
Methadone Requires the Co-Activation of μ-Opioid and Toll-Like-4 Receptors to Produce Extracellular DNA Traps in Bone-Marrow-Derived Mast Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
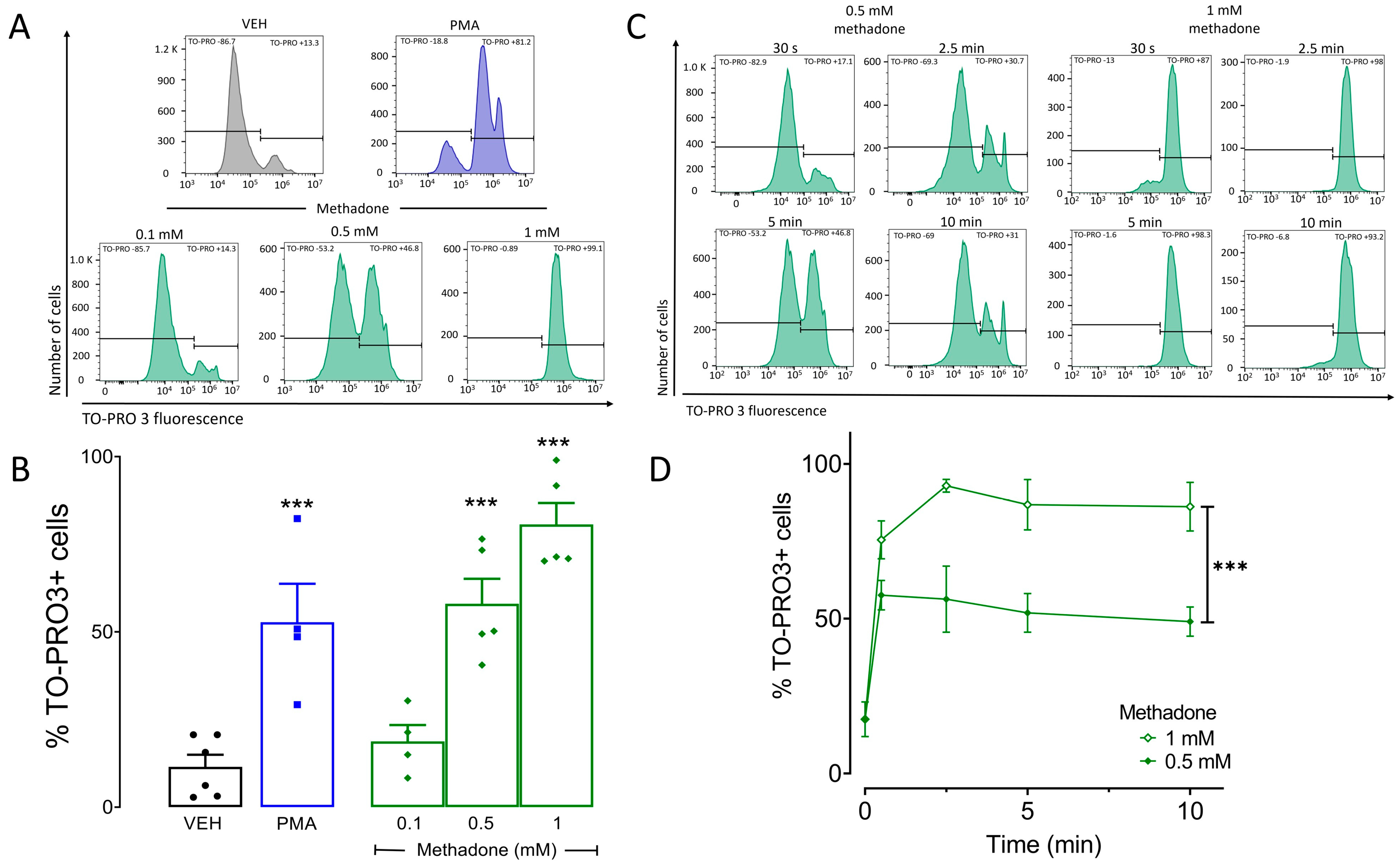
2.1. Methadone Induces Cell Death in BMMCs in A Concentration-Dependent Manner
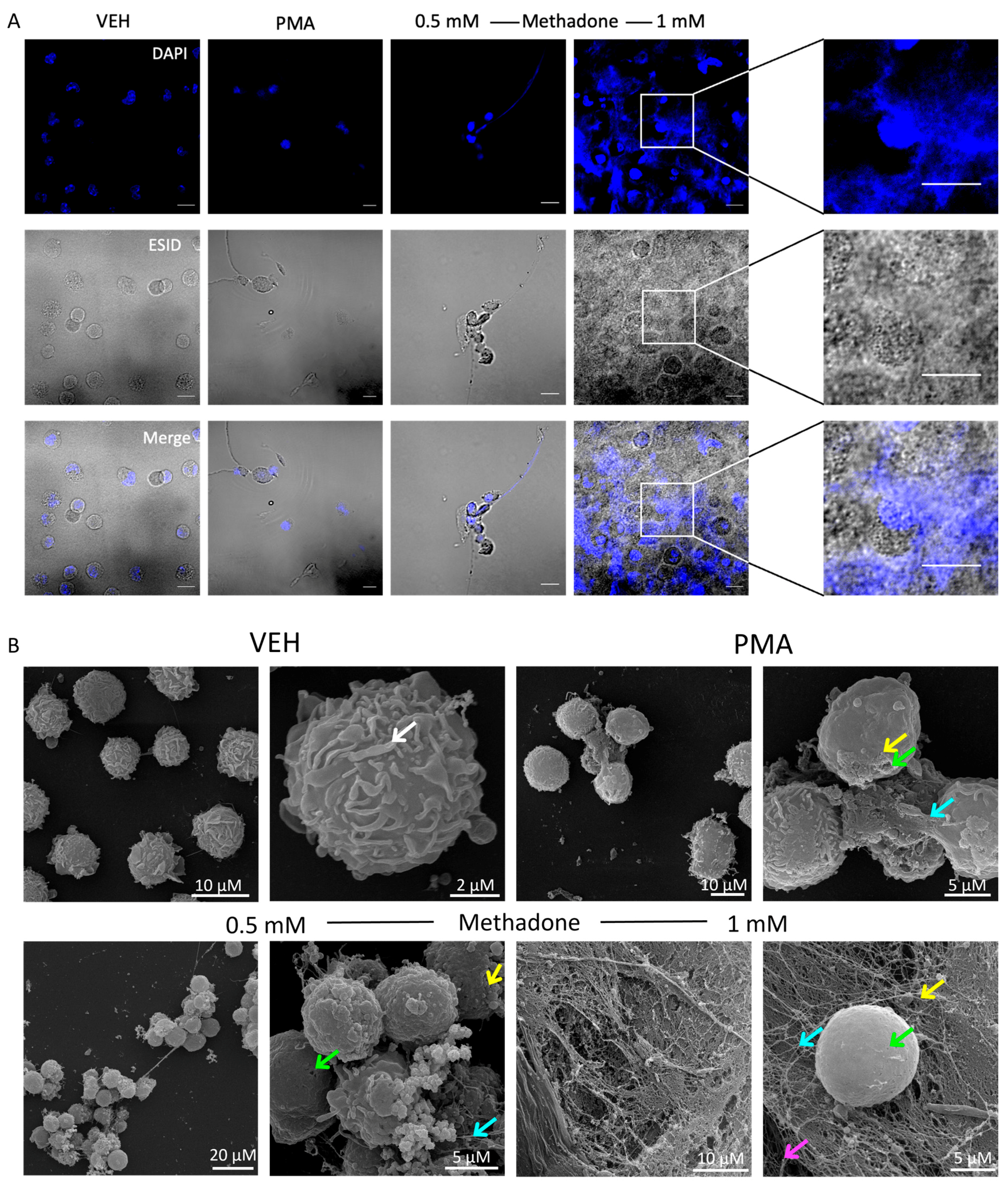
2.2. Methadone Induces Cell Death through the Formation of DNA Extracellular Trap (MCETosis) in BMMCs
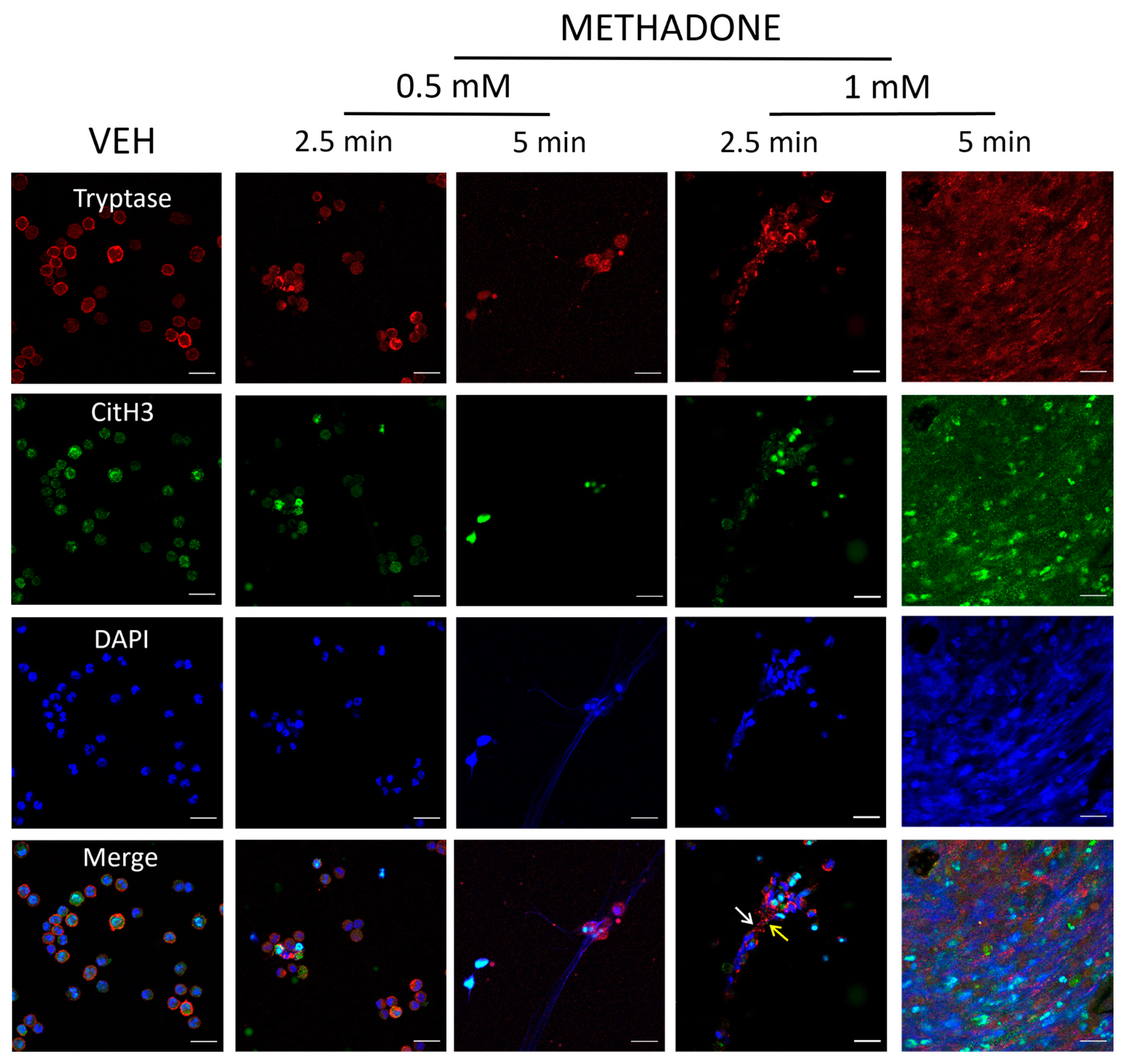
2.3. Methadone-Induced MCETs Contain CitH3 and Tryptase
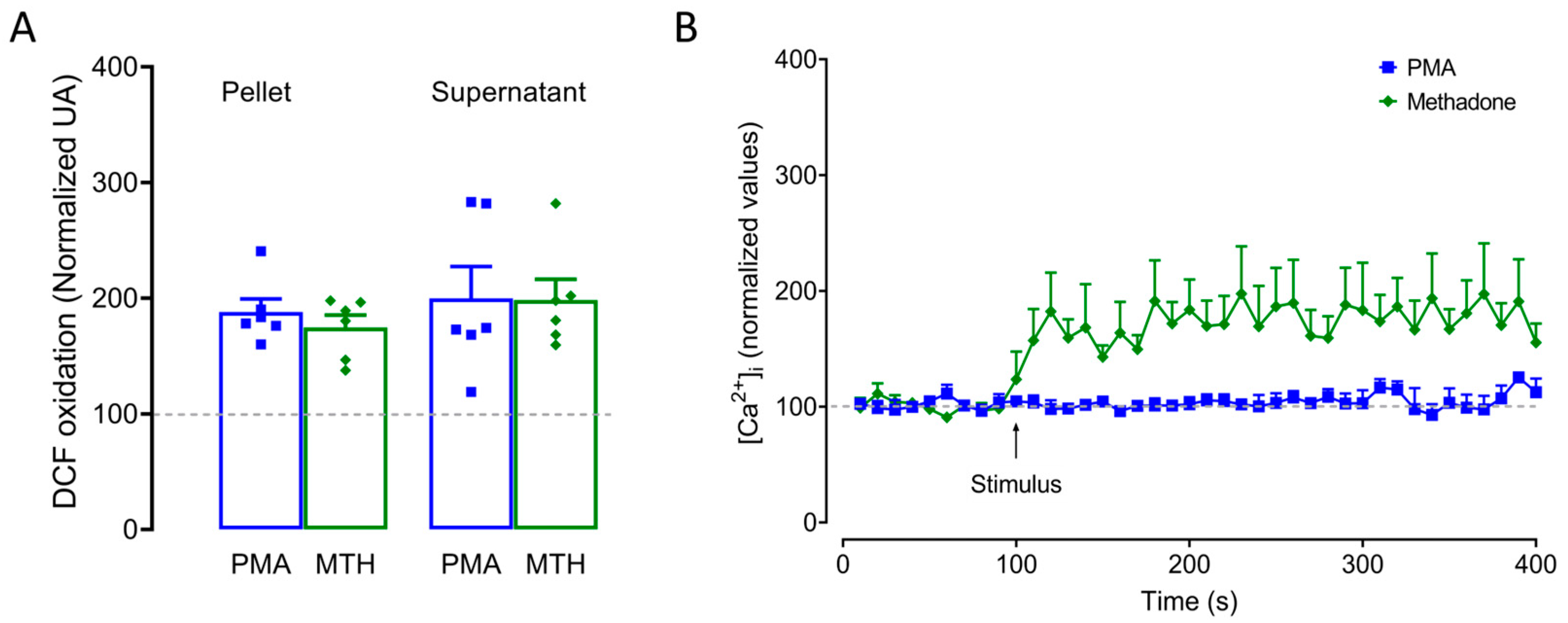
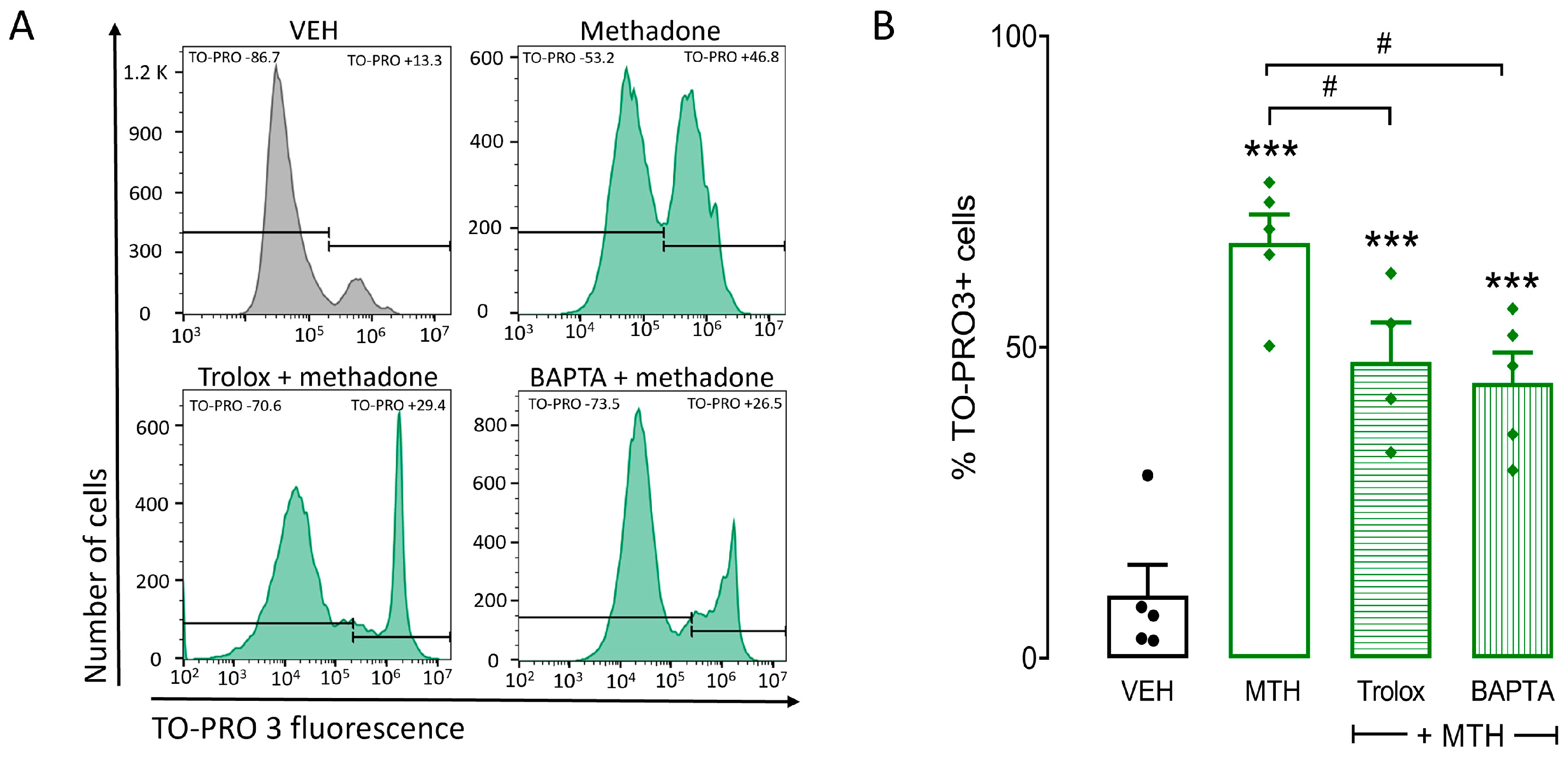
2.4. ROS Production and Intracellular Ca2+ Increase Are Critical for Methadone-Induced Cell Death in BMMCs
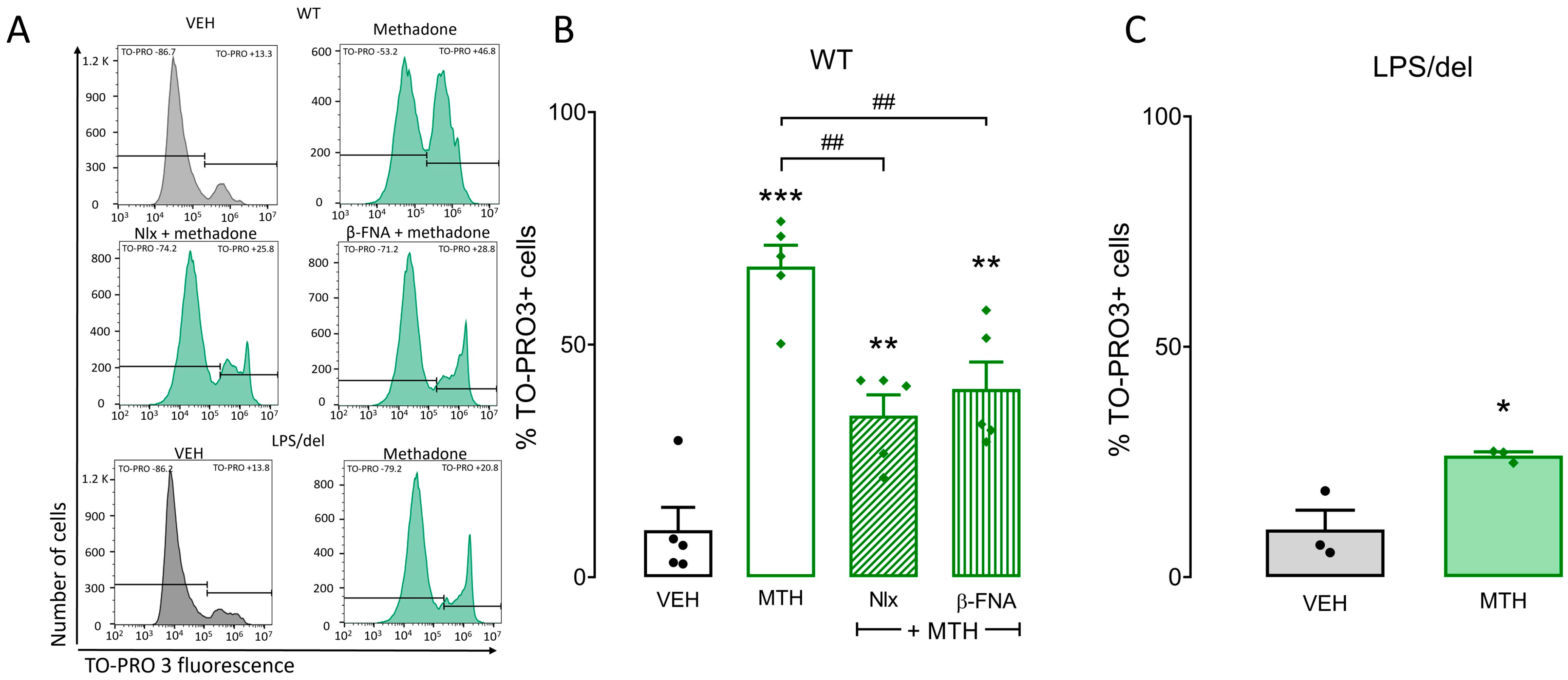
2.5. μ-Opioid and TLR4 Receptors Participate in Methadone-Induced Cell Death in BMMCs
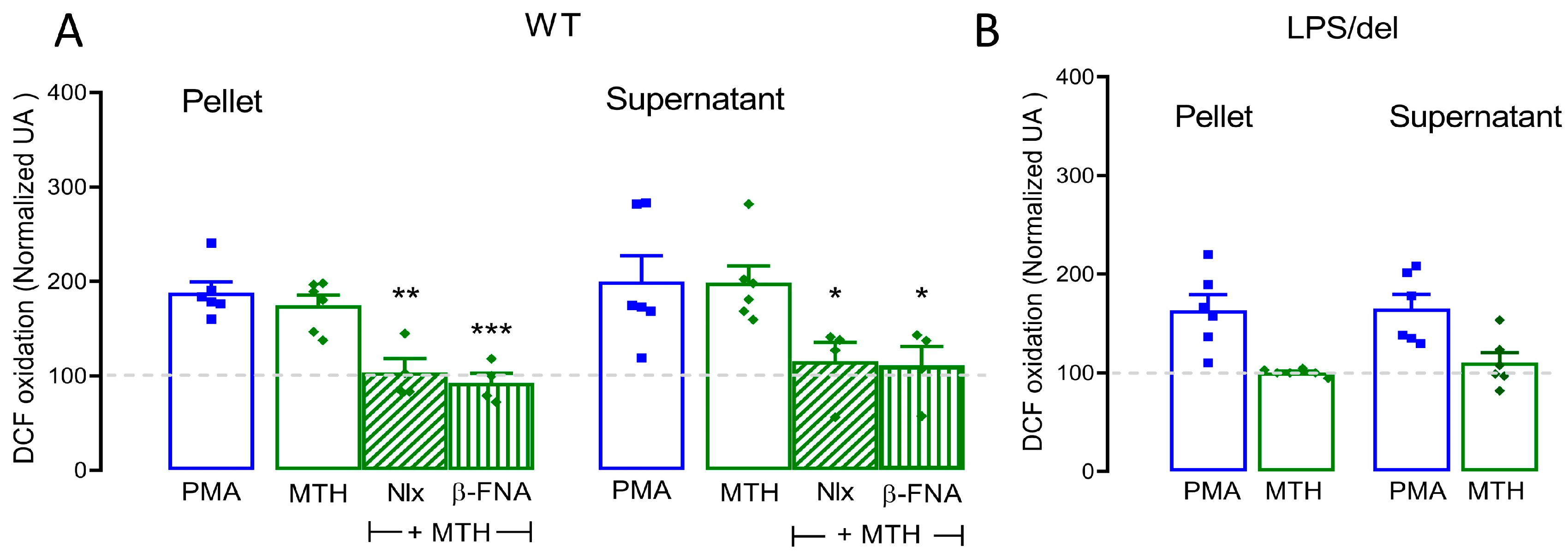
2.6. µ-Opioid Receptors Require TLR4 Receptors to Mediate Methadone-Induced ROS Production
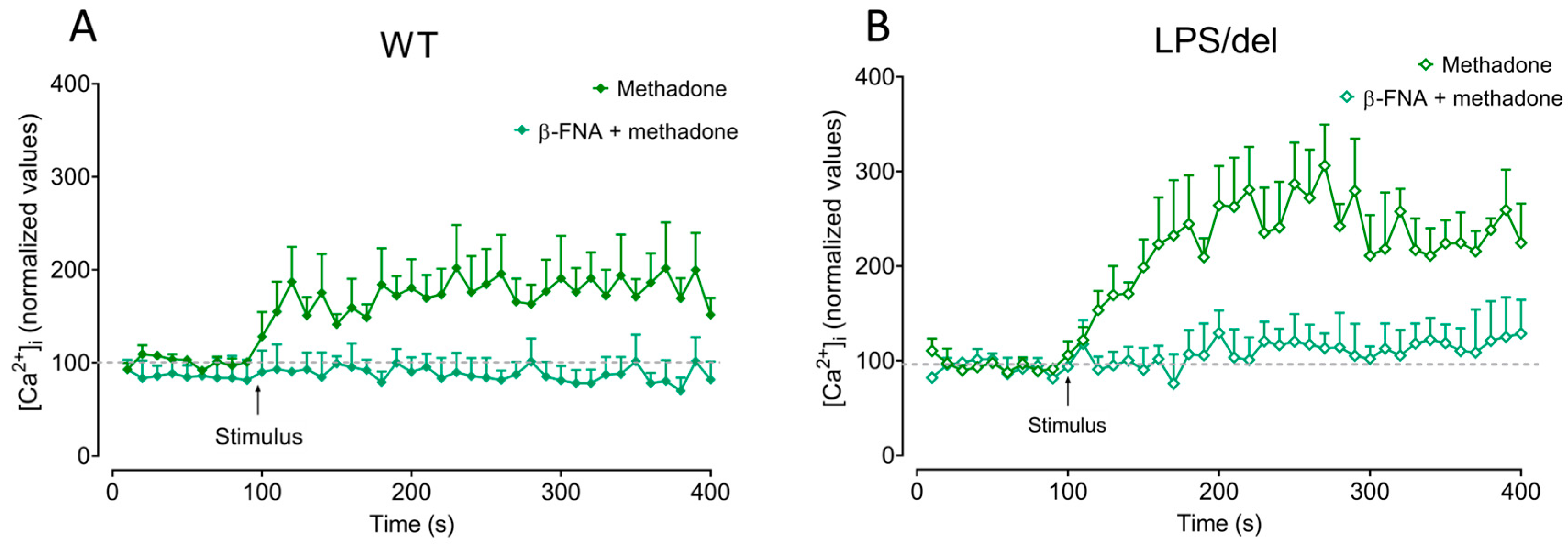
2.7. µ-Opioid Receptors Induce Calcium Mobilization in BMMCs
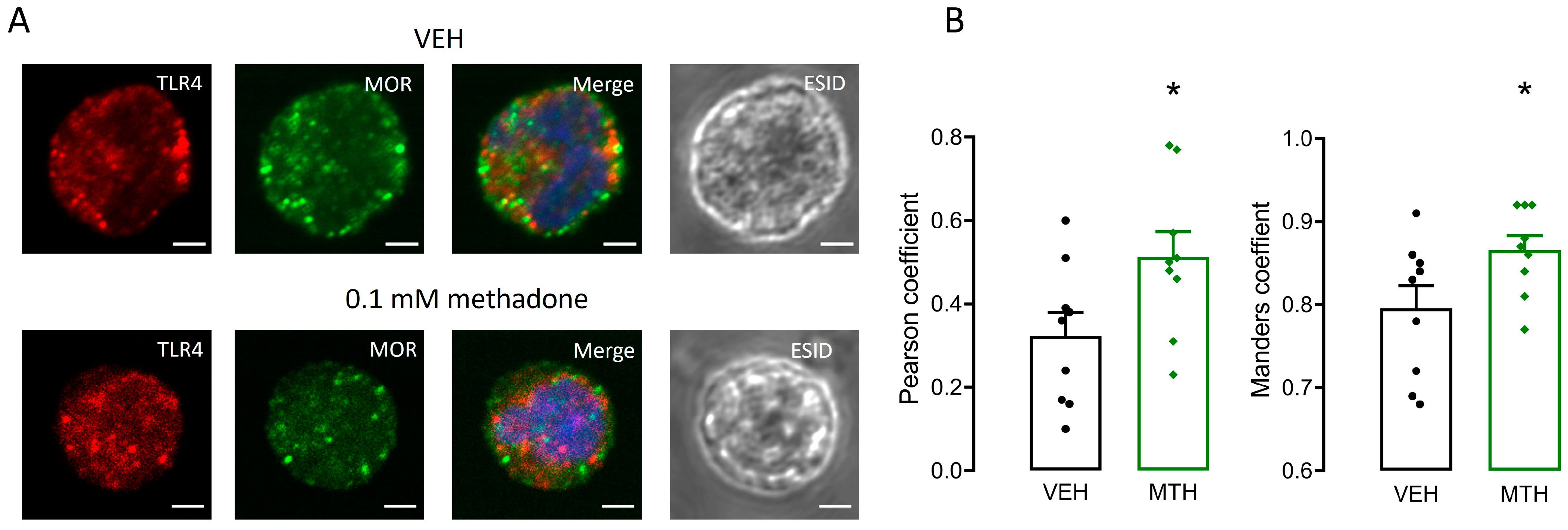
2.8. µ-Opioid and TLR4 Receptors Share Intracellular Localization in BMMCs
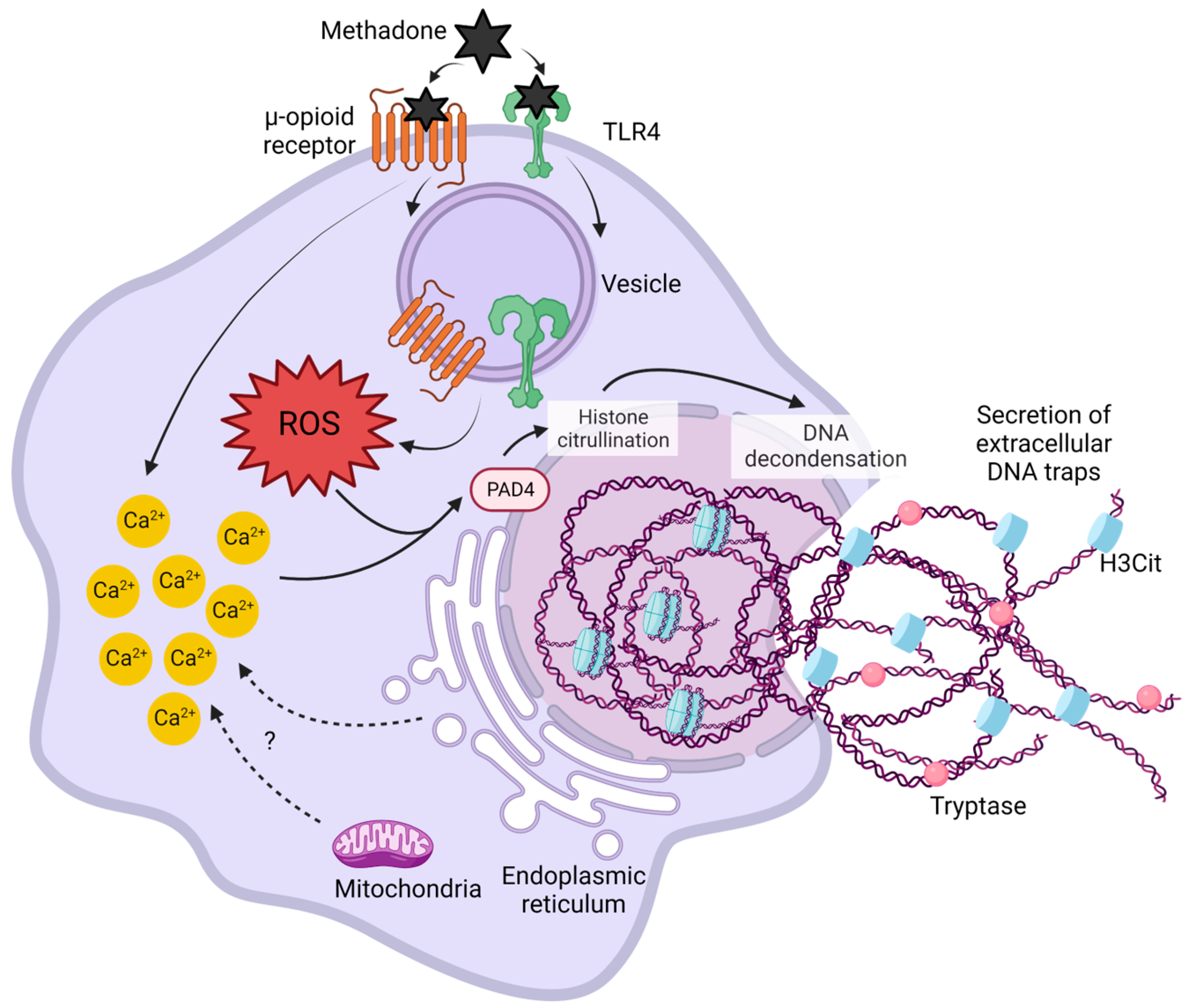
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs, Reagents, and Antibodies
4.3. BMMC Cultures
4.4. Viability Test
4.5. Immunofluorescence
4.6. Scanning Electron Microscopy
4.7. Determination of ROS Production
4.8. Determination of Intracellular Calcium
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volkow, N.; Benveniste, H.; Mclellan, A.T. Annual Review of Medicine Use and Misuse of Opioids in Chronic Pain. Annu. Rev. Med. 2017, 69, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Fredheim, O.M.S.; Moksnes, K.; Borchgrevink, P.C.; Kaasa, S.; Dale, O. Clinical pharmacology of methadone for pain. Acta Anaesthesiol. Scand. 2008, 52, 879–889. [Google Scholar] [CrossRef]
- Ma, J.; Bao, Y.-P.; Wang, R.-J.; Su, M.-F.; Liu, M.-X.; Li, J.-Q.; Degenhardt, L.; Farrell, M.; Blow, F.C.; Ilgen, M.; et al. Effects of medication-assisted treatment on mortality among opioids users: A systematic review and meta-analysis. Mol. Psychiatry 2019, 24, 1868–1883. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Ma, P.; Lu, H.; Liu, S.; Cao, Q. Fentanyl Suppresses the Survival of CD4+ T Cells Isolated from Human Umbilical Cord Blood through Inhibition of IKKs-mediated NF-κB Activation. Scand. J. Immunol. 2017, 85, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Mazahery, C.; Benson, B.L.; Cruz-Lebrón, A.; Levine, A.D. Chronic Methadone Use Alters the CD8+ T Cell Phenotype In Vivo and Modulates Its Responsiveness Ex Vivo to Opioid Receptor and TCR Stimuli. J. Immunol. 2020, 204, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Quang-Cantagrel, N.D.; Wallace, M.S.; Ashar, N.; Mathews, C. Long-term methadone treatment: Effect on CD4+ lymphocyte counts and HIV-1 plasma RNA level in patients with HIV infection. Eur. J. Pain 2001, 5, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Rojavin, M.; Szabo, I.; Bussiere, J.L.; Rogers, T.J.; Adler, M.W.; Eisenstein, T.K. Morphine treatment in vitro or in vivo decreases phagocytic functions of murine macrophages. Life Sci. 1993, 53, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Tian, S.; Guo, C.; Douglas, S.D.; Ho, W. Methadone Enhances Human Immunodeficiency Virus Infectionof Human Immune Cells. J. Infect. Dis. 2014, 1, 118–122. [Google Scholar] [CrossRef]
- Börner, C.; Lanciotti, S.; Koch, T.; Höllt, V.; Kraus, J. Mu opioid receptor agonist-selective regulation of interleukin-4 in T lymphocytes. J. Neuroimmunol. 2013, 263, 35–42. [Google Scholar] [CrossRef]
- Kafami, L.; Etesami, I.; Felfeli, M.; Enayati, N.; Ghiaghi, R.; Aminian, A.; Dehpour, A. Methadone diminishes neuroinflammation and disease severity in EAE through modulating T cell function. J. Neuroimmunol. 2013, 255, 39–44. [Google Scholar] [CrossRef]
- Klimas, N.G.; Blaney, N.T.; Morgan, R.O.; Chitwood, D.; Milles, K.; Lee, H.; Fletcher, M. Immune Function and Anti-HTLV-I/II Status in Anti-HIV-l-Negative Intravenous Drug Users Receiving Methadone. Am. J. Med. 1991, 90, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Carranza-Aguilar, C.J.; Hernández-Mendoza, A.; Mejias-Aponte, C.; Rice, K.C.; Morales, M.; González-Espinosa, C.; Cruz, S.L. Morphine and Fentanyl Repeated Administration Induces Different Levels of NLRP3-Dependent Pyroptosis in the Dorsal Raphe Nucleus of Male Rats via Cell-Specific Activation of TLR4 and Opioid Receptors. Cell. Mol. Neurobiol. 2022, 42, 677–694. [Google Scholar] [CrossRef]
- Lee, J.; Rosales, J.L.; Byun, H.-G.; Lee, K.-Y. d,l-Methadone causes leukemic cell apoptosis via an OPRM1-triggered increase in IP3R-mediated ER Ca2+ release and decrease in Ca2+ efflux, elevating [Ca2+]i. Sci. Rep. 2021, 11, 1009. [Google Scholar] [CrossRef]
- Friesen, C.; Bacher, S.; Hormann, I.; Roscher, M.; Miltner, E. Cytotoxic effects of opioids on cancer cell lines. Int. J. Clin. Pharmacol. Ther. 2011, 49, 60–62. [Google Scholar]
- Michalska, M.; Schultze-Seemann, S.; Kuckuck, I.; Katzenwadel, A.; Wolf, P. Impact of methadone on cisplatin treatment of bladder cancer cells. Anticancer. Res. 2018, 38, 1369–1375. [Google Scholar] [CrossRef]
- Theile, D.; Mikus, G. Methadone against cancer: Lost in translation. Int. J. Cancer 2018, 143, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of μ-opioid receptors: Desensitization, Phosphorylation, Internalization, and Tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.R.; Zhang, Y.; Shridhar, M.; Evans, J.H.; Buchanan, M.M.; Zhao, T.X.; Slivka, P.F.; Coats, B.D.; Rezvani, N.; Wieseler, J.; et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav. Immun. 2010, 24, 83–95. [Google Scholar] [CrossRef]
- John, A.L.S.; Abraham, S.N. Innate Immunity and Its Regulation by Mast Cells. J. Immunol. 2013, 190, 4458–4463. [Google Scholar] [CrossRef]
- Galli, S.J.; Gaudenzio, N.; Tsai, M. Mast Cells in Inflammation and Disease: Recent Progress and Ongoing Concerns. Annu. Rev. Immunol. 2020, 38, 49–77. [Google Scholar] [CrossRef]
- Möllerherm, H.; von Köckritz-Blickwede, M.; Branitzki-Heinemann, K. Antimicrobial activity of mast cells: Role and relevance of extracellular DNA traps. Front. Immunol. 2016, 7, 265. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; de Boer, O.J.; Mackaaij, C.; Pabittei, D.R.; de Winter, R.J.; Li, X.; van der Wal, A.C. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J. Pathol. 2019, 247, 505–512. [Google Scholar] [CrossRef]
- Goldmann, O.; Medina, E. The expanding world of extracellular traps: Not only neutrophils but much more. Front. Immunol. 2012, 3, 420. [Google Scholar] [CrossRef] [PubMed]
- Madera-Salcedo, I.K.; Cruz, S.L.; Gonzalez-Espinosa, C. Morphine Prevents Lipopolysaccharide-Induced TNF Secretion in Mast Cells Blocking I B Kinase Activation and SNAP-23 Phosphorylation: Correlation with the Formation of a β-Arrestin/TRAF6 Complex. J. Immunol. 2013, 191, 3400–3409. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Riquer, Z.P.; Segura-Villalobos, D.; Ramírez-Moreno, I.G.; Rodríguez, M.J.P.; Lamas, M.; Gonzalez-Espinosa, C. Signal Transduction Pathways Activated by Innate Immunity in Mast Cells: Translating Sensing of Changes into Specific Responses. Cells 2020, 9, 2411. [Google Scholar] [CrossRef] [PubMed]
- Perez-Alvarez, S.; Cuenca-Lopez, M.D.; de Mera, R.M.M.-F.; Puerta, E.; Karachitos, A.; Bednarczyk, P.; Kmita, H.; Aguirre, N.; Galindo, M.F.; Jordán, J. Methadone induces necrotic-like cell death in SH-SY5Y cells by an impairment of mitochondrial ATP synthesis. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2010, 1802, 1036–1047. [Google Scholar] [CrossRef]
- Li, M.-C.; Yu, J.-H.; Yu, S.-S.; Chi, Y.-Y.; Xiang, Y.-B. MicroRNA-873 Inhibits Morphine-Induced Macrophage Apoptosis by Elevating A20 Expression. Pain Med. 2015, 16, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Navarro, M.; Leyva-Paredes, K.; Donis-Maturano, L.; Rodríguez-López, G.M.; Soria-Castro, R.; García-Pérez, B.E.; Puebla-Osorio, N.; Ullrich, S.E.; Luna-Herrera, J.; Flores-Romo, L.; et al. Mycobacterium tuberculosis catalase inhibits the formation of mast cell extracellular traps. Front. Immunol. 2018, 9, 1161. [Google Scholar] [CrossRef] [PubMed]
- von KöCkritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Espinosa, C.; Madera-Salcedo, I.K.; Molina-Martínez, L.M.; Martínez-Cuevas, F.L. Opioids and the Immune System; Cruz, O.S., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 249–285. [Google Scholar] [CrossRef]
- Franchi, S.; Moschetti, G.; Amodeo, G.; Sacerdote, P. Do All Opioid Drugs Share the Same Immunomodulatory Properties? A Review From Animal and Human Studies. Front. Immunol. 2019, 10, 2914. [Google Scholar] [CrossRef]
- Gabr, M.M.; Saeed, I.; Miles, J.A.; Ross, B.P.; Shaw, P.N.; Hollmann, M.W.; Parat, M.-O. Interaction of opioids with tlr4—Mechanisms and ramifications. Cancers 2021, 13, 5274. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.R.; Hutchinson, M.R.; Rice, K.C.; Maier, S.F. The “Toll” of Opioid-Induced Glial Activation: Improving the Clinical Efficacy of Opioids by Targeting Glia. Trends Pharmacol. Sci. 2009, 30, 581–591. [Google Scholar] [CrossRef]
- Friesen, C.; Roscher, M.; Hormann, I.; Fichtner, I.; Alt, A.; Hilger, R.A.; Debatin, K.-M.; Miltner, E. Cell death sensitization of leukemia cells by opioid receptor activation. Oncotarget 2013, 4, 677–690. [Google Scholar] [CrossRef]
- Friesen, C.; Roscher, M.; Alt, A.; Miltner, E. Methadone, commonly used as maintenance medication for outpatient treatment of opioid dependence, kills leukemia cells and overcomes chemoresistance. Cancer Res 2008, 68, 6059–6064. [Google Scholar] [CrossRef]
- Singh, A.; Jayanthan, A.; Farran, A.; Elwi, A.N.; Kim, S.-W.; Farran, P.; Narendran, A. Induction of apoptosis in pediatric acute lymphoblastic leukemia (ALL) cells by the therapeutic opioid methadone and effective synergy with Bcl-2 inhibition. Leuk. Res. 2011, 35, 1649–1657. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Lokensgard, J.R.; Peterson, P.K. Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 2002, 42, 829–836. [Google Scholar] [CrossRef]
- McCoy, M.T.; Jayanthi, S.; Cadet, J.L. Potassium channels and their potential roles in substance use disorders. Int. J. Mol. Sci. 2021, 22, 1249. [Google Scholar] [CrossRef]
- Zünkler, B.J.; Wos-Maganga, M. Comparison of the effects of methadone and heroin on human ether-à-go-go-related gene channels. Cardiovasc. Toxicol. 2010, 10, 161–165. [Google Scholar] [CrossRef]
- Eap, C.B.; Crettol, S.; Rougier, J.-S.; Schläpfer, J.; Grilo, L.S.; Déglon, J.-J.; Besson, J.; Croquette-Krokar, M.; Carrupt, P.-A.; Abriel, H. Stereoselective block of hERG channel by (S)-methadone and QT interval prolongation in CYP2B6 slow metabolizers. Clin. Pharmacol. Ther. 2007, 81, 719–728. [Google Scholar] [CrossRef]
- Pourhassanali, N.; Zarbakhsh, S.; Miladi-Gorji, H. Morphine dependence and withdrawal-induced changes in mouse Sertoli cell (TM4) line: Evaluation of apoptotic, inflammatory and oxidative stress biomarkers. Reprod. Toxicol. 2021, 105, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Zamani, N.; Osgoei, L.T.; Aliaghaei, A.; Zamani, N.; Hassanian-Moghaddam, H. Chronic exposure to methadone induces activated microglia and astrocyte and cell death in the cerebellum of adult male rats. Metab. Brain Dis. 2023, 38, 323–338. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Yeoh, B.S.; Xiao, X.; Golonka, R.M.; Singh, V.; Wang, Y.; Vijay-Kumar, M. PAD4-dependent NETs generation are indispensable for intestinal clearance of Citrobacter rodentium. Mucosal Immunol. 2019, 12, 761–771. [Google Scholar] [CrossRef]
- Hidalgo, A.; Libby, P.; Soehnlein, O.; Aramburu, I.V.; Papayannopoulos, V.; Silvestre-Roig, C. Neutrophil extracellular traps: From physiology to pathology. Cardiovasc. Res. 2022, 118, 2737–2753. [Google Scholar] [CrossRef]
- Chen, T.; Li, Y.; Sun, R.; Hu, H.; Liu, Y.; Herrmann, M.; Zhao, Y.; Muñoz, L.E. Receptor-Mediated NETosis on Neutrophils. Front. Immunol. 2021, 12, 775267. [Google Scholar] [CrossRef]
- Agrawal, I.; Sharma, N.; Saxena, S.; Arvind, S.; Chakraborty, D.; Chakraborty, D.B.; Jha, D.; Ghatak, S.; Epari, S.; Gupta, T.; et al. Dopamine induces functional extracellular traps in microglia. iScience 2021, 24, 101968. [Google Scholar] [CrossRef]
- Celik, M.; Labuz, D.; Henning, K.; Busch-Dienstfertig, M.; Gaveriaux-Ruff, C.; Kieffer, B.L.; Zimmer, A.; Machelska, H. Leukocyte opioid receptors mediate analgesia via Ca2+-regulated release of opioid peptides. Brain Behav. Immun. 2016, 57, 227–242. [Google Scholar] [CrossRef]
- Petry, A.; Görlach, A. Regulation of NADPH Oxidases by G Protein-Coupled Receptors. Antioxid. Redox Signal. 2019, 30, 74–94. [Google Scholar] [CrossRef] [PubMed]
- Gavériaux-Ruff, C.; Matthes, H.W.D.; Peluso, J.; Kieffer, B.L. Abolition of morphine-immunosuppression in mice lacking the μ-opioid receptor gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6326–6330. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Nagase, H.; Okuzumi, A.; Nishiyama, C. Delta Opioid Receptor Agonists Ameliorate Colonic Inflammation by Modulating Immune Responses. Front. Immunol. 2021, 12, 730706. [Google Scholar] [CrossRef]
- Wang, X.; Loram, L.C.; Ramos, K.; de Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R.; et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Shavit, Y.; Grace, P.M.; Rice, K.C.; Maier, S.F.; Watkins, L.R. Exploring the neuroimmunopharmacology of opioids: An integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol. Rev. 2011, 63, 772–810. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.R.; Coats, B.D.; Lewis, S.S.; Zhang, Y.; Sprunger, D.B.; Rezvani, N.; Baker, E.M.; Jekich, B.M.; Wieseler, J.L.; Somogyi, A.A.; et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav. Immun. 2008, 22, 1178–1189. [Google Scholar] [CrossRef]
- Stevens, C.; Aravind, S.; Das, S.; Davis, R. Pharmacological characterization of LPS and opioid interactions at the toll-like receptor 4. Br. J. Pharmacol. 2013, 168, 1421–1429. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, M.; Chen, C.; Liu, L.; Wei, X.; Zeng, S. Toll-Like Receptor 4 (TLR4)/Opioid Receptor Pathway Crosstalk and Impact on Opioid Analgesia, Immune Function, and Gastrointestinal Motility. Front. Immunol. 2020, 11, 1455. [Google Scholar] [CrossRef]
- Cuitavi, J.; Torres-Pérez, J.V.; Lorente, J.D.; Campos-Jurado, Y.; Andrés-Herrera, P.; Polache, A.; Agustín-Pavón, C.; Hipólito, L. Crosstalk between Mu-Opioid receptors and neuroinflammation: Consequences for drug addiction and pain. Neurosci. Biobehav. Rev. 2023, 145, 105011. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Nhu, Q.M.; Toshchakov, V.Y.; Piao, W.; Medvedev, A.E.; Hollenberg, M.D.; Fasano, A.; Vogel, S.N. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: A novel paradigm for receptor cooperativity. J. Biol. Chem. 2008, 283, 24314–24325. [Google Scholar] [CrossRef]
- Bucci, M.; Vellecco, V.; Harrington, L.; Brancaleone, V.; Roviezzo, F.; Raso, G.M.; Ianaro, A.; Lungarella, G.; De Palma, R.; Meli, R.; et al. Crosstalk between toll-like receptor 4 (TLR4) and proteinase-activated receptor 2 (PAR2) is involved in vascular function. Br. J. Pharmacol. 2013, 168, 411–420. [Google Scholar] [CrossRef]
- du Sert, N.P.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Perez-Alvarez, S.; Iglesias-Guimarais, V.; Solesio, M.E.; de Mera, R.M.M.-F.; Yuste, V.J.; Galindo, M.F.; Jordán, J. Methadone induces CAD degradation and AIF-mediated necrotic-like cell death in neuroblastoma cells. Pharmacol. Res. 2011, 63, 352–360. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cuevas, F.L.; Cruz, S.L.; González-Espinosa, C. Methadone Requires the Co-Activation of μ-Opioid and Toll-Like-4 Receptors to Produce Extracellular DNA Traps in Bone-Marrow-Derived Mast Cells. Int. J. Mol. Sci. 2024, 25, 2137. https://doi.org/10.3390/ijms25042137
Martínez-Cuevas FL, Cruz SL, González-Espinosa C. Methadone Requires the Co-Activation of μ-Opioid and Toll-Like-4 Receptors to Produce Extracellular DNA Traps in Bone-Marrow-Derived Mast Cells. International Journal of Molecular Sciences. 2024; 25(4):2137. https://doi.org/10.3390/ijms25042137
Chicago/Turabian StyleMartínez-Cuevas, Frida L., Silvia L. Cruz, and Claudia González-Espinosa. 2024. "Methadone Requires the Co-Activation of μ-Opioid and Toll-Like-4 Receptors to Produce Extracellular DNA Traps in Bone-Marrow-Derived Mast Cells" International Journal of Molecular Sciences 25, no. 4: 2137. https://doi.org/10.3390/ijms25042137
APA StyleMartínez-Cuevas, F. L., Cruz, S. L., & González-Espinosa, C. (2024). Methadone Requires the Co-Activation of μ-Opioid and Toll-Like-4 Receptors to Produce Extracellular DNA Traps in Bone-Marrow-Derived Mast Cells. International Journal of Molecular Sciences, 25(4), 2137. https://doi.org/10.3390/ijms25042137