
Comorbidity Genes of Alzheimer’s Disease and Type 2 Diabetes Associated with Memory and Cognitive Function


Abstract
:1. Introduction
2. Biological Mechanisms of Alzheimer’s Disease and Type 2 Diabetes
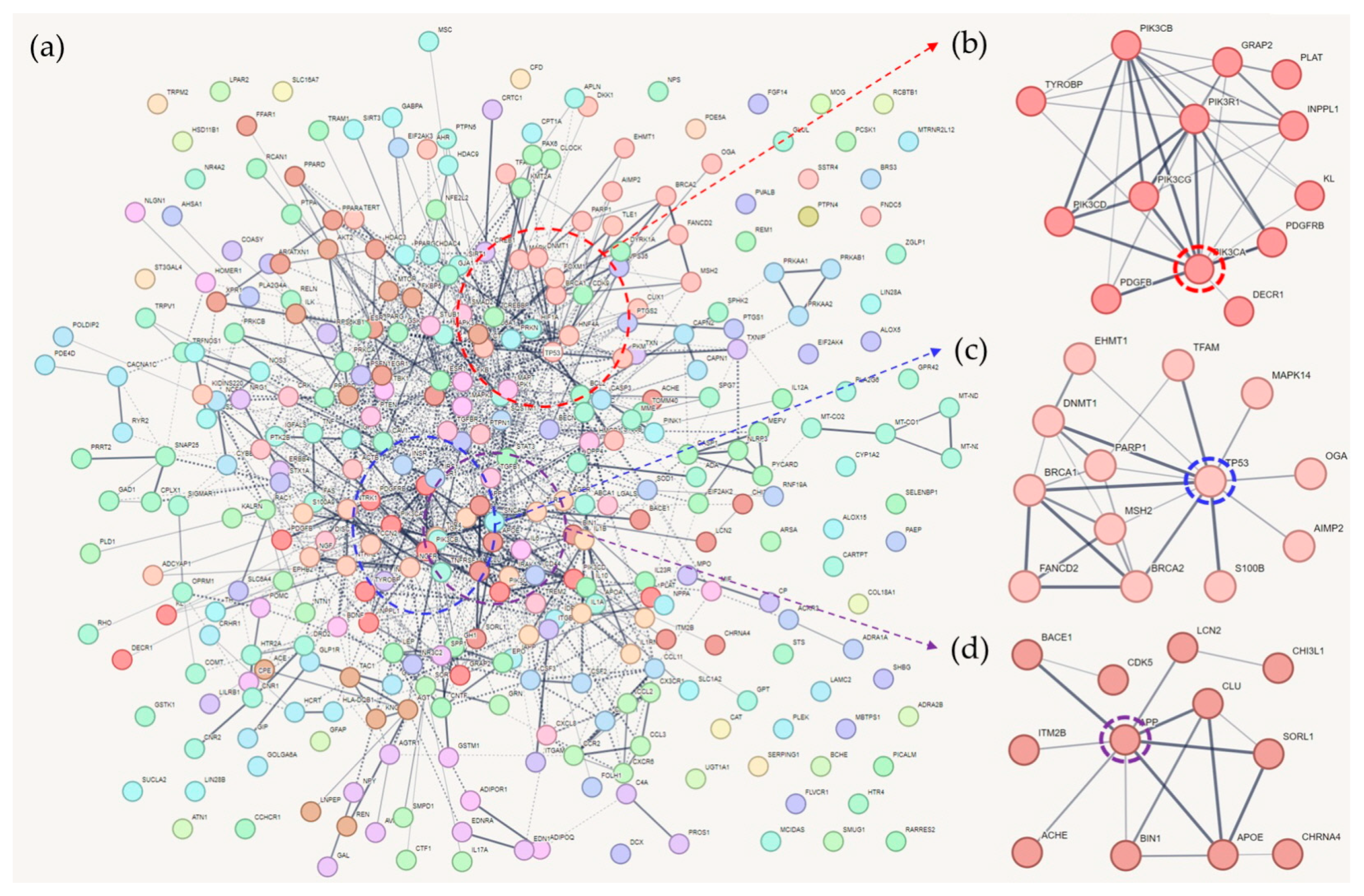
3. Gene Clusters of Common Genes That Are Associated with AD, T2DM, and Memory Function
3.1. Cluster 1 (CL1)
3.2. Cluster 2 (CL2)
3.3. Cluster 3 (CL3)
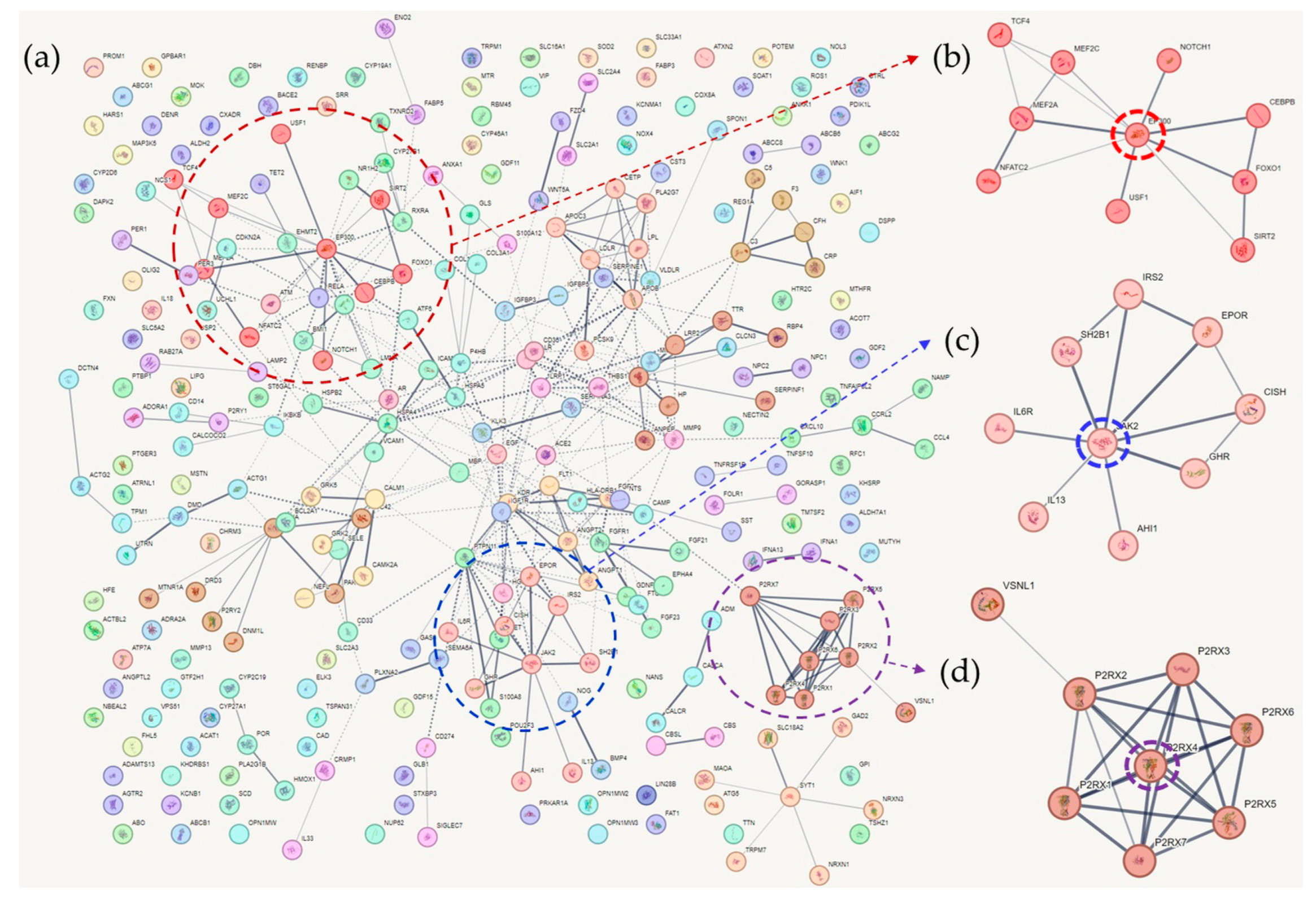
4. Clusters of Cognitive Function-Associated Genes
4.1. Cluster 1
4.2. Cluster 2
4.3. Cluster 3
5. Discussion
6. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 2014, 5, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kodamullil, A.T.; Hofmann-Apitius, M. Comorbidity Analysis between Alzheimer’s Disease and Type 2 Diabetes Mellitus (T2DM) Based on Shared Pathways and the Role of T2DM Drugs. J. Alzheimer’s Dis. 2017, 60, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Diogo, D.; Tian, C.; Franklin, C.S.; Alanne-Kinnunen, M.; March, M.; Spencer, C.C.A.; Vangjeli, C.; Weale, M.E.; Mattsson, H.; Kilpeläinen, E.; et al. Phenome-wide association studies across large population cohorts support drug target validation. Nat. Commun. 2018, 9, 4285. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Curtis, D.; Bandyopadhyay, S. Mini-review: Role of the PI3K/Akt pathway and tyrosine phosphatases in Alzheimer’s disease susceptibility. Ann. Hum. Genet. 2020, 85, 1–6. [Google Scholar] [CrossRef]
- Maffei, A.; Lembo, G.; Carnevale, D. PI3Kinases in Diabetes Mellitus and Its Related Complications. Int. J. Mol. Sci. 2018, 19, 4098. [Google Scholar] [CrossRef]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef]
- Giese, K.P.; Mizuno, K. The roles of protein kinases in learning and memory. Learn. Mem. 2013, 20, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Marin, P.; Yan, M.; Li, L.; Mao, B.; Li, H.; Li, S.Y.T.; Mruk, D.; Silvestrini, B.; Lian, Q.; et al. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef] [PubMed]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Xu, H.; Jia, J. Immune-Related Hub Genes and the Competitive Endogenous RNA Network in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1255–1265. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.; Liu, J.; Song, J.; Zhang, S.; Chen, L.; Zhang, M. 20 S-Protopanaxatriol improves cognitive function of Alzheimer’s disease by promoting endogenous neurogenesis. Food Funct. 2023, 14, 4191–4203. [Google Scholar] [CrossRef]
- Du, Y.; Guo, J.; Zhou, Y.; Yan, S.; Xu, B.; Wang, Y.; Lu, D.; Ma, Z.; Chen, Q.; Tang, Q.; et al. Revealing the Mechanisms of Byu dMar 25 in the Treatment of Alzheimer’s Disease through Network Pharmacology, Molecular Docking, and In Vivo Experiment. ACS Omega 2023, 8, 25066–25080. [Google Scholar] [CrossRef]
- Owolabi, B.O.; Ojo, O.O.; Srinivasan, D.K.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H.A. Glucoregulatory, endocrine and morphological effects of [P5K]hymenochirin-1B in mice with diet-induced glucose intolerance and insulin resistance. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016, 389, 769–781. [Google Scholar] [CrossRef]
- Musale, V.; Moffett, R.C.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Beneficial actions of the [A14K] analog of the frog skin peptide PGLa-AM1 in mice with obesity and degenerative diabetes: A mechanistic study. Peptides 2020, 136, 170472. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, C.; Dechartres, A.; Mariot, V.; Lotton, C.; Trainor, C.; Del Giudice, E.M.; Meyre, D.; Bieche, I.; Laurendeau, I.; Froguel, P.; et al. Association Analysis Indicates That a Variant GATA-Binding Site in the PIK3CB Promoter Is a Cis-Acting Expression Quantitative Trait Locus for This Gene and Attenuates Insulin Resistance in Obese Children. Diabetes 2008, 57, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Le Stunff, C.; Meirhaeghe, A.; Dechartres, A.; Ferrieres, J.; Basdevant, A.; Boitard, C.; Amouyel, P.; Bougnères, P. In obese and non-obese adults, the cis-regulatory rs361072 promoter variant of PIK3CB is associated with insulin resistance not with type 2 diabetes. Mol. Genet. Metab. 2009, 96, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Ribel-Madsen, R.; Poulsen, P.; Holmkvist, J.; Mortensen, B.; Grarup, N.; Friedrichsen, M.; Jørgensen, T.; Lauritzen, T.; Wojtaszewski, J.F.; Pedersen, O.; et al. Impact of rs361072 in the Phosphoinositide 3-Kinase p110β Gene on Whole-Body Glucose Metabolism and Subunit Protein Expression in Skeletal Muscle. Diabetes 2010, 59, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bai, J.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Downregulation of PIK3CB Involved in Alzheimer’s Disease via Apoptosis, Axon Guidance, and FoxO Signaling Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 1260161. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Shu, H.; Ye, Q.; Wang, Z.; Xie, C.; Yuan, B.; Zhang, Z.; Bai, F. Brain insulin resistance deteriorates cognition by altering the topological features of brain networks. NeuroImage: Clin. 2017, 13, 280–287. [Google Scholar] [CrossRef]
- Wójcik, M.; Mac-Marcjanek, K.; Woźniak, L.A.; Nadel, I.; Lewiński, A.; Cypryk, K. Związek podwyższonej ekspresji leukocytarnej kinazy 3-fosfatydyloinozytolu delta z cukrzycą ciążową (GDM). Endokrynol. Polska 2014, 65, 17–24. [Google Scholar] [CrossRef]
- Ma, R.; Deng, X.-L.; Aleteng, Q.-Q.; Li, L.; Zhu, J. Genome-Wide Transcriptome Analysis in Type 2 Diabetes Patients Treated by Sitagliptin. Diabetes, Metab. Syndr. Obesity: Targets Ther. 2022, 15, 1761–1770. [Google Scholar] [CrossRef]
- Malodobra, M.; Pilecka, A.; Gworys, B.; Adamiec, R. Single nucleotide polymorphisms within functional regions of genes implicated in insulin action and association with the insulin resistant phenotype. Mol. Cell. Biochem. 2010, 349, 187–193. [Google Scholar] [CrossRef]
- Kaneko, K.; Ueki, K.; Takahashi, N.; Hashimoto, S.; Okamoto, M.; Awazawa, M.; Okazaki, Y.; Ohsugi, M.; Inabe, K.; Umehara, T.; et al. Class IA Phosphatidylinositol 3-Kinase in Pancreatic β Cells Controls Insulin Secretion by Multiple Mechanisms. Cell Metab. 2010, 12, 619–632. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, B.; Li, J.; Li, Y.; Zhang, M.; Ban, B. SHORT syndrome in two Chinese girls: A case report and review of the literature. Mol. Genet. Genom. Med. 2020, 8, e1385. [Google Scholar] [CrossRef]
- Masunaga, Y.; Fujisawa, Y.; Muramatsu, M.; Ono, H.; Inoue, T.; Fukami, M.; Kagami, M.; Saitsu, H.; Ogata, T. Insulin resistant diabetes mellitus in SHORT syndrome: Case report and literature review. Endocr. J. 2021, 68, 111–117. [Google Scholar] [CrossRef]
- Chung, B.; Gibson, W. Autosomal dominant PIK3R1 mutations cause SHORT syndrome. Clin. Genet. 2013, 85, 228–229. [Google Scholar] [CrossRef]
- Curtis, D.; Bakaya, K.; Sharma, L.; Bandyopadhyay, S. Weighted burden analysis of exome-sequenced late-onset Alzheimer’s cases and controls provides further evidence for a role for PSEN1 and suggests involvement of the PI3K/Akt/GSK-3β and WNT signalling pathways. Ann. Hum. Genet. 2020, 84, 291–302. [Google Scholar] [CrossRef]
- Qian, X.-H.; Liu, X.-L.; Chen, S.-D.; Tang, H.-D. Identification of Immune Hub Genes Associated with Braak Stages in Alzheimer’s Disease and Their Correlation of Immune Infiltration. Front. Aging Neurosci. 2022, 14, 887168. [Google Scholar] [CrossRef]
- Shen, S.; Wang, F.; Fernandez, A.; Hu, W. Role of platelet-derived growth factor in type II diabetes mellitus and its complications. Diabetes Vasc. Dis. Res. 2020, 17. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Alzheimer-associated neuronal thread protein mediated cell death is linked to impaired insulin signaling. J. Alzheimer’s Dis. 2004, 6, 231–242. [Google Scholar] [CrossRef]
- Yeboah, J.; Sane, D.C.; Crouse, J.R.; Herrington, D.M.; Bowden, D.W. Low Plasma Levels of FGF-2 and PDGF-BB Are Associated with Cardiovascular Events in Type II Diabetes Mellitus (Diabetes Heart Study). Dis. Markers 2007, 23, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Bessa, S.S.-E.; Hussein, T.A.; Morad, M.A.; Amer, A.M. Urinary Platelet-Derived Growth Factor-BB as an Early Marker of Nephropathy in Patients with Type 2 Diabetes: An Egyptian Study. Ren. Fail. 2012, 34, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Saffi, G.T.; Vasefi, M.S.; Choi, Y.; Kruk, J.S.; Ahmed, N.; Gondora, N.; Mielke, J.; Leonenko, Z.; Beazely, M.A. Amyloid-β Inhibits PDGFβ Receptor Activation and Prevents PDGF-BBInduced Neuroprotection. Curr. Alzheimer Res. 2018, 15, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Westenberger, A.; Sobrido, M.J.; García-Murias, M.; Domingo, A.; Sears, R.L.; Lemos, R.R.; Ordoñez-Ugalde, A.; Nicolas, G.; Cunha, J.E.G.d.; et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet. 2013, 45, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Arts, F.A.; Velghe, A.I.; Stevens, M.; Renauld, J.; Essaghir, A.; Demoulin, J. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J. Cell. Mol. Med. 2014, 19, 239–248. [Google Scholar] [CrossRef]
- Tsolaki, E.; Csincsik, L.; Xue, J.; Lengyel, I.; Bertazzo, S. Nuclear and cellular, micro and nano calcification in Alzheimer’s disease patients and correlation to phosphorylated Tau. Acta Biomater. 2022, 143, 138–144. [Google Scholar] [CrossRef]
- Abate, G.; Frisoni, G.B.; Bourdon, J.-C.; Piccirella, S.; Memo, M.; Uberti, D. The pleiotropic role of p53 in functional/dysfunctional neurons: Focus on pathogenesis and diagnosis of Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 160. [Google Scholar] [CrossRef]
- Buizza, L.; Cenini, G.; Lanni, C.; Ferrari-Toninelli, G.; Prandelli, C.; Govoni, S.; Buoso, E.; Racchi, M.; Barcikowska, M.; Styczynska, M.; et al. Conformational Altered p53 as an Early Marker of Oxidative Stress in Alzheimer’s Disease. PLoS ONE 2012, 7, e29789. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.; Martínez, C.; Coto, E.; Clarimón, J.; Lleó, A.; Illán-Gala, I.; Dols-Icardo, O.; Borroni, B.; Almeida, M.R.; van der Zee, J.; et al. Genetic variation in APOE, GRN, and TP53 are phenotype modifiers in frontotemporal dementia. Neurobiol. Aging 2020, 99, 99.e15–99.e22. [Google Scholar] [CrossRef]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer’s disease inverse relationship: An age-associated diverging derailment of shared pathways. Mol. Psychiatry 2020, 26, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, A.; Czapski, G.A.; Adamczyk, A.; Gajkowska, B.; Strosznajder, J.B. A novel mechanism of non-Aβ component of Alzheimer’s disease amyloid (NAC) neurotoxicity. Interplay between p53 protein and cyclin-dependent kinase 5 (Cdk5). Neurochem. Int. 2011, 58, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Amor-Gutiérrez, O.; Costa-Rama, E.; Arce-Varas, N.; Martínez-Rodríguez, C.; Novelli, A.; Fernández-Sánchez, M.T.; Costa-García, A. Competitive electrochemical immunosensor for the detection of unfolded p53 protein in blood as biomarker for Alzheimer’s disease. Anal. Chim. Acta 2019, 1093, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.-Q.; Luo, T.-T.; Luo, S.-C.; Wang, J.-Q.; Wang, S.-M.; Bai, Y.-H.; Yang, Y.-L.; Wang, Y.-Y. p53 and mitochondrial dysfunction: Novel insight of neurodegenerative diseases. J. Bioenerg. Biomembr. 2016, 48, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; He, B.; Pan, Y.; Xu, Y.; Zhu, C.; Tang, Z.; Bao, Q.; Tian, F.; Wang, S. Association between polymorphisms in RAPGEF1, TP53, NRF1 and type 2 diabetes in Chinese Han population. Diabetes Res. Clin. Pract. 2011, 91, 171–176. [Google Scholar] [CrossRef]
- Guo, D.; Fang, L.; Yu, X.; Wang, C.; Wang, Y.; Guo, W. Different Roles of TP53 Codon 72 Polymorphism in Type 2 Diabetes and Its Complications: Evidence from a Case-Control Study on a Chinese Han Population. Int. J. Gen. Med. 2021, 14, 4259–4268. [Google Scholar] [CrossRef]
- Burgdorf, K.S.; Grarup, N.; Justesen, J.M.; Harder, M.N.; Witte, D.R.; Jørgensen, T.; Sandbæk, A.; Lauritzen, T.; Madsbad, S.; Hansen, T.; et al. Studies of the Association of Arg72Pro of Tumor Suppressor Protein p53 with Type 2 Diabetes in a Combined Analysis of 55,521 Europeans. PLoS ONE 2011, 6, e15813. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, H.; Zhang, F.; Zhang, M.; Wang, Q.; Wang, J. The common genes involved in the pathogenesis of Alzheimer’s disease and type 2 diabetes and their implication for drug repositioning. Neuropharmacology 2023, 223, 109327. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; E Murphy, M. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef]
- Nakanishi, A.; Minami, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. BRCA1 and p53 Tumor Suppressor Molecules in Alzheimer’s Disease. Int. J. Mol. Sci. 2015, 16, 2879–2892. [Google Scholar] [CrossRef]
- Wezyk, M.; Zekanowski, C. Role of BRCA1 in Neuronal Death in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.R.T.; Santos, A.C.F.; Farfel, J.M.; Grinberg, L.T.; Ferretti, R.E.L.; Campos, A.H.J.F.M.; Cunha, I.W.; Begnami, M.D.; Rocha, R.M.; Carraro, D.M.; et al. Repair of Oxidative DNA Damage, Cell-Cycle Regulation and Neuronal Death May Influence the Clinical Manifestation of Alzheimer’s Disease. PLoS ONE 2014, 9, e99897. [Google Scholar] [CrossRef]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef]
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant Accumulation of BRCA1 in Alzheimer Disease and Other Tauopathies. J. Neuropathol. Exp. Neurol. 2019, 79, 22–33. [Google Scholar] [CrossRef]
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau Aggregates in Human Tauopathies. Brain Sci. 2019, 10, 7. [Google Scholar] [CrossRef]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654. [Google Scholar] [CrossRef]
- Brunet, J.; Vazquez-Martin, A.; Colomer, R.; Graña-Suarez, B.; Martin-Castillo, B.; Menendez, J.A. BRCA1 and acetyl-CoA carboxylase: The metabolic syndrome of breast cancer. Mol. Carcinog. 2007, 47, 157–163. [Google Scholar] [CrossRef]
- Karachanak-Yankova, S.; Dimova, R.; Nikolova, D.; Nesheva, D.; Koprinarova, M.; Maslyankov, S.; Tafradjiska, R.; Gateva, P.; Velizarova, M.; Hammoudeh, Z.; et al. Epigenetic alterations in patients with type 2 diabetes mellitus. Balk. J. Med. Genet. 2015, 18, 15–24. [Google Scholar] [CrossRef]
- Mori, T.; Asano, T.; Town, T. Targeting S100B in Cerebral Ischemia and in Alzheimer’s Disease. Cardiovasc. Psychiatry Neurol. 2010, 2010, 687067. [Google Scholar] [CrossRef]
- Serbinek, D.; Ullrich, C.; Pirchl, M.; Hochstrasser, T.; Schmidt-Kastner, R.; Humpel, C. S100b Counteracts Neurodegeneration of Rat Cholinergic Neurons in Brain Slices after Oxygen-Glucose Deprivation. Cardiovasc. Psychiatry Neurol. 2010, 2010, 106123. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 Signaling Rescues Cognition, Attenuates Tau Pathology, and Restores Neuronal β-Catenin Pathway Function in an Alzheimer’s Disease Model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Vieira, A.; Michels, M.; Borges, H.; Goulart, A.; Fernandes, F.; Dominguini, D.; Ritter, C.; Dal-Pizzol, F. Effects of S100B neutralization on the long-term cognitive impairment and neuroinflammatory response in an animal model of sepsis. Neurochem. Int. 2020, 142, 104906. [Google Scholar] [CrossRef]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. BioMed Res. Int. 2015, 2015, 508342. [Google Scholar] [CrossRef]
- Leclerc, E.; Sturchler, E.; Vetter, S.W. The S100B/RAGE Axis in Alzheimer’s Disease. Cardiovasc. Psychiatry Neurol. 2010, 2010, 539581. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Albrecht, M.A.; Takechi, R.; Giles, C.; James, A.P.; Foster, J.K.; Mamo, J.C.L. The Serum Concentration of the Calcium Binding Protein S100B is Positively Associated with Cognitive Performance in Older Adults. Front. Aging Neurosci. 2013, 5, 61. [Google Scholar] [CrossRef]
- Chaves, M.L.; Camozzato, A.L.; Ferreira, E.D.; Piazenski, I.; Kochhann, R.; Dall’Igna, O.; Mazzini, G.S.; O Souza, D.; Portela, L.V. Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J. Neuroinflamm. 2010, 7, 6. [Google Scholar] [CrossRef]
- Lee, B.W.; Chae, H.Y.; Kwon, S.J.; Park, S.Y.; Ihm, J.; Ihm, S.H. RAGE ligands induce apoptotic cell death of pancreatic beta-cells via oxidative stress. Int. J. Mol. Med. 2010, 26, 813–818. [Google Scholar] [PubMed]
- Afarideh, M.; Esteghamati, V.Z.; Ganji, M.; Heidari, B.; Esteghamati, S.; Lavasani, S.; Ahmadi, M.; Tafakhori, A.; Nakhjavani, M.; Esteghamati, A. Associations of Serum S100B and S100P with the Presence and Classification of Diabetic Peripheral Neuropathy in Adults With Type 2 Diabetes: A Case-Cohort Study. Can. J. Diabetes 2019, 43, 336–344.e2. [Google Scholar] [CrossRef]
- Yu, H.; Li, H.; Liu, X.; Du, X.; Deng, B. Levels of serum S100B are associated with cognitive dysfunction in patients with type 2 diabetes. Aging 2020, 12, 4193–4203. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C.; Liu, Z.H.; Dai, D.P.; Ding, F.H.; Pan, W.Q.; Fang, Y.H.; Zhang, Q.; et al. AGE/RAGE produces endothelial dysfunction in coronary arterioles in Type 2 diabetic mice. Am. J. Physiol. Circ. Physiol. 2008, 295, H491–H498. [Google Scholar] [CrossRef]
- Kesner, R.P.; Hui, X.; Sommer, T.; Wright, C.; Barrera, V.R.; Fanselow, M.S. The role of postnatal neurogenesis in supporting remote memory and spatial metric processing. Hippocampus 2014, 24, 1663–1671. [Google Scholar] [CrossRef]
- Benoit, J.D.; Rakic, P.; Frick, K.M. Prenatal stress induces spatial memory deficits and epigenetic changes in the hippocampus indicative of heterochromatin formation and reduced gene expression. Behav. Brain Res. 2015, 281, 1–8. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef]
- Mitchnick, K.A.; Creighton, S.; O’Hara, M.; Kalisch, B.E.; Winters, B.D. Differential contributions of de novo and maintenance DNA methyltransferases to object memory processing in the rat hippocampus and perirhinal cortex—A double dissociation. Eur. J. Neurosci. 2014, 41, 773–786. [Google Scholar] [CrossRef]
- Pi, T.; Wei, S.; Jiang, Y.; Shi, J.-S. High Methionine Diet-Induced Alzheimer’s Disease like Symptoms Are Accompanied by 5-Methylcytosine Elevated Levels in the Brain. Behav. Neurol. 2021, 2021, 6683318. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Li, C.; Gao, Y.; Meng, P.; Ji, S.; Xu, Y.; Mao, Y.; Wang, H.; Tian, J. The tyrosine kinase inhibitor LPM4870108 impairs learning and memory and induces transcriptomic and gene-specific DNA methylation changes in rats. Arch. Toxicol. 2022, 96, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Tannorella, P.; Stoccoro, A.; Tognoni, G.; Petrozzi, L.; Salluzzo, M.G.; Ragalmuto, A.; Siciliano, G.; Haslberger, A.; Bosco, P.; Bonuccelli, U.; et al. Methylation analysis of multiple genes in blood DNA of Alzheimer’s disease and healthy individuals. Neurosci. Lett. 2015, 600, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-Specific Epigenetic Drift in Late-Onset Alzheimer’s Disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Pezzi, J.C.; Ens, C.M.B.; Borba, E.M.; Schumacher-Schuh, A.F.; de Andrade, F.M.; Chaves, M.L.F.; Fiegenbaum, M.; Camozzato, A.L. DNA methyltransferase haplotype is associated with Alzheimer’s disease. Neurosci. Lett. 2014, 579, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renström, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA Methylation and Decreased Expression of PDX-1 in Pancreatic Islets from Patients with Type 2 Diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Guruprasad, K.P.; Satyamoorthy, K.; Joshi, M.B. Interleukin-6 determines protein stabilization of DNA methyltransferases and alters DNA promoter methylation of genes associated with insulin signaling and angiogenesis. Mod. Pathol. 2018, 98, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Choi, S.-K.; Galán, M.; Bishop, A.; Umezawa, K.; Trebak, M.; Belmadani, S.; Matrougui, K. Enhanced NF-κB Activity Impairs Vascular Function Through PARP-1–, SP-1–, and COX-2–Dependent Mechanisms in Type 2 Diabetes. Diabetes 2013, 62, 2078–2087. [Google Scholar] [CrossRef]
- Kassan, M.; Choi, S.-K.; Galán, M.; Trebak, M.; Belmadani, S.; Matrougui, K. Nuclear factor kappa B inhibition improves conductance artery function in type 2 diabetic mice. Diabetes/Metabolism Res. Rev. 2015, 31, 39–49. [Google Scholar] [CrossRef]
- Zakaria, E.M.; El-Bassossy, H.M.; El-Maraghy, N.N.; Ahmed, A.F.; Ali, A.A. PARP-1 inhibition alleviates diabetic cardiac complications in experimental animals. Eur. J. Pharmacol. 2016, 791, 444–454. [Google Scholar] [CrossRef]
- Xia, Q.; Lu, S.; Ostrovsky, J.; E McCormack, S.; Falk, M.J.; A Grant, S.F. PARP-1 Inhibition Rescues Short Lifespan in Hyperglycemic C. Elegans and Improves GLP-1 Secretion in Human Cells. Aging Dis. 2018, 9, 17–30. [Google Scholar] [CrossRef]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp. Cell Res. 2018, 373, 112–118. [Google Scholar] [CrossRef]
- Cui, N.-H.; Yang, J.-M.; Liu, X.; Wang, X.-B. Poly(ADP-Ribose) Polymerase Activity and Coronary Artery Disease in Type 2 Diabetes Mellitus. Arter. Thromb. Vasc. Biol. 2020, 40, 2516–2526. [Google Scholar] [CrossRef]
- Reda, E.; Hassaneen, S.; El-Abhar, H.S. Novel Trajectories of Bromocriptine Antidiabetic Action: Leptin-IL-6/ JAK2/p-STAT3/SOCS3, p-IR/p-AKT/GLUT4, PPAR-γ/Adiponectin, Nrf2/PARP-1, and GLP-1. Front. Pharmacol. 2018, 9, 771. [Google Scholar] [CrossRef]
- Li, R.; Sun, X.; Li, P.; Li, W.; Zhao, L.; Zhu, L.; Zhu, S. GLP-1-Induced AMPK Activation Inhibits PARP-1 and Promotes LXR-Mediated ABCA1 Expression to Protect Pancreatic β-Cells Against Cholesterol-Induced Toxicity Through Cholesterol Efflux. Front. Cell Dev. Biol. 2021, 9, 646113. [Google Scholar] [CrossRef]
- Sclip, A.; Antoniou, X.; Colombo, A.; Camici, G.G.; Pozzi, L.; Cardinetti, D.; Feligioni, M.; Veglianese, P.; Bahlmann, F.H.; Cervo, L.; et al. c-Jun N-terminal Kinase Regulates Soluble Aβ Oligomers and Cognitive Impairment in AD Mouse Model*. J. Biol. Chem. 2011, 286, 43871–43880. [Google Scholar] [CrossRef]
- España, J.; Valero, J.; Miñano-Molina, A.J.; Masgrau, R.; Martín, E.; Guardia-Laguarta, C.; Lleó, A.; Giménez-Llort, L.; Rodríguez-Alvarez, J.; Saura, C.A. β-Amyloid Disrupts Activity-Dependent Gene Transcription Required for Memory through the CREB Coactivator CRTC1. J. Neurosci. 2010, 30, 9402–9410. [Google Scholar] [CrossRef]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.-S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.-H. Retinoic Acid Attenuates β-Amyloid Deposition and Rescues Memory Deficits in an Alzheimer’s Disease Transgenic Mouse Model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Tsai, C.-W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-Mediated Wnt Signaling Contributes to Synaptic Abnormalities and Amyloid Pathology in Alzheimer’s Disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; D’Adamio, L. Memory Deficits of British Dementia Knock-In Mice Are Prevented by Aβ-Precursor Protein Haploinsufficiency. J. Neurosci. 2012, 32, 5481–5485. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.-F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed]
- Pousinha, P.A.; Mouska, X.; Bianchi, D.; Temido-Ferreira, M.; Rajão-Saraiva, J.; Gomes, R.; Fernandez, S.P.; Salgueiro-Pereira, A.R.; Gandin, C.; Raymond, E.F.; et al. The Amyloid Precursor Protein C-Terminal Domain Alters CA1 Neuron Firing, Modifying Hippocampus Oscillations and Impairing Spatial Memory Encoding. Cell Rep. 2019, 29, 317–331.e5. [Google Scholar] [CrossRef]
- Tu, Z.; Keller, M.P.; Zhang, C.; Rabaglia, M.E.; Greenawalt, D.M.; Yang, X.; Wang, I.-M.; Dai, H.; Bruss, M.D.; Lum, P.Y.; et al. Integrative Analysis of a Cross-Loci Regulation Network Identifies App as a Gene Regulating Insulin Secretion from Pancreatic Islets. PLoS Genet. 2012, 8, e1003107. [Google Scholar] [CrossRef] [PubMed]
- Casini, P.; Cadavez, L.; Visa, M.; Montane, J.; Novials, A.; Alcarraz-Vizán, G.; Servitja, J.-M. Inhibition of BACE2 counteracts hIAPP-induced insulin secretory defects in pancreatic β-cells. FASEB J. 2014, 29, 95–104. [Google Scholar] [CrossRef]
- Southan, C. BACE2 as a new diabetes target: A patent review (2010–2012). Expert. Opin. Ther. Pat. 2013, 23, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Rulifson, I.C.; Cao, P.; Miao, L.; Kopecky, D.; Huang, L.; White, R.D.; Samayoa, K.; Gardner, J.; Wu, X.; Chen, K.; et al. Identification of Human Islet Amyloid Polypeptide as a BACE2 Substrate. PLoS ONE 2016, 11, e0147254. [Google Scholar] [CrossRef] [PubMed]
- Probst, G.; Xu, Y.Z. Small-molecule BACE1 inhibitors: A patent literature review (2006–2011). Expert. Opin. Ther. Pat. 2012, 22, 511–540. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, F.; Kusakabe, K.I.; Hsiao, C.C.; Gijsen, H.J.M. Small-molecule BACE1 inhibitors: A patent literature review (2011 to 2020). Expert Opin. Ther. Pat. 2021, 31, 25–52. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and Regulation of Apolipoprotein E (ApoE) Expression in the CNS in Mice with Targeting of Green Fluorescent Protein Gene to the ApoE Locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Ebbert, M.T.W.; Baker, K.E.; Cook, C.; Wang, X.; Sens, J.P.; Kocher, J.-P.; Petrucelli, L.; Fryer, J.D. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 2018, 215, 2235–2245. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Legleiter, J.; Han, X.; Fryer, J.D.; Kowalewski, T.; Holtzman, D.M. ABCA1 Is Required for Normal Central Nervous System ApoE Levels and for Lipidation of Astrocyte-secreted apoE. J. Biol. Chem. 2004, 279, 40987–40993. [Google Scholar] [CrossRef]
- Neuner, S.M.; Wilmott, L.A.; Hoffmann, B.R.; Mozhui, K.; Kaczorowski, C.C. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav. Brain Res. 2017, 322, 288–298. [Google Scholar] [CrossRef]
- Lee, L.C.; Goh, M.Q.L.; Koo, E.H. Transcriptional regulation of APP by apoE: To boldly go where no isoform has gone before. BioEssays 2017, 39. [Google Scholar] [CrossRef]
- Gezen-Ak, D.; Atasoy, I.L.; Candaş, E.; Alaylıoğlu, M.; Dursun, E. The Transcriptional Regulatory Properties of Amyloid Beta 1–42 may Include Regulation of Genes Related to Neurodegeneration. NeuroMol. Med. 2018, 20, 363–375. [Google Scholar] [CrossRef]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.-H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.-T.; et al. REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer’s Disease. Cell Rep. 2019, 26, 1112–1127.e9. [Google Scholar] [CrossRef]
- Yin, J.; Nielsen, M.; Carcione, T.; Li, S.; Shi, J. Apolipoprotein E regulates mitochondrial function through the PGC-1α-sirtuin 3 pathway. Aging 2019, 11, 11148–11156. [Google Scholar] [CrossRef]
- Madrid, L.; Moreno-Grau, S.; Ahmad, S.; González-Pérez, A.; de Rojas, I.; Xia, R.; Adami, P.V.M.; García-González, P.; Kleineidam, L.; Yang, Q.; et al. Multiomics integrative analysis identifies APOE allele-specific blood biomarkers associated to Alzheimer’s disease etiopathogenesis. Aging 2021, 13, 9277–9329. [Google Scholar] [CrossRef] [PubMed]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.-Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Wu, D.; Chi, N.-F.; Chen, P.-C.; Liao, Y.-P.; Chiu, H.-W.; Hu, C.-J. Effects of the Apolipoprotein E ε4 Allele on Functional MRI during n-Back Working Memory Tasks in Healthy Middle-Aged Adults. Am. J. Neuroradiol. 2012, 34, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Kerchner, G.A.; Berdnik, D.; Shen, J.C.; Bernstein, J.D.; Fenesy, M.C.; Deutsch, G.K.; Wyss-Coray, T.; Rutt, B.K. APOE ε4 worsens hippocampal CA1 apical neuropil atrophy and episodic memory. Neurology 2014, 82, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Mikos, A.E.; Piryatinsky, I.; Tremont, G.; Malloy, P.F. The APOE ε4 Allele Is Associated with Increased Frontally Mediated Neurobehavioral Symptoms in Amnestic MCI. Alzheimer Dis. Assoc. Disord. 2013, 27, 109–115. [Google Scholar] [CrossRef]
- Luck, T.; Then, F.S.; Luppa, M.; Schroeter, M.L.; Arélin, K.; Burkhardt, R.; Thiery, J.; Löffler, M.; Villringer, A.; Riedel-Heller, S.G. Association of the apolipoprotein E genotype with memory performance and executive functioning in cognitively intact elderly. Neuropsychology 2015, 29, 382–387. [Google Scholar] [CrossRef]
- Chang, P.; Li, X.; Ma, C.; Zhang, S.; Liu, Z.; Chen, K.; Ai, L.; Chang, J.; Zhang, Z. The Effects of an APOE Promoter Polymorphism on Human White Matter Connectivity during Non-Demented Aging. J. Alzheimer’s Dis. 2016, 55, 77–87. [Google Scholar] [CrossRef]
- Salvadó, G.; Ferreira, D.; Operto, G.; Cacciaglia, R.; Falcon, C.; Minguillon, C.; Groot, C.; van der Flier, W.M.; Barkhof, F.; Scheltens, P.; et al. The protective gene dose effect of the APOEε2 allele on gray matter volume in cognitively unimpaired individuals. Alzheimer’s Dement. 2021, 18, 1383–1395. [Google Scholar] [CrossRef]
- El-Lebedy, D.; Raslan, H.M.; Mohammed, A.M. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 12. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; Weng, R.; Gu, X.; Zhong, Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc. Disord. 2019, 19, 213. [Google Scholar] [CrossRef]
- Ma, S.-W.; Benzie, I.F.; Yeung, V.T. Type 2 diabetes mellitus and its renal complications in relation to apolipoprotein E gene polymorphism. Transl. Res. 2008, 152, 134–142. [Google Scholar] [CrossRef]
- Perron, P.; Brisson, D.; Santuré, M.; Blackburn, P.; Bergeron, J.; Vohl, M.C.; Després, J.P.; Gaudet, D. Apolipoprotein E and lipoprotein lipase gene polymorphisms interaction on the atherogenic combined expression of hypertriglyceridemia and hyperapobetalipoproteinemia phenotypes. J. Endocrinol. Investig. 2007, 30, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Guangda, X.; Linshuang, Z.; Jie, H.; Ling, Y.; Huijuan, X. Apo e4 allele is associated with endothelium-dependent arterial dilation in women with type 2 diabetes. Diabetes Res. Clin. Pract. 2006, 72, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Monastiriotis, C.; Papanas, N.; Trypsianis, G.; Karanikola, K.; Veletza, S.; Maltezos, E. The ε4 Allele of the APOE Gene Is Associated With More Severe Peripheral Neuropathy in Type 2 Diabetic Patients. Angiology 2012, 64, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Mulugeta, A.; Zhou, A.; Hyppönen, E. Apolipoprotein E (APOE) genotype-associated disease risks: A phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine 2020, 59, 102954. [Google Scholar] [CrossRef]
- Gao, C.; Fu, X.; Chu, Q.; Li, J.; Shu, G. Relationship Between the ApoE Gene Polymorphism and Type 2 Diabetes Mellitus Complications. Genet. Test. Mol. Biomarkers 2021, 25, 111–115. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef] [PubMed]
- Belloy, M.E.; Andrews, S.J.; Le Guen, Y.; Cuccaro, M.; Farrer, L.A.; Napolioni, V.; Greicius, M.D. APOE Genotype and Alzheimer Disease Risk Across Age, Sex, and Population Ancestry. JAMA Neurol. 2023, 80, 1284. [Google Scholar] [CrossRef]
- Kawashima, Y.; Chen, J.; Sun, H.; Lann, D.; Hajjar, R.J.; Yakar, S.; LeRoith, D. Apolipoprotein E deficiency abrogates insulin resistance in a mouse model of type 2 diabetes mellitus. Diabetologia 2009, 52, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhao, H.-L.; Sui, Y.; He, L.; Lee, H.-M.; Lai, F.M.; Tong, P.C.; Chan, J.C. Histopathological Correlations of Islet Amyloidosis with Apolipoprotein E Polymorphisms in Type 2 Diabetic Chinese Patients. Pancreas 2013, 42, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Tomono, Y.; Iwai, M.; Inaba, S.; Mogi, M.; Horiuchi, M. Blockade of AT1 Receptor Improves Adipocyte Differentiation in Atherosclerotic and Diabetic Models. Am. J. Hypertens. 2008, 21, 206–212. [Google Scholar] [CrossRef]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-analysis Confirms CR1, CLU, and PICALM as Alzheimer Disease Risk Loci and Reveals Interactions with APOE Genotypes. Arch. Neurol. 2010, 67, 1473–1484. [Google Scholar] [CrossRef]
- Thambisetty, M.; Simmons, A.; Velayudhan, L.; Hye, A.; Campbell, J.; Zhang, Y.; Wahlund, L.-O.; Westman, E.; Kinsey, A.; Güntert, A.; et al. Association of Plasma Clusterin Concentration with Severity, Pathology, and Progression in Alzheimer Disease. Arch. Gen. Psychiatry 2010, 67, 739–748. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Belbin, O.; Hunter, T.A.; Ma, L.; Bisceglio, G.D.; Zou, F.; Crook, J.E.; Pankratz, V.S.; Dickson, D.W.; Graff-Radford, N.R.; et al. Replication of CLU, CR1, and PICALM Associations with Alzheimer Disease. Arch. Neurol. 2010, 67, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Corneveaux, J.J.; Myers, A.J.; Allen, A.N.; Pruzin, J.J.; Ramirez, M.; Engel, A.; Nalls, M.A.; Chen, K.; Lee, W.; Chewning, K.; et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum. Mol. Genet. 2010, 19, 3295–3301. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.-Y.; Yu, J.-T.; Cui, W.-Z.; Zhong, X.-L.; Wu, Z.-C.; Zhang, Q.; Tan, L. Blood Clusterin Levels, rs9331888 Polymorphism, and the Risk of Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 29, 515–519. [Google Scholar] [CrossRef]
- Schrijvers, E.M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Plasma Clusterin and the Risk of Alzheimer Disease. JAMA 2011, 305, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Schjeide, B.-M.M.; Schnack, C.; Lambert, J.-C.; Lill, C.M.; Kirchheiner, J.; Tumani, H.; Otto, M.; Tanzi, R.E.; Lehrach, H.; Amouyel, P.; et al. The Role of Clusterin, Complement Receptor 1, and Phosphatidylinositol Binding Clathrin Assembly Protein in Alzheimer Disease Risk and Cerebrospinal Fluid Biomarker Levels. Arch. Gen. Psychiatry 2011, 68, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.-C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E6962–E6971. [Google Scholar] [CrossRef]
- Robbins, J.P.; Perfect, L.; Ribe, E.M.; Maresca, M.; Dangla-Valls, A.; Foster, E.M.; Killick, R.; Nowosiad, P.; Reid, M.J.; Polit, L.D.; et al. Clusterin Is Required for β-Amyloid Toxicity in Human iPSC-Derived Neurons. Front. Neurosci. 2018, 12, 504. [Google Scholar] [CrossRef]
- Wojtas, A.M.; Sens, J.P.; Kang, S.S.; Baker, K.E.; Berry, T.J.; Kurti, A.; Daughrity, L.; Jansen-West, K.R.; Dickson, D.W.; Petrucelli, L.; et al. Astrocyte-derived clusterin suppresses amyloid formation in vivo. Mol. Neurodegener. 2020, 15, 71. [Google Scholar] [CrossRef]
- Liu, Z.; Chao, J.; Wang, C.; Sun, G.; Roeth, D.; Liu, W.; Chen, X.; Li, L.; Tian, E.; Feng, L.; et al. Astrocytic response mediated by the CLU risk allele inhibits OPC proliferation and myelination in a human iPSC model. Cell Rep. 2023, 42, 112841. [Google Scholar] [CrossRef]
- Stevens, B.W.; DiBattista, A.M.; Rebeck, G.W.; Green, A.E. A gene−brain−cognition pathway for the effect of an Alzheimer׳s risk gene on working memory in young adults. Neuropsychologia 2014, 61, 143–149. [Google Scholar] [CrossRef]
- Alfimova, M.; Kondratyev, N.; Golov, A.; Golimbet, V. Relationship between Alzheimer’s disease-associated SNPs within the CLU gene, local DNA methylation and episodic verbal memory in healthy and schizophrenia subjects. Psychiatry Res. 2018, 272, 380–386. [Google Scholar] [CrossRef]
- Chen, L.H.; Mak, T.S.H.; Fan, Y.; Ho, D.T.Y.; Sham, P.C.; Chu, L.W.; Song, Y.-Q. Associations between CLU polymorphisms and memory performance: The role of serum lipids in Alzheimer’s disease. J. Psychiatr. Res. 2020, 129, 281–288. [Google Scholar] [CrossRef]
- Ferencz, B.; Laukka, E.J.; Welmer, A.-K.; Kalpouzos, G.; Angleman, S.; Keller, L.; Graff, C.; Lövdén, M.; Bäckman, L. The benefits of staying active in old age: Physical activity counteracts the negative influence of PICALM, BIN1, and CLU risk alleles on episodic memory functioning. Psychol. Aging 2014, 29, 440–449. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, Z.; Khoury, N.; Betley, M.J.; Lehallier, B.; Willoughby, D.; Olsson, N.; Yang, A.C.; Hahn, O.; Lu, N.; Vest, R.T.; et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 2021, 600, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, O.; Labonté, J.; Albrecht, B.; Wieczorek, D.; Lechno, S.; Zechner, U.; Haaf, T. Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein–Taybi syndrome. Am. J. Med. Genet. Part A 2009, 152A, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes 2021, 12, 968. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Stockton, M.E.; Eisinger, B.E.; Zhao, Y.; Miller, J.L.; Bhuiyan, I.; Gao, Y.; Wu, Z.; Peng, J.; Zhao, X. Reducing histone acetylation rescues cognitive deficits in a mouse model of Fragile X syndrome. Nat. Commun. 2018, 9, 2494. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Deng, Y.; Yu, D.; Cao, H.; Wang, L.; Liu, L.; Yu, C.; Zhang, Y.; Guo, X.; Yu, G. Histone Acetyltransferase p300 Mediates Histone Acetylation of PS1 and BACE1 in a Cellular Model of Alzheimer’s Disease. PLoS ONE 2014, 9, e103067. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Tan, B.; Cheng, Y. P300 Inhibition Improves Cell Apoptosis and Cognition Impairment Induced by Sevoflurane Through Regulating IL-17A Activation. World Neurosurg. 2021, 154, e566–e571. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Wang, C.; Tang, Y.; Mok, S.-A.; Tsai, R.M.; Rojas, J.C.; Karydas, A.; Miller, B.L.; Boxer, A.L.; et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener. 2020, 15, 2. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, J.; Chen, Q.-N.; Lyu, A.-K.; Chen, J.-L.; Sun, Y.; Lyu, Q.; Zhao, Y.-X.; Guo, A.; Liao, Z.-Y.; et al. Type 2 diabetes-induced overactivation of P300 contributes to skeletal muscle atrophy by inhibiting autophagic flux. Life Sci. 2020, 258, 118243. [Google Scholar] [CrossRef] [PubMed]
- Long, H.-Z.; Cheng, Y.; Zhou, Z.-W.; Luo, H.-Y.; Wen, D.-D.; Gao, L.-C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef]
- Talchai, C.; Lin, H.V.; Kitamura, T.; Accili, D. Genetic and biochemical pathways of β-cell failure in type 2 diabetes. Diabetes Obes. Metab. 2009, 11, 38–45. [Google Scholar] [CrossRef]
- Kitamura, T. The role of FOXO1 in β-cell failure and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 615–623. [Google Scholar] [CrossRef]
- Li, X.; Wan, T.; Li, Y. Role of FoxO1 in regulating autophagy in type 2 diabetes mellitus (Review). Exp. Ther. Med. 2021, 22, 707. [Google Scholar] [CrossRef]
- Benchoula, K.; Arya, A.; Parhar, I.S.; Hwa, W.E. FoxO1 signaling as a therapeutic target for type 2 diabetes and obesity. Eur. J. Pharmacol. 2020, 891, 173758. [Google Scholar] [CrossRef] [PubMed]
- Moll, L.; Schubert, M. The Role of Insulin and Insulin-Like Growth Factor-1/FoxO-Mediated Transcription for the Pathogenesis of Obesity-Associated Dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 384094. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef]
- Paroni, G.; Seripa, D.; D’Onofrio, G.; Gravina, C.; Urbano, M.; Fontana, A.; Cascavilla, L.; Pilotto, A.; Greco, A.; Pellegrini, F. FOXO1 locus and acetylcholinesterase inhibitors in elderly patients with Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 1783–1791. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.; Liao, Y.; Luo, C.; Zou, D.; Wei, X.; Huang, Q.; Wu, Y. MiR-181a influences the cognitive function of epileptic rats induced by pentylenetetrazol. Int. J. Clin. Exp. Pathol. 2015, 8, 12861–12868. [Google Scholar]
- Wu, Q.; Yuan, X.; Bai, J.; Han, R.; Li, Z.; Zhang, H.; Xiu, R. MicroRNA-181a protects against pericyte apoptosis via directly targeting FOXO1: Implication for ameliorated cognitive deficits in APP/PS1 mice. Aging 2019, 11, 6120–6133. [Google Scholar] [CrossRef]
- Shigemizu, D.; Akiyama, S.; Higaki, S.; Sugimoto, T.; Sakurai, T.; Boroevich, K.A.; Sharma, A.; Tsunoda, T.; Ochiya, T.; Niida, S.; et al. Prognosis prediction model for conversion from mild cognitive impairment to Alzheimer’s disease created by integrative analysis of multi-omics data. Alzheimer’s Res. Ther. 2020, 12, 145. [Google Scholar] [CrossRef]
- Oh, S.Y.; Ellenstein, A.; Chen, C.-D.; Hinman, J.D.; Berg, E.A.; Costello, C.E.; Yamin, R.; Neve, R.L.; Abraham, C.R. Amyloid precursor protein interacts with notch receptors. J. Neurosci. Res. 2005, 82, 32–42. [Google Scholar] [CrossRef]
- Boo, J.H.; Sohn, J.H.; Kim, J.E.; Song, H.; Mook-Jung, I. Rac1 changes the substrate specificity of γ-secretase between amyloid precursor protein and Notch1. Biochem. Biophys. Res. Commun. 2008, 372, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.-N.; Park, J.-S.; Gwon, A.-R.; Arumugam, T.V.; Jo, D.-G. Alzheimer’s disease and Notch signaling. Biochem. Biophys. Res. Commun. 2009, 390, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavalu, K.; Choi, S.H.; Zhang, X.; Sisodia, S.S. Presenilin 1 Mutants Impair the Self-Renewal and Differentiation of Adult Murine Subventricular Zone-Neuronal Progenitors via Cell-Autonomous Mechanisms Involving Notch Signaling. J. Neurosci. 2010, 30, 6903–6915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, G.; Liu, H.; Chang, H.; Wilson, J.X. Folic acid enhances Notch signaling, hippocampal neurogenesis, and cognitive function in a rat model of cerebral ischemia. Nutr. Neurosci. 2012, 15, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Mao, X.; Xie, L.; Ding, M.; Shao, B.; Jin, K. Notch1 signaling modulates neuronal progenitor activity in the subventricular zone in response to aging and focal ischemia. Aging Cell 2013, 12, 978–987. [Google Scholar] [CrossRef]
- Leal, M.C.; Surace, E.I.; Holgado, M.P.; Ferrari, C.C.; Tarelli, R.; Pitossi, F.; Wisniewski, T.; Castaño, E.M.; Morelli, L. Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: Implications for cellular Aβ metabolism. Biochim. et Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 227–235. [Google Scholar] [CrossRef]
- Brai, E.; Raio, N.A.; Alberi, L. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 64. [Google Scholar] [CrossRef]
- Kapoor, A.; Nation, D.A. Role of Notch signaling in neurovascular aging and Alzheimer’s disease. Semin. Cell Dev. Biol. 2020, 116, 90–97. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective effect of melatonin on soluble Aβ1–42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimer’s Res. Ther. 2016, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Chaijaroonkhanarak, W.; Prachaney, P.; Pannangrong, W.; Leksomboon, R.; Chaichun, A.; Wigmore, P.; Welbat, J.U. Effects of Asiatic Acid on Spatial Working Memory and Cell Proliferation in the Adult Rat Hippocampus. Nutrients 2015, 7, 8413–8423. [Google Scholar] [CrossRef]
- Xue, F.; Chen, Y.-C.; Zhou, C.-H.; Wang, Y.; Cai, M.; Yan, W.-J.; Wu, R.; Wang, H.-N.; Peng, Z.-W. Risperidone ameliorates cognitive deficits, promotes hippocampal proliferation, and enhances Notch signaling in a murine model of schizophrenia. Pharmacol. Biochem. Behav. 2017, 163, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, P.; He, X.; Hong, M.; Liu, F. Micro ribonucleic acid-363 regulates the phosphatidylinositol 3-kinase/threonine protein kinase axis by targeting NOTCH1 and forkhead box C2, leading to hepatic glucose and lipids metabolism disorder in type 2 diabetes mellitus. J. Diabetes Investig. 2021, 13, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Li, X.; Yang, J.; Liu, H.; Zhang, Y.; Yang, X.; Yang, S.; Chang, B.; Chen, L.; Chang, B. Salsalate Prevents β-Cell Dedifferentiation in OLETF Rats with Type 2 Diabetes through Notch1 Pathway. Aging Dis. 2019, 10, 719–730. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-β Induces Hepatic Insulin Resistance In Vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef]
- Chen, Z.; Morris, D.L.; Jiang, L.; Liu, Y.; Rui, L. SH2B1 in β-cells promotes insulin expression and glucose metabolism in mice. Mol. Endocrinol. 2014, 28, 696–705. [Google Scholar] [CrossRef]
- Lu, L.; Ye, X.; Yao, Q.; Lu, A.; Zhao, Z.; Ding, Y.; Meng, C.; Yu, W.; Du, Y.; Cheng, J. Egr2 enhances insulin resistance via JAK2/STAT3/SOCS-1 pathway in HepG2 cells treated with palmitate. Gen. Comp. Endocrinol. 2018, 260, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Zhang, H.; Li, S.; Tian, H. Glucagon-like peptide 1 receptor agonist ameliorates the insulin resistance function of islet β cells via the activation of PDX-1/JAK signaling transduction in C57/BL6 mice with high-fat diet-induced diabetes. Int. J. Mol. Med. 2017, 39, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Brosius, F.C.; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zheng, Q.; Wang, Y.; Lin, J.; Wang, H.; Liu, R.; Yan, M.; Chen, X.; Yang, J.; Chen, X. Renoprotective Effect of the Recombinant Anti-IL-6R Fusion Proteins by Inhibiting JAK2/STAT3 Signaling Pathway in Diabetic Nephropathy. Front. Pharmacol. 2021, 12, 681424. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.J.; Natarajan, C.; Sriram, S.; Muthian, G. Signaling through JAK2-STAT5 pathway is essential for IL-3-induced activation of microglia. Glia 2003, 45, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Sasabe, J.; Terashita, K.; Shimoda, M.; Matsuoka, M.; Aiso, S. Amyloid-β causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry 2008, 14, 206–222. [Google Scholar] [CrossRef]
- Marwarha, G.; Prasanthi, J.R.; Schommer, J.; Dasari, B.; Ghribi, O. Molecular interplay between leptin, insulin-like growth factor-1, and β-amyloid in organotypic slices from rabbit hippocampus. Mol. Neurodegener. 2011, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- He, G.-L.; Luo, Z.; Shen, T.-T.; Li, P.; Yang, J.; Luo, X.; Chen, C.-H.; Gao, P.; Yang, X.-S. Inhibition of STAT3- and MAPK-dependent PGE2 synthesis ameliorates phagocytosis of fibrillar β-amyloid peptide (1-42) via EP2 receptor in EMF-stimulated N9 microglial cells. J. Neuroinflamm. 2016, 13, 296. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.-Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.-A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Rebuffat, S.A.; Gasa, R.; Gomis, R. Targeting type 2 diabetes: Lessons from a knockout model of insulin receptor substrate 2. Can. J. Physiol. Pharmacol. 2014, 92, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.; Huang, G.C.; Amiel, S.; Jones, P.M.; Persaud, S.J. Identification of Insulin Signaling Elements in Human β-Cells. Diabetes 2006, 55, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Lingohr, M.K.; Briaud, I.; Dickson, L.M.; McCuaig, J.F.; Alárcon, C.; Wicksteed, B.L.; Rhodes, C.J. Specific Regulation of IRS-2 Expression by Glucose in Rat Primary Pancreatic Islet β-Cells. J. Biol. Chem. 2006, 281, 15884–15892. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Barbosa-Sampaio, H.; Jones, P.M.; Persaud, S.J.; Muller, D.S. The CaMK4/CREB/IRS-2 Cascade Stimulates Proliferation and Inhibits Apoptosis of β-Cells. PLoS ONE 2012, 7, e45711. [Google Scholar] [CrossRef] [PubMed]
- Demozay, D.; Tsunekawa, S.; Briaud, I.; Shah, R.; Rhodes, C.J. Specific Glucose-Induced Control of Insulin Receptor Substrate-2 Expression Is Mediated via Ca2+-Dependent Calcineurin/NFAT Signaling in Primary Pancreatic Islet β-Cells. Diabetes 2011, 60, 2892–2902. [Google Scholar] [CrossRef]
- Wei, K.; Piecewicz, S.M.; McGinnis, L.M.; Taniguchi, C.M.; Wiegand, S.J.; Anderson, K.; Chan, C.W.-M.; Mulligan, K.X.; Kuo, D.; Yuan, J.; et al. A liver Hif-2α–Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat. Med. 2013, 19, 1331–1337. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 2013, 19, 1325–1330. [Google Scholar] [CrossRef]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; I Choudhury, A.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Aβ accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’connor, R.; Ravid, R.; O’neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Chua, L.-M.; Lim, M.-L.; Chong, P.-R.; Hu, Z.P.; Cheung, N.S.; Wong, B.-S. Impaired Neuronal Insulin Signaling Precedes Aβ42 Accumulation in Female AβPPsw/PS1ΔE9 Mice. J. Alzheimer’s Dis. 2012, 29, 783–791. [Google Scholar] [CrossRef]
- Ochiai, T.; Sano, T.; Nagayama, T.; Kubota, N.; Kadowaki, T.; Wakabayashi, T.; Iwatsubo, T. Differential involvement of insulin receptor substrate (IRS)-1 and IRS-2 in brain insulin signaling is associated with the effects on amyloid pathology in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 159, 105510. [Google Scholar] [CrossRef]
- Alves-Borba, L.; Espinosa-Fernández, V.; Canseco-Rodríguez, A.; Sánchez-Pérez, A.M. ABA Supplementation Rescues IRS2 and BDNF mRNA Levels in a Triple-Transgenic Mice Model of Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2023, 7, 1007–1013. [Google Scholar] [CrossRef]
- Wang, M.; Song, H.; Jia, J. Interleukin-6 receptor gene polymorphisms were associated with sporadic Alzheimer’s disease in Chinese Han. Brain Res. 2010, 1327, 1–5. [Google Scholar] [CrossRef]
- Sasayama, D.; Hori, H.; Teraishi, T.; Hattori, K.; Ota, M.; Matsuo, J.; Kawamoto, Y.; Kinoshita, Y.; Amano, N.; Kunugi, H. Association of cognitive performance with interleukin-6 receptor Asp358Ala polymorphism in healthy adults. J. Neural Transm. 2011, 119, 313–318. [Google Scholar] [CrossRef]
- Haddick, P.C.; Larson, J.L.; Rathore, N.; Bhangale, T.R.; Phung, Q.T.; Srinivasan, K.; Hansen, D.V.; Lill, J.R.; Pericak-Vance, M.A.; Haines, J.; et al. A Common Variant of IL-6R is Associated with Elevated IL-6 Pathway Activity in Alzheimer’s Disease Brains. J. Alzheimer’s Dis. 2017, 56, 1037–1054. [Google Scholar] [CrossRef]
- Quillen, D.; Hughes, T.M.; Craft, S.; Howard, T.; Register, T.; Suerken, C.; Hawkins, G.A.; Milligan, C. Levels of Soluble Interleukin 6 Receptor and Asp358Ala Are Associated With Cognitive Performance and Alzheimer Disease Biomarkers. Neurol.-Neuroimmunol. Neuroinflamm. 2023, 10. [Google Scholar] [CrossRef]
- Elcioğlu, H.K.; Aslan, E.; Ahmad, S.; Alan, S.; Salva, E.; Elcioglu, H.; Kabasakal, L. Tocilizumab’s effect on cognitive deficits induced by intracerebroventricular administration of streptozotocin in Alzheimer’s model. Mol. Cell. Biochem. 2016, 420, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Rifai, N.; Hu, F.B. Interleukin-6 Receptor Gene Variations, Plasma Interleukin-6 Levels, and Type 2 Diabetes in U.S. Women. Diabetes 2007, 56, 3075–3081. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Z.; Chu, W.; Hale, T.; Cooper, J.J.; Elbein, S.C. Molecular Screening and Association Analyses of the Interleukin 6 Receptor Gene Variants with Type 2 Diabetes, Diabetic Nephropathy, and Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2005, 90, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Hamid, Y.H.; Urhammer, S.A.; Jensen, D.P.; Glümer, C.; Borch-Johnsen, K.; Jørgensen, T.; Hansen, T.; Pedersen, O. Variation in the Interleukin-6 Receptor Gene Associates with Type 2 Diabetes in Danish Whites. Diabetes 2004, 53, 3342–3345. [Google Scholar] [CrossRef]
- Wu, X.; Yu, T.; Ji, N.; Huang, Y.; Gao, L.; Shi, W.; Yan, Y.; Li, H.; Ma, L.; Wu, K.; et al. IL6R inhibits viability and apoptosis of pancreatic beta-cells in type 2 diabetes mellitus via regulation by miR-22 of the JAK/STAT signaling pathway. Diabetes Metab. Syndr. Obesity: Targets Ther. 2019, 12, 1645–1657. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2. [Google Scholar] [CrossRef]
- Hua, J.; de Paco, E.G.; Linck, N.; Maurice, T.; Desrumaux, C.; Manoury, B.; Rassendren, F.; Ulmann, L. Microglial P2X4 receptors promote ApoE degradation and contribute to memory deficits in Alzheimer’s disease. Cell. Mol. Life Sci. 2023, 80, 138. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Muraleva, N.A.; Kolosova, N.G. Association of cerebrovascular dysfunction with the development of Alzheimer’s disease-like pathology in OXYS rats. BMC Genom. 2018, 19, 51–63. [Google Scholar] [CrossRef]
- Varma, R.; Chai, Y.; Troncoso, J.; Gu, J.; Xing, H.; Stojilkovic, S.S.; Mattson, M.P.; Haughey, N.J. Amyloid-β Induces a Caspase-mediated Cleavage of P2X4 to Promote Purinotoxicity. NeuroMol. Med. 2009, 11, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Glas, R.; Sauter, N.S.; Schulthess, F.T.; Shu, L.; Oberholzer, J.; Maedler, K. Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia 2009, 52, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Chiozzi, P.; Morelli, A.; Adinolfi, E.; Rizzo, R.; Baricordi, O.R.; Di Virgilio, F. Enhanced P2X7 Activity in Human Fibroblasts From Diabetic Patients. Arter. Thromb. Vasc. Biol. 2004, 24, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.N.; Poon, W.; Lyssenko, V.; Groop, L.; Nichols, B.; Wilmot, M.; Robson, S.; Enjyoji, K.; Herman, M.A.; Hu, C.; et al. Variation in Glucose Homeostasis Traits Associated with P2RX7 Polymorphisms in Mice and Humans. J. Clin. Endocrinol. Metab. 2015, 100, E688–E696. [Google Scholar] [CrossRef] [PubMed]
- Uresti-Rivera, E.E.; García-Jacobo, R.E.; Méndez-Cabañas, J.A.; Gaytan-Medina, L.E.; Cortez-Espinosa, N.; Portales-Pérez, D.P.; González-Amaro, R.; Enciso-Moreno, J.A.; García-Hernández, M.H. The presence of the 1068 G>A variant of P2X7 receptors is associated to an increase in IL-1Ra levels, insulin secretion and pancreatic β-cell function but not with glycemic control in type 2 diabetes patients. Gene 2018, 652, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 Mediates Superoxide Production in Primary Microglia and Is Up-regulated in a Transgenic Mouse Model of Alzheimer’s Disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef]
- Rampe, D.; Wang, L.; Ringheim, G.E. P2X7 receptor modulation of β-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J. Neuroimmunol. 2004, 147, 56–61. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of Microglia by Amyloid β Requires P2X7 Receptor Expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef]
- Beltran-Lobo, P.; Hughes, M.M.; Troakes, C.; Croft, C.L.; Rupawala, H.; Jutzi, D.; Ruepp, M.-D.; Jimenez-Sanchez, M.; Perkinton, M.S.; Kassiou, M.; et al. P2X7R influences tau aggregate burden in human tauopathies and shows distinct signalling in microglia and astrocytes. Brain Behav. Immun. 2023, 114, 414–429. [Google Scholar] [CrossRef]
- Sun, L.; Gao, J.; Zhao, M.; Cui, J.; Li, Y.; Yang, X.; Jing, X.; Wu, Z. A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behav. Brain Res. 2015, 291, 315–324. [Google Scholar] [CrossRef]
- Ruan, Z.; Delpech, J.-C.; Kalavai, S.V.; Van Enoo, A.A.; Hu, J.; Ikezu, S.; Ikezu, T. P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol. Neurodegener. 2020, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Groblewska, M.; Muszyński, P.; Wojtulewska-Supron, A.; Kulczyńska-Przybik, A.; Mroczko, B. The Role of Visinin-Like Protein-1 in the Pathophysiology of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 17–32. [Google Scholar] [CrossRef]
- Schnurra, I.; Bernstein, H.-G.; Riederer, P.; Braunewell, K.-H. The Neuronal Calcium Sensor Protein VILIP-1 Is Associated with Amyloid Plaques and Extracellular Tangles in Alzheimer’s Disease and Promotes Cell Death and Tau Phosphorylation in Vitro: A Link between Calcium Sensors and Alzheimer’s Disease? Neurobiol. Dis. 2001, 8, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Sweet, R.; Sims, R.; Harold, D.; Russo, G.; Abraham, R.; Stretton, A.; Jones, N.; Gerrish, A.; Chapman, J.; et al. Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol. Psychiatry 2011, 17, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, Y.; Gong, D. A meta-analysis of cerebrospinal fluid visinin-like protein-1 in alzheimer’s disease patients relative to healthy controls and mild cognitive impairment patients. Neurosciences 2017, 22, 94–101. [Google Scholar] [CrossRef]
- Leko, M.B.; Borovečki, F.; Dejanović, N.; Hof, P.R.; Šimić, G. Predictive Value of Cerebrospinal Fluid Visinin-Like Protein-1 Levels for Alzheimer’s Disease Early Detection and Differential Diagnosis in Patients with Mild Cognitive Impairment. J. Alzheimer’s Dis. 2016, 50, 765–778. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Zboch, M.; Muszyński, P.; Zajkowska, A.; Borawska, R.; Szmitkowski, M.; Kornhuber, J.; Lewczuk, P. Evaluation of Visinin-Like Protein 1 Concentrations in the Cerebrospinal Fluid of Patients with Mild Cognitive Impairment as a Dynamic Biomarker of Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1031–1037. [Google Scholar] [CrossRef]
- Tarawneh, R.; Lee, J.-M.; Ladenson, J.; Morris, J.; Holtzman, D. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology 2012, 78, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.F.; Zhang, Y.; Kang, Y.; Wang, Q.; Gaisano, H.Y.; Braunewell, K.-H.; Chan, C.B.; Wheeler, M.B. The Neuronal Ca2+ Sensor Protein Visinin-like Protein-1 Is Expressed in Pancreatic Islets and Regulates Insulin Secretion. J. Biol. Chem. 2006, 281, 21942–21953. [Google Scholar] [CrossRef] [PubMed]
- Atla, G.; Cuenca-Ardura, M.; Beucher, A.; Crouch, D.J.M.; Garcia-Hurtado, J.; Moran, I.; Irimia, M.; Prasad, R.B.; Gloyn, A.L.; Marselli, L.; et al. Genetic regulation of RNA splicing in human pancreatic islets. Genome Biol. 2022, 23, 196. [Google Scholar] [CrossRef] [PubMed]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene set analysis: Challenges, opportunities, and future research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- Brohée, S.; van Helden, J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinf. 2006, 7, 488. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| GOBP 1 | Odds Ratio | p Value |
|---|---|---|
| RESPONSE TO OXYGEN CONTAINING COMPOUND | 16.06 | 1.97 × 10−301 |
| POSITIVE REGULATION OF MULTICELLULAR ORGANISMAL PROCESS | 13.616 | 1.24 × 10−240 |
| RESPONSE TO ENDOGENOUS STIMULUS | 12.81 | 2.40 × 10−234 |
| CELLULAR RESPONSE TO OXYGEN CONTAINING COMPOUND | 15.04 | 3.76 × 10−224 |
| POSITIVE REGULATION OF SIGNALING | 11.70 | 1.76 × 10−222 |
| REGULATION OF TRANSPORT | 11.53 | 3.81 × 10−222 |
| REGULATION OF CELL DEATH | 11.97 | 5.53 × 10−222 |
| APOPTOTIC PROCESS | 10.91 | 1.28 × 10−219 |
| HOMEOSTATIC PROCESS | 11.63 | 1.17 × 10−216 |
| REGULATION OF CELL POPULATION PROLIFERATION | 11.11 | 6.84 × 10−212 |
| KEGG 1 Pathway | Odds Ratio | p Value |
|---|---|---|
| PATHWAYS IN CANCER | 21.62 | 9.22 × 10−41 |
| NEUROTROPHIN SIGNALING PATHWAY | 36.62 | 9.14 × 10−32 |
| LEISHMANIA INFECTION | 59.33 | 2.17 × 10−30 |
| TOLL-LIKE RECEPTOR SIGNALING PATHWAY | 41.26 | 1.18 × 10−29 |
| CYTOKINE–CYTOKINE–RECEPTOR INTERACTION | 18.45 | 1.46 × 10−28 |
| FOCAL ADHESION | 22.04 | 2.64 × 10−27 |
| MAPK SIGNALING PATHWAY | 17.61 | 3.42 × 10−27 |
| COLORECTAL CANCER | 59.67 | 1.10 × 10−26 |
| APOPTOSIS | 42.63 | 1.43 × 10−26 |
| PROSTATE CANCER | 41.36 | 2.49 × 10−26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.B. Comorbidity Genes of Alzheimer’s Disease and Type 2 Diabetes Associated with Memory and Cognitive Function. Int. J. Mol. Sci. 2024, 25, 2211. https://doi.org/10.3390/ijms25042211
Cho SB. Comorbidity Genes of Alzheimer’s Disease and Type 2 Diabetes Associated with Memory and Cognitive Function. International Journal of Molecular Sciences. 2024; 25(4):2211. https://doi.org/10.3390/ijms25042211
Chicago/Turabian StyleCho, Seong Beom. 2024. "Comorbidity Genes of Alzheimer’s Disease and Type 2 Diabetes Associated with Memory and Cognitive Function" International Journal of Molecular Sciences 25, no. 4: 2211. https://doi.org/10.3390/ijms25042211
APA StyleCho, S. B. (2024). Comorbidity Genes of Alzheimer’s Disease and Type 2 Diabetes Associated with Memory and Cognitive Function. International Journal of Molecular Sciences, 25(4), 2211. https://doi.org/10.3390/ijms25042211