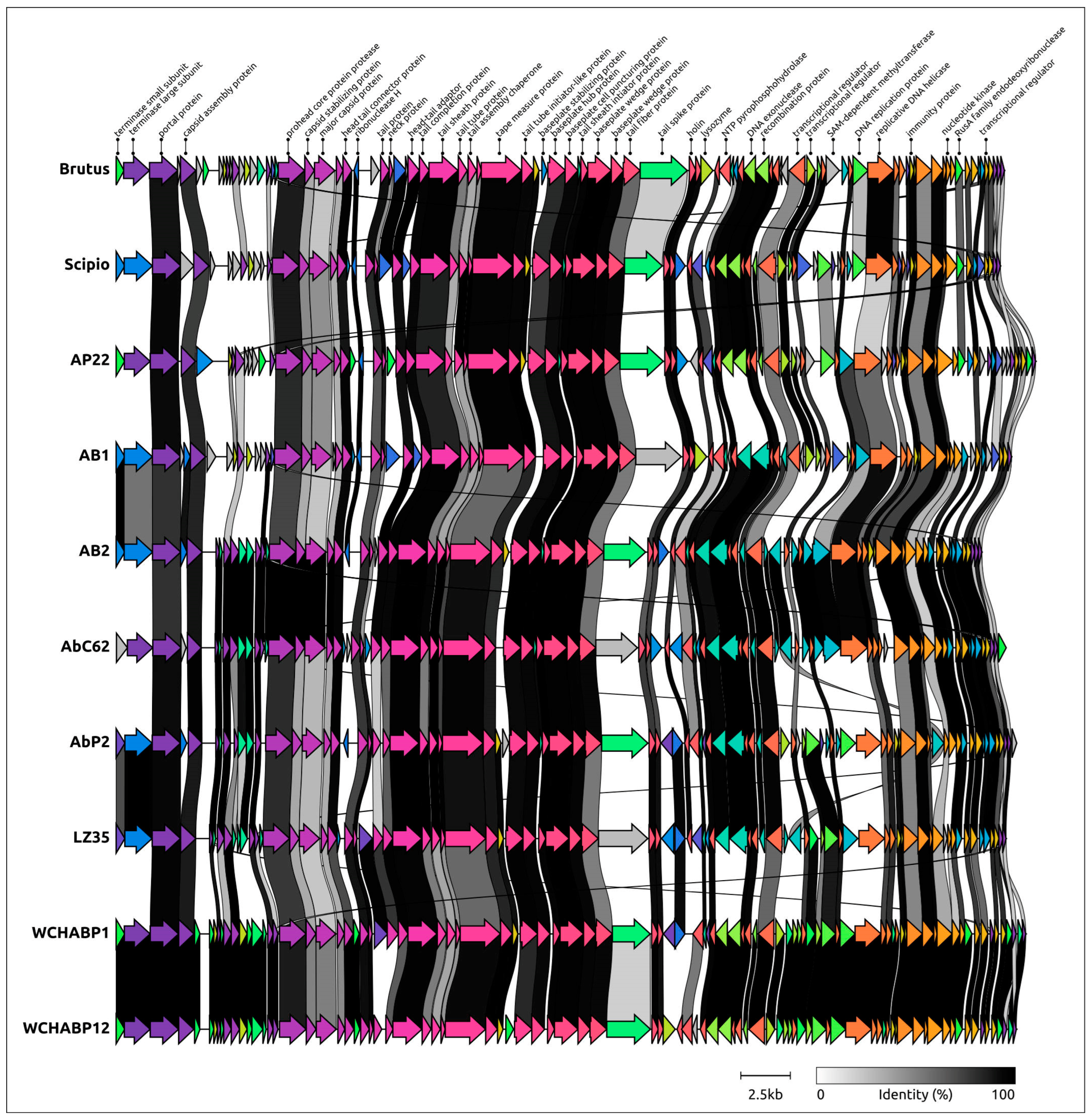
2.5. Genome Architecture and Pangenome Analysis
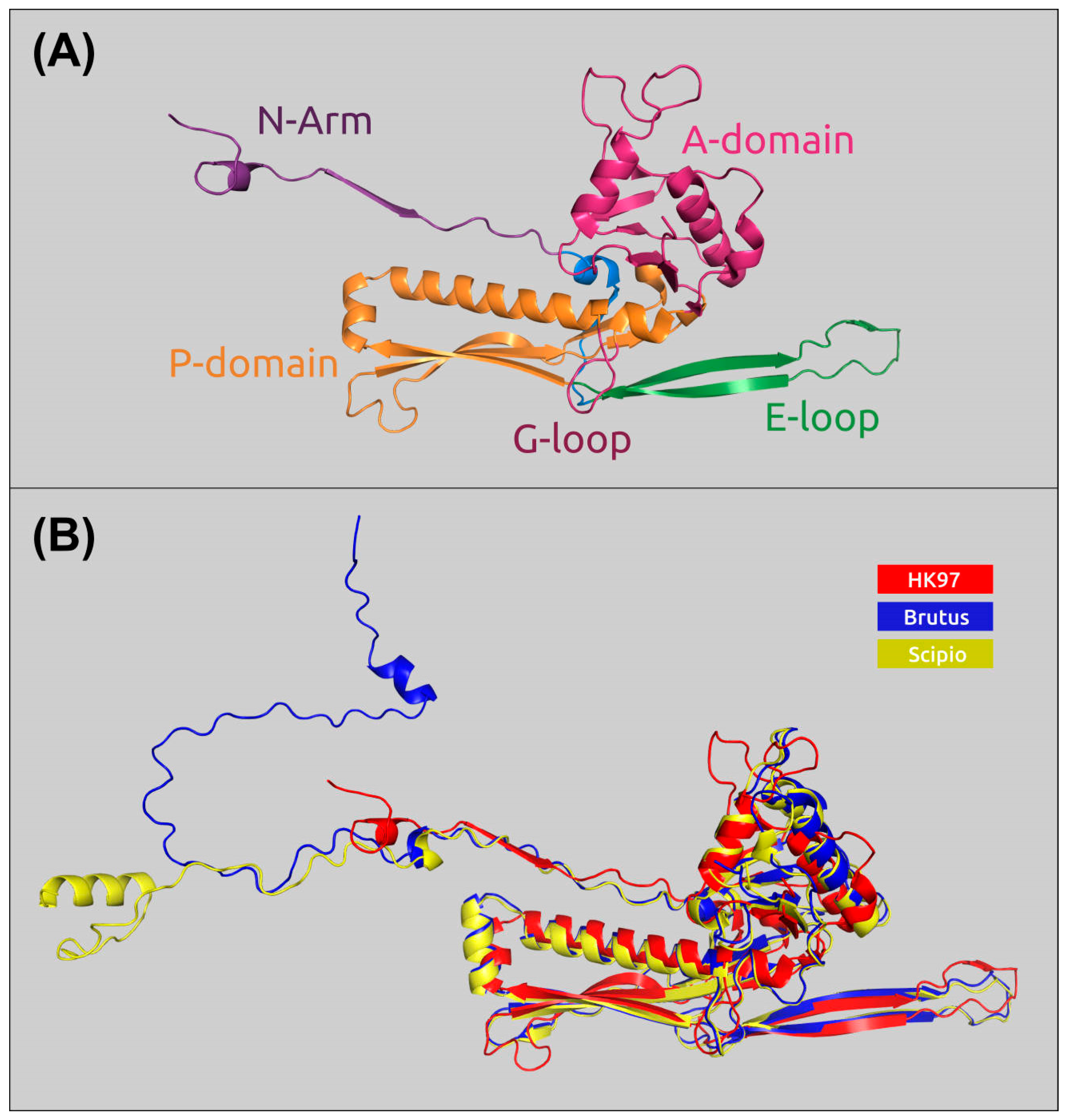
2.6. Receptor-Binding Proteins
2.7. The Mechanism of Enzymatic Activity of TSP Scipio_gp39
3. Discussion
4. Materials and Methods
4.1. Phage Isolation, Propagation, and Purification
4.2. Phage Adsorption and One-Step Growth Experiments
4.3. Electron Microscopy
4.4. Phage DNA Isolation and Sequencing
4.5. Phage Genome Annotation and Analysis
4.6. Phylogenetic Analysis
4.7. Protein Analysis and Structure Prediction
4.8. Cloning, Expression, and Purification of the Recombinant TSP
4.9. Isolation of the K82 Capsular Polysaccharide
4.10. Depolymerisation of the K82 CPS by Recombinant Tailspike Depolymerase
4.11. NMR Spectroscopy
4.12. Mass Spectroscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Herelle, F. On an invisible microbe antagonistic toward dysenteric bacilli: Brief Note by Mr. F. D’Herelle, Presented by Mr. Roux. 1917. Res. Microbiol. 2007, 158, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; De Roock, S.; Opal, S.M. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. In the beginning…2. Bacteriophage 2011, 1, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zhu, T. Potential of therapeutic bacteriophages in nosocomial infection management. Front. Microbiol. 2021, 12, 638094. [Google Scholar] [CrossRef] [PubMed]
- Ayoub Moubareck, C.; Hammoudi Halat, D. Insights into Acinetobacter baumannii: A review of microbiological, virulence, and resistance traits in a threatening nosocomial pathogen. Antibiotics 2020, 9, 119. [Google Scholar] [CrossRef]
- Roy, S.; Chowdhury, G.; Mukhopadhyay, A.K.; Dutta, S.; Basu, S. Convergence of biofilm formation and antibiotic resistance in Acinetobacter baumannii infection. Front. Med. 2022, 9, 793615. [Google Scholar] [CrossRef]
- Tan, Y.; Su, J.; Fu, M.; Zhang, H.; Zeng, H. Recent advances in phage-based therapeutics for multi-drug resistant Acinetobacter baumannii. Bioengineering 2022, 10, 35. [Google Scholar] [CrossRef]
- Lee, C.-R.; Lee, J.H.; Park, M.; Park, K.S.; Bae, I.K.; Kim, Y.B.; Cha, C.-J.; Jeong, B.C.; Lee, S.H. Biology of Acinetobacter baumannii: Pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front. Cell. Infect. Microbiol. 2017, 7, 55. [Google Scholar] [CrossRef]
- Oliveira, H.; Domingues, R.; Evans, B.; Sutton, J.M.; Adriaenssens, E.M.; Turner, D. Genomic diversity of bacteriophages infecting the genus Acinetobacter. Viruses 2022, 14, 181. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Shneider, M.M.; Popova, A.V.; Kasimova, A.A.; Senchenkova, S.N.; Shashkov, A.S.; Chizhov, A.O. Mechanisms of Acinetobacter baumannii capsular polysaccharide cleavage by phage depolymerases. Biochemistry 2020, 85, 567–574. [Google Scholar] [CrossRef]
- Kasimova, A.A.; Kenyon, J.J.; Arbatsky, N.P.; Shashkov, A.S.; Popova, A.V.; Knirel, Y.A.; Hall, R.M. Structure of the K82 capsular polysaccharide from Acinetobacter baumannii LUH5534 containing a D-galactose 4,6-pyruvic acid acetal. Biochemistry 2018, 83, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Shashkov, A.S.; Cahill, S.M.; Arbatsky, N.P.; Westacott, A.C.; Kasimova, A.A.; Shneider, M.M.; Popova, A.V.; Shagin, D.A.; Shelenkov, A.A.; Mikhailova, Y.V.; et al. Acinetobacter baumannii K116 capsular polysaccharide structure is a hybrid of the K14 and revised K37 structures. Carbohydr. Res. 2019, 484, 107774. [Google Scholar] [CrossRef] [PubMed]
- Tsurimoto, T.; Matsubara, K. Purified bacteriophage lambda O protein binds to four repeating sequences at the lambda replication origin. Nucleic Acids Res. 1981, 9, 1789–1799. [Google Scholar] [CrossRef]
- Kreuzer, K.N.; Brister, J.R. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: A review in the virology journal series on bacteriophage T4 and its relatives. Virol. J. 2010, 7, 358. [Google Scholar] [CrossRef] [PubMed]
- Filutowicz, M.; Rakowski, S.A. Regulatory implications of protein assemblies at the gamma origin of plasmid R6K—A review. Gene 1998, 223, 195–204. [Google Scholar] [CrossRef]
- Jiang, L.; Tan, J.; Hao, Y.; Wang, Q.; Yan, X.; Wang, D.; Tuo, L.; Wei, Z.; Huang, G. Isolation and characterization of a novel myophage Abp9 against pandrug resistant Acinetobacater baumannii. Front. Microbiol. 2020, 11, 506068. [Google Scholar] [CrossRef]
- Huang, L.; Huang, S.; Jiang, L.; Tan, J.; Yan, X.; Gou, C.; Chen, X.; Xiang, L.; Wang, D.; Huang, G.; et al. Characterisation and sequencing of the novel phage Abp95, which is effective against multi-genotypes of carbapenem-resistant Acinetobacter baumannii. Sci. Rep. 2023, 13, 188. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, K.; Chen, L.; Luo, S.; Fan, H.; Tong, Y. Biological characterization and genomic analysis of Acinetobacter baumannii phage BUCT628. Arch. Virol. 2022, 167, 1471–1474. [Google Scholar] [CrossRef]
- Han, K.; Zhu, Y.; Li, F.; Li, M.; An, X.; Song, L.; Fan, H.; Tong, Y. Genomic analysis of Acinetobacter phage BUCT629, a newly isolated member of the genus Obolenskvirus. Arch. Virol. 2022, 167, 1197–1199. [Google Scholar] [CrossRef]
- Evseev, P.; Gornostal, E.; Shneider, M.; Mikhaylova, Y.; Shelenkov, A.; Popova, A.; Miroshnikov, K. A novel Acinetobacter phage Cato: Lytic myovirus containing tailspike depolymerase. In Proceedings of the 2022 Ural-Siberian Conference on Computational Technologies in Cognitive Science, Genomics and Biomedicine (CSGB), Novosibirsk, Russia, 7–8 July 2022; pp. 110–114. [Google Scholar] [CrossRef]
- Cha, K.; Oh, H.K.; Jang, J.Y.; Jo, Y.; Kim, W.K.; Ha, G.U.; Ko, K.S.; Myung, H. Characterization of two novel bacteriophages infecting multidrug-resistant (MDR) Acinetobacter baumannii and evaluation of their therapeutic efficacy in vivo. Front. Microbiol. 2018, 9, 696. [Google Scholar] [CrossRef]
- Kim, J.H.; Oh, C.; Choresca, C.H.; Shin, S.P.; Han, J.E.; Jun, J.W.; Heo, S.-J.; Kang, D.-H.; Park, S.C. Complete genome sequence of bacteriophage phiAC-1 infecting Acinetobacter soli strain KZ-1. J. Virol. 2012, 86, 13131–13132. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, P.; Zhu, Y.; Huang, Y.; Gao, M.; Yuan, X.; Niu, W.; Liu, H.; Fan, H.; Qin, Y.; et al. Identification of a novel Acinetobacter baumannii phage-derived depolymerase and its therapeutic application in mice. Front. Microbiol. 2020, 11, 1407. [Google Scholar] [CrossRef]
- Wintachai, P.; Voravuthikunchai, S.P. Characterization of novel lytic Myoviridae phage infecting multidrug-resistant Acinetobacter baumannii and synergistic antimicrobial efficacy between phage and sacha inchi oil. Pharmaceuticals 2022, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Mi, Z.; Huang, Y.; Yuan, X.; Niu, W.; Wang, Y.; Hua, Y.; Fan, H.; Bai, C.; Tong, Y. Characterization, sequencing and comparative genomic analysis of vB_AbaM-IME-AB2, a novel lytic bacteriophage that infects multidrug-resistant Acinetobacter baumannii clinical isolates. BMC Microbiol. 2014, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, X.; Shi, Y.; Yin, S.; Shen, W.; Chen, J.; Chen, Y.; Chen, Y.; You, B.; Gong, Y.; et al. Characterization and genome annotation of a newly detected bacteriophage infecting multidrug-resistant Acinetobacter baumannii. Arch. Virol. 2019, 164, 1527–1533. [Google Scholar] [CrossRef]
- Popova, A.V.; Zhilenkov, E.L.; Myakinina, V.P.; Krasilnikova, V.M.; Volozhantsev, N.V. Isolation and characterization of wide host range lytic bacteriophage AP22 infecting Acinetobacter baumannii. FEMS Microbiol. Lett. 2012, 332, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Huang, H.; Wu, X.; Hao, Y.; Sun, Y. Complete genome sequence of lytic bacteriophage LZ35 Infecting Acinetobacter baumannii isolates. Genome Announc. 2016, 4, e01104-16. [Google Scholar] [CrossRef]
- Zhou, W.; Feng, Y.; Zong, Z. Two new lytic bacteriophages of the Myoviridae family against carbapenem-resistant Acinetobacter baumannii. Front. Microbiol. 2018, 9, 850. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Xu, J.; Wang, D.; Gui, M.; Xiang, Y. Structural assembly of the tailed bacteriophage Φ29. Nat. Commun. 2019, 10, 2366. [Google Scholar] [CrossRef]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an integrated approach involving AlphaFold predictions for the evolutionary taxonomy of Duplodnaviria viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Duda, R.L.; Hempel, J.; Michel, H.; Shabanowitz, J.; Hunt, D.; Hendrix, R.W. Structural tduring bacteriophage HK97 Head Assembly. J. Mol. Biol. 1995, 247, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Do we have the hole story yet? Curr. Opin. Microbiol. 2013, 16, 790–797. [Google Scholar] [CrossRef]
- Zheng, Y.; Struck, D.K.; Dankenbring, C.A.; Young, R. Evolutionary dominance of holin lysis systems derives from superior genetic malleability. Microbiol. Read. Engl. 2008, 154, 1710–1718. [Google Scholar] [CrossRef]
- Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of phage tail sheath protein. Viruses 2022, 14, 1148. [Google Scholar] [CrossRef] [PubMed]
- Narajczyk, M.; Barańska, S.; Węgrzyn, A.; Węgrzyn, G. Switch from θ to σ replication of bacteriophage λ DNA: Factors involved in the process and a model for its regulation. Mol. Genet. Genom. 2007, 278, 65–74. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef]
- Leprince, A.; Mahillon, J. Phage adsorption to gram-positive bacteria. Viruses 2023, 15, 196. [Google Scholar] [CrossRef]
- Oliveira, H.; Costa, A.R.; Konstantinides, N.; Ferreira, A.; Akturk, E.; Sillankorva, S.; Nemec, A.; Shneider, M.; Dötsch, A.; Azeredo, J. Ability of phages to infect Acinetobacter calcoaceticus-Acinetobacter baumannii complex species through acquisition of different pectate lyase depolymerase domains. Environ. Microbiol. 2017, 19, 5060–5077. [Google Scholar] [CrossRef]
- Lee, I.-M.; Tu, I.-F.; Yang, F.-L.; Ko, T.-P.; Liao, J.-H.; Lin, N.-T.; Wu, C.-Y.; Ren, C.-T.; Wang, A.H.-J.; Chang, C.-M.; et al. Structural basis for fragmenting the exopolysaccharide of Acinetobacter baumannii by bacteriophage ΦAB6 tailspike protein. Sci. Rep. 2017, 7, 42711. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.V.; Shneider, M.M.; Arbatsky, N.P.; Kasimova, A.A.; Senchenkova, S.N.; Shashkov, A.S.; Dmitrenok, A.S.; Chizhov, A.O.; Mikhailova, Y.V.; Shagin, D.A.; et al. Specific interaction of novel Friunavirus phages encoding tailspike depolymerases with corresponding Acinetobacter baumannii capsular types. J. Virol. 2021, 95, e01714-20. [Google Scholar] [CrossRef] [PubMed]
- Kasimova, A.A.; Arbatsky, N.P.; Timoshina, O.Y.; Shneider, M.M.; Shashkov, A.S.; Chizhov, A.O.; Popova, A.V.; Hall, R.M.; Kenyon, J.J.; Knirel, Y.A. The K26 capsular polysaccharide from Acinetobacter baumannii KZ-1098: Structure and cleavage by a specific phage depolymerase. Int. J. Biol. Macromol. 2021, 191, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Shchurova, A.S.; Shneider, M.M.; Arbatsky, N.P.; Shashkov, A.S.; Chizhov, A.O.; Skryabin, Y.P.; Mikhaylova, Y.V.; Sokolova, O.S.; Shelenkov, A.A.; Miroshnikov, K.A.; et al. Novel Acinetobacter baumannii myovirus TaPaz encoding two tailspike depolymerases: Characterization and host-recognition strategy. Viruses 2021, 13, 978. [Google Scholar] [CrossRef] [PubMed]
- Timoshina, O.Y.; Kasimova, A.A.; Shneider, M.M.; Matyuta, I.O.; Nikolaeva, A.Y.; Evseev, P.V.; Arbatsky, N.P.; Shashkov, A.S.; Chizhov, A.O.; Shelenkov, A.A.; et al. Friunavirus phage-encoded depolymerases specific to different capsular types of Acinetobacter baumannii. Int. J. Mol. Sci. 2023, 24, 9100. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, J.J.; Hall, R.M. Variation in the complex carbohydrate biosynthesis loci of Acinetobacter baumannii Genomes. PLoS ONE 2013, 8, e62160. [Google Scholar] [CrossRef]
- Ge, H.; Hu, M.; Zhao, G.; Du, Y.; Xu, N.; Chen, X.; Jiao, X. The “Fighting Wisdom and Bravery” of tailed phage and host in the process of adsorption. Microbiol. Res. 2020, 230, 126344. [Google Scholar] [CrossRef]
- Klumpp, J.; Dunne, M.; Loessner, M.J. A perfect fit: Bacteriophage receptor-binding proteins for diagnostic and therapeutic applications. Curr. Opin. Microbiol. 2023, 71, 102240. [Google Scholar] [CrossRef]
- Sun, L.; You, J.; Li, D.; Zhang, Z.; Qin, X.; Pang, W.; Li, P.; Han, Q.; Li, Y.; Huang, Z.; et al. Variants of a putative baseplate wedge protein extend the host range of Pseudomonas phage K8. Microbiome 2023, 11, 18. [Google Scholar] [CrossRef]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef]
- Stummeyer, K.; Schwarzer, D.; Claus, H.; Vogel, U.; Gerardy-Schahn, R.; Mühlenhoff, M. Evolution of bacteriophages infecting encapsulated bacteria: Lessons from Escherichia coli K1-specific phages. Mol. Microbiol. 2006, 60, 1123–1135. [Google Scholar] [CrossRef]
- Moura de Sousa, J.A.; Pfeifer, E.; Touchon, M.; Rocha, E.P.C. Causes and consequences of bacteriophage diversification via genetic exchanges across lifestyles and bacterial taxa. Mol. Biol. Evol. 2021, 38, 2497–2512. [Google Scholar] [CrossRef]
- Botstein, D. A Theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484–490. [Google Scholar] [CrossRef]
- Christie, G.E.; Calendar, R. Bacteriophage P2. Bacteriophage 2016, 6, e1145782. [Google Scholar] [CrossRef]
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of bacteriophage P1. J. Bacteriol. 2004, 186, 7032–7068. [Google Scholar] [CrossRef] [PubMed]
- Ravin, N.V. N15: The linear phage-plasmid. Plasmid 2011, 65, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or Lysogenic’. FEMS Microbiol. Lett. 2016, 363, fnw047. [Google Scholar] [CrossRef] [PubMed]
- Gummalla, V.S.; Zhang, Y.; Liao, Y.-T.; Wu, V.C.H. The role of temperate phages in bacterial pathogenicity. Microorganisms 2023, 11, 541. [Google Scholar] [CrossRef]
- Brady, A.; Felipe-Ruiz, A.; Gallego del Sol, F.; Marina, A.; Quiles-Puchalt, N.; Penadés, J.R. Molecular basis of lysis–lysogeny decisions in gram-positive phages. Annu. Rev. Microbiol. 2021, 75, 563–581. [Google Scholar] [CrossRef]
- De Paepe, M.; Hutinet, G.; Son, O.; Amarir-Bouhram, J.; Schbath, S.; Petit, M.-A. Temperate phages acquire DNA from defective prophages by relaxed homologous recombination: The role of Rad52-like recombinases. PLoS Genet. 2014, 10, e1004181. [Google Scholar] [CrossRef]
- Juhala, R.J.; Ford, M.E.; Duda, R.L.; Youlton, A.; Hatfull, G.F.; Hendrix, R.W. Genomic sequences of bacteriophages HK97 and HK022: Pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 2000, 299, 27–51. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; Casjens, S. The origins and ongoing evolution of viruses. Trends Microbiol. 2000, 8, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; ISBN 0-87969-309-6. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a Program to Detect tRNA Genes and tmRNA Genes in Nucleotide Sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap. Js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Edler, D.; Klein, J.; Antonelli, A.; Silvestro, D. raxmlGUI 2.0: A graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol. Evol. 2021, 12, 373–377. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. Biorxiv 2021. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Westphal, O.; Jann, K. Bacterial lipopolysaccharides extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 1965, 5, 83–91. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Phage | NCBI Accession | Sequence Length, b.p. | % GC | ORF # | Reference |
|---|---|---|---|---|---|---|
| 1 | Acinetobacter phage Abp9 | MN166083.1 | 44,820 | 37.70% | 88 | [17] |
| 2 | Acinetobacter phage Abp95 | MZ618622.1 | 43,176 | 37.80% | 84 | [18] |
| 3 | Acinetobacter phage Arbor | ON237674.1 | 45,041 | 37.40% | 85 | |
| 4 | Acinetobacter phage Bphi-R1888 | MN516422.1 | 44,590 | 37.90% | 83 | |
| 5 | Acinetobacter phage Bphi-R2919 | MN516421.1 | 44,227 | 37.80% | 82 | |
| 9 | Acinetobacter phage Brutus | ON036882.1 | 44,931 | 37.40% | 91 | |
| 7 | Acinetobacter phage BUCT628 | MZ593728.1 | 44,935 | 37.50% | 86 | [19] |
| 8 | Acinetobacter phage BUCT629 | MZ712044.1 | 46,325 | 37.60% | 88 | [20] |
| 9 | Acinetobacter phage Cato | OM471864.1 | 45,188 | 37.40% | 87 | [21] |
| 10 | Acinetobacter phage PBAB08 | MG366114.1 | 42,312 | 38.60% | 110 | [22] |
| 11 | Acinetobacter phage phiAC-1 | NC_028995.1 | 43,216 | 38.50% | 82 | [23] |
| 12 | Acinetobacter phage Scipio | ON036883.1 | 44,602 | 37.60% | 84 | |
| 13 | Acinetobacter phage SHI | ON480525.1 | 44,180 | 37.60% | 91 | |
| 14 | Acinetobacter phage vB_AbaM_BP10 | OP585104.1 | 44,443 | 37.30% | 90 | |
| 15 | Acinetobacter phage vB_AbaM_IME284 | MH853787.1 | 43,557 | 38.30% | 86 | |
| 16 | Acinetobacter phage vB_AbaM_IME285 | MH853786.1 | 45,063 | 37.90% | 84 | [24] |
| 17 | Acinetobacter phage vB_AbaM_IME512 | MH853788.1 | 46,499 | 37.60% | 91 | |
| 18 | Acinetobacter phage vB_AbM_WUPSU | OL743187.1 | 44,241 | 37.20% | 82 | [25] |
| 19 | Acinetobacter phage YMC11/12/R1215 | KP861231.1 | 44,866 | 37.60% | 85 | |
| 20 | Acinetobacter phage YMC11/12/R2315 | NC_028855.1 | 44,846 | 37.60% | 86 | |
| 21 | Assembly HumMal, Ma_F259_Contig_623 | CYGL01000085.1 | 45,317 | 37.80% | 83 | |
| 22 | Myoviridae sp. isolate ct6H824 | BK017052.1 | 42,126 | 37.70% | 85 | |
| 23 | Obolenskvirus AB1 | NC_042028.1 | 45,159 | 37.70% | 85 | [26] |
| 24 | Obolenskvirus AB2 | NC_041857.1 | 43,665 | 37.50% | 82 | [26] |
| 25 | Obolenskvirus AbC62 | NC_024785.1 | 44,844 | 37.60% | 85 | |
| 26 | Obolenskvirus AbP2 | NC_041998.1 | 45,373 | 37.80% | 87 | [27] |
| 27 | Obolenskvirus AP22 | NC_017984.1 | 46,387 | 37.70% | 91 | [28] |
| 28 | Obolenskvirus LZ35 | NC_031117.1 | 44,885 | 37.90% | 82 | [29] |
| 29 | Obolenskvirus WCHABP1 | NC_041966.1 | 45,888 | 37.60% | 92 | [30] |
| 30 | Obolenskvirus WCHABP12 | NC_041924.1 | 45,415 | 37.60% | 93 | [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.V.; Shneider, M.M.; Kolupaeva, L.V.; Kasimova, A.A.; Timoshina, O.Y.; Perepelov, A.V.; Shpirt, A.M.; Shelenkov, A.A.; Mikhailova, Y.V.; Suzina, N.E.; et al. New Obolenskvirus Phages Brutus and Scipio: Biology, Evolution, and Phage-Host Interaction. Int. J. Mol. Sci. 2024, 25, 2074. https://doi.org/10.3390/ijms25042074
Evseev PV, Shneider MM, Kolupaeva LV, Kasimova AA, Timoshina OY, Perepelov AV, Shpirt AM, Shelenkov AA, Mikhailova YV, Suzina NE, et al. New Obolenskvirus Phages Brutus and Scipio: Biology, Evolution, and Phage-Host Interaction. International Journal of Molecular Sciences. 2024; 25(4):2074. https://doi.org/10.3390/ijms25042074
Chicago/Turabian StyleEvseev, Peter V., Mikhail M. Shneider, Lyubov V. Kolupaeva, Anastasia A. Kasimova, Olga Y. Timoshina, Andrey V. Perepelov, Anna M. Shpirt, Andrey A. Shelenkov, Yulia V. Mikhailova, Natalia E. Suzina, and et al. 2024. "New Obolenskvirus Phages Brutus and Scipio: Biology, Evolution, and Phage-Host Interaction" International Journal of Molecular Sciences 25, no. 4: 2074. https://doi.org/10.3390/ijms25042074
APA StyleEvseev, P. V., Shneider, M. M., Kolupaeva, L. V., Kasimova, A. A., Timoshina, O. Y., Perepelov, A. V., Shpirt, A. M., Shelenkov, A. A., Mikhailova, Y. V., Suzina, N. E., Knirel, Y. A., Miroshnikov, K. A., & Popova, A. V. (2024). New Obolenskvirus Phages Brutus and Scipio: Biology, Evolution, and Phage-Host Interaction. International Journal of Molecular Sciences, 25(4), 2074. https://doi.org/10.3390/ijms25042074