
Trained Innate Immunity in Animal Models of Cardiovascular Diseases

 ,
,
Abstract
:1. A Brief Introduction into Trained Innate Immunity
2. Stimuli and Mechanisms That Mediate Trained Innate Immunity
3. Trained Innate Immunity and Cardiovascular Diseases
4. Interconnections between Trained Immunity and Adaptive Immune Responses
5. Trained Innate Immunity in Animal Models of Cardiovascular Diseases

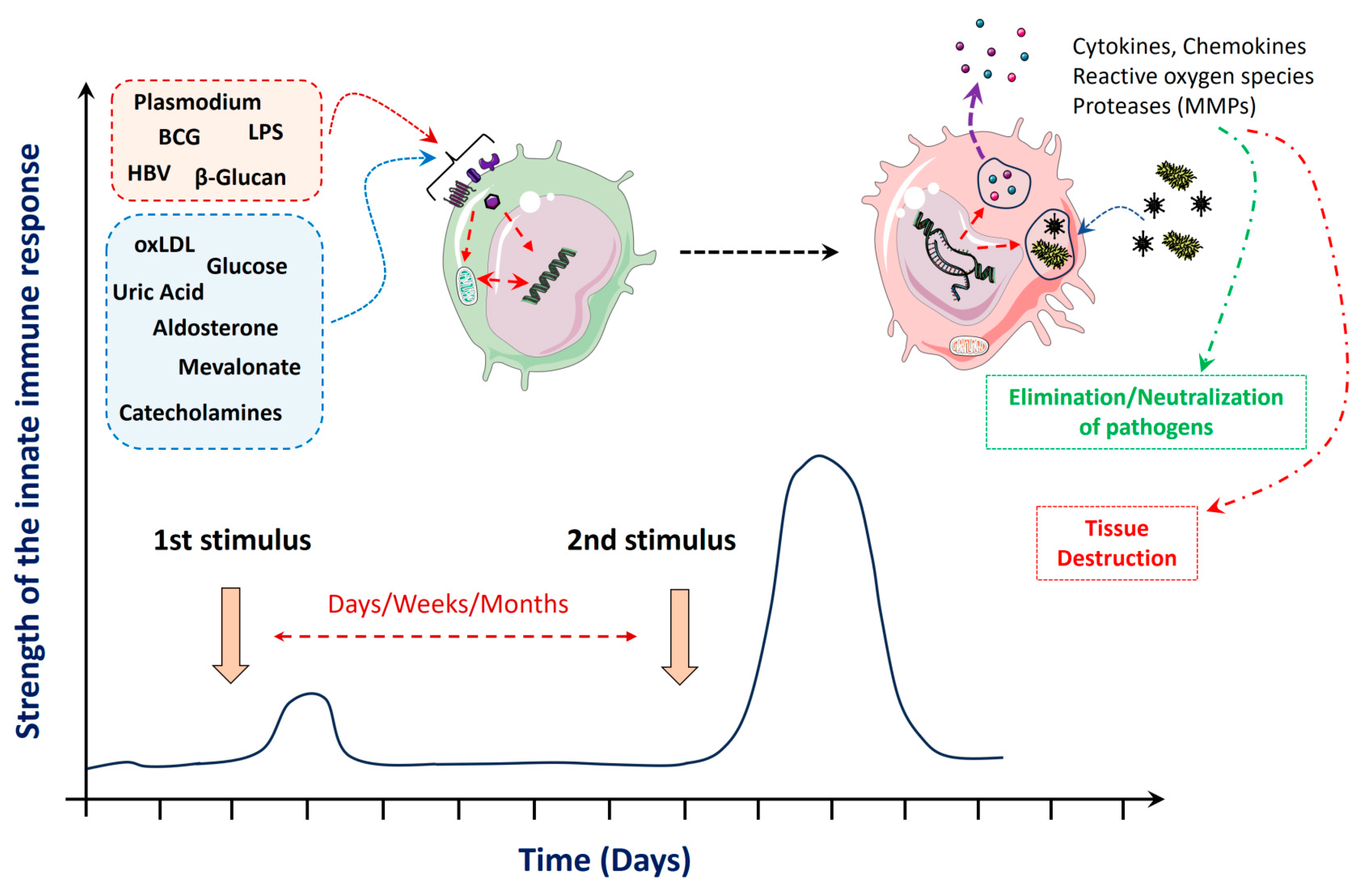{kind=link}
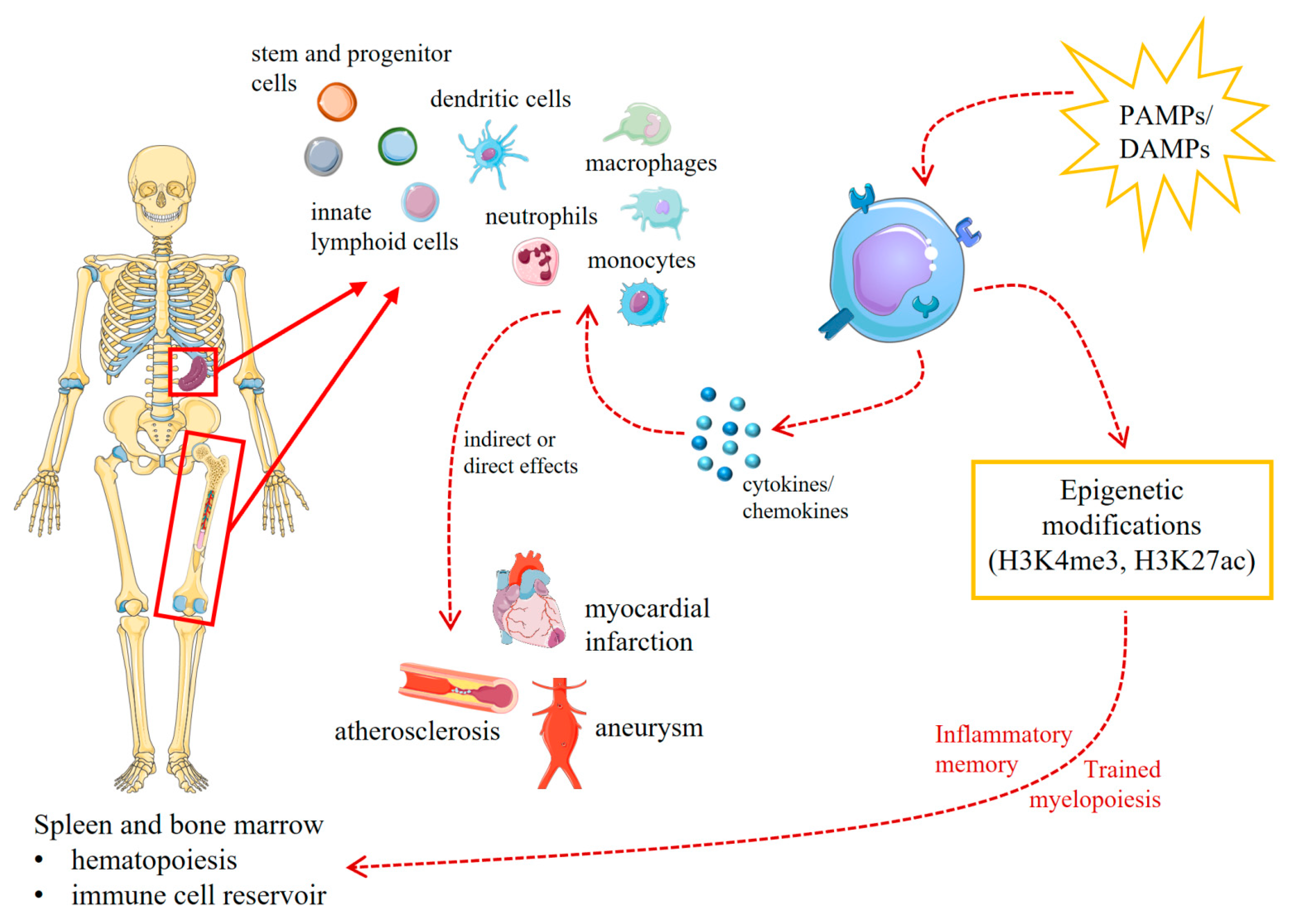{kind=link}
| Species | Disease/Effect | TI Trigger | 2nd Stimulus | Biological Effect/Mechanism | Evidence for Trained Immunity | References |
|---|---|---|---|---|---|---|
| Mouse (LDLr-/-) | Atherosclerosis (increased) | Hypercholesterolemia (LDLr-/-) | High-fat diet (HFD) |
|
| [71] |
| Mouse (LDLr-/-) | Atherosclerosis (increased) | Hypercholesterolemia (LDLr-/-) | Western diet (WD) |
|
| [72] |
| Mouse (C57BL/6 and LDLr-/-) | Atherosclerosis (increased) | Hyperglycemia (streptoztocin-induced diabetes) | Western diet (WD) |
|
| [73] |
| Mouse (ApoE-/-) | Atherosclerosis (increased) | Super low-dose lipopolysaccharide (5 ng/kg BW) | Western, high-fat diet (HFD) |
|
| [74] |
| Mouse (ApoE-/-) | Abdominal aortic aneurysms | High-fat diet (HFD) | Angiotensin II (Ang II) |
|
| [75,76] |
| Mouse | Stroke (increased) | High salt (HS) | Stroke |
|
| [62] |
| Mouse | Cancer | Breast cancer (E0771 cells) | Myocardial infarction (MI) |
|
| [77] |
5.1. Atherosclerosis
5.2. Abdominal Aortic Aneurysms
5.3. Stroke
5.4. Myocardial Infarction as as Trigger for Trained Innate Immune Suppression
6. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.; Wilming, L.; Rand, V.; Lovering, R.C.; Bruford, E.A.; Khodiyar, V.K.; Lush, M.J.; Povey, S.; Talbot, C.C.; Wright, M.W.; et al. Gene Map of the Extended Human MHC. Nat. Rev. Genet. 2004, 5, 889–899. [Google Scholar] [CrossRef]
- Strawbridge, A.B.; Blum, J.S. Autophagy in MHC Class II Antigen Processing. Curr. Opin. Immunol. 2007, 19, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.R. CD8+ T Cells: The Past and Future of Immune Regulation. Cell. Immunol. 2020, 357, 104212. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Oukka, M.; Kuchroo, V.; Bettelli, E. Th17 Cells: Effector T Cells with Inflammatory Properties. Semin. Immunol. 2007, 19, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C.M. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- Lipscomb, M.F.; Masten, B.J. Dendritic Cells: Immune Regulators in Health and Disease. Physiol. Rev. 2002, 82, 97–130. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Tercan, H.; Riksen, N.P.; Joosten, L.A.B.; Netea, M.G.; Bekkering, S. Trained Immunity: Long-Term Adaptation in Innate Immune Responses. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.-S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef] [PubMed]
- Riksen, N.P.; Bekkering, S.; Mulder, W.J.M.; Netea, M.G. Trained Immunity in Atherosclerotic Cardiovascular Disease. Nat. Rev. Cardiol. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Domínguez-Andrés, J.; Joosten, L.A.B.; Riksen, N.P.; Netea, M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021, 39, 667–693. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Trained Innate Immunity. Immunol. Res. 2021, 69, 1–7. [Google Scholar] [CrossRef]
- Bigot, J.; Guillot, L.; Guitard, J.; Ruffin, M.; Corvol, H.; Chignard, M.; Hennequin, C.; Balloy, V. Respiratory Epithelial Cells Can Remember Infection: A Proof-of-Concept Study. J. Infect. Dis. 2020, 221, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zeng, X.; Peng, L.; Xiang, C.; Zhou, Y.; Zhang, X.; Zhang, J.; Wang, N.; Guo, G.; Li, Y.; et al. Vaccination Induces Rapid Protection against Bacterial Pneumonia via Training Alveolar Macrophage in Mice. eLife 2021, 10, e69951. [Google Scholar] [CrossRef]
- Zughaier, S.M.; Rouquette-Loughlin, C.E.; Shafer, W.M. Identification of a Neisseria Gonorrhoeae Histone Deacetylase: Epigenetic Impact on Host Gene Expression. Pathog. Basel Switz. 2020, 9, 132. [Google Scholar] [CrossRef]
- Barton, E.S.; White, D.W.; Cathelyn, J.S.; Brett-McClellan, K.A.; Engle, M.; Diamond, M.S.; Miller, V.L.; Virgin, H.W. Herpesvirus Latency Confers Symbiotic Protection from Bacterial Infection. Nature 2007, 447, 326–329. [Google Scholar] [CrossRef]
- Hong, M.; Sandalova, E.; Low, D.; Gehring, A.J.; Fieni, S.; Amadei, B.; Urbani, S.; Chong, Y.-S.; Guccione, E.; Bertoletti, A. Trained Immunity in Newborn Infants of HBV-Infected Mothers. Nat. Commun. 2015, 6, 6588. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.-J.; Wijmenga, C.; et al. Candida albicans Infection Affords Protection against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef]
- Hole, C.R.; Wager, C.M.L.; Castro-Lopez, N.; Campuzano, A.; Cai, H.; Wozniak, K.L.; Wang, Y.; Wormley, F.L. Induction of Memory-like Dendritic Cell Responses in Vivo. Nat. Commun. 2019, 10, 2955. [Google Scholar] [CrossRef] [PubMed]
- Schrum, J.E.; Crabtree, J.N.; Dobbs, K.R.; Kiritsy, M.C.; Reed, G.W.; Gazzinelli, R.T.; Netea, M.G.; Kazura, J.W.; Dent, A.E.; Fitzgerald, K.A.; et al. Cutting Edge: Plasmodium falciparum Induces Trained Innate Immunity. J. Immunol. Baltim. Md 1950 2018, 200, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Moorlag, S.J.C.F.M.; Novakovic, B.; Li, Y.; Wang, S.-Y.; Oosting, M.; Kumar, V.; Xavier, R.J.; Wijmenga, C.; Joosten, L.A.B.; et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 2018, 23, 89–100.e5. [Google Scholar] [CrossRef] [PubMed]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.B.; Jacobs, C.; Xavier, R.J.; van der Meer, J.W.M.; van Crevel, R.; Netea, M.G. BCG-Induced Trained Immunity in NK Cells: Role for Non-Specific Protection to Infection. Clin. Immunol. Orlando Fla 2014, 155, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Walk, J.; de Bree, L.C.J.; Graumans, W.; Stoter, R.; van Gemert, G.-J.; van de Vegte-Bolmer, M.; Teelen, K.; Hermsen, C.C.; Arts, R.J.W.; Behet, M.C.; et al. Outcomes of Controlled Human Malaria Infection after BCG Vaccination. Nat. Commun. 2019, 10, 874. [Google Scholar] [CrossRef]
- Pennington, S.H.; Ferreira, D.M.; Caamaño-Gutiérrez, E.; Reiné, J.; Hewitt, C.; Hyder-Wright, A.D.; Gordon, S.B.; Gordon, M.A. Nonspecific Effects of Oral Vaccination with Live-Attenuated Salmonella Typhi Strain Ty21a. Sci. Adv. 2019, 5, eaau6849. [Google Scholar] [CrossRef] [PubMed]
- Aaby, P.; Samb, B.; Simondon, F.; Seck, A.M.; Knudsen, K.; Whittle, H. Non-Specific Beneficial Effect of Measles Immunisation: Analysis of Mortality Studies from Developing Countries. BMJ 1995, 311, 481–485. [Google Scholar] [CrossRef]
- Rieckmann, A.; Villumsen, M.; Sørup, S.; Haugaard, L.K.; Ravn, H.; Roth, A.; Baker, J.L.; Benn, C.S.; Aaby, P. Vaccinations against Smallpox and Tuberculosis Are Associated with Better Long-Term Survival: A Danish Case-Cohort Study 1971-2010. Int. J. Epidemiol. 2017, 46, 695–705. [Google Scholar] [CrossRef]
- Andersen, A.; Fisker, A.B.; Nielsen, S.; Rodrigues, A.; Benn, C.S.; Aaby, P. National Immunization Campaigns With Oral Polio Vaccine May Reduce All-Cause Mortality: An Analysis of 13 Years of Demographic Surveillance Data From an Urban African Area. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 72, e596–e603. [Google Scholar] [CrossRef]
- Debisarun, P.A.; Gössling, K.L.; Bulut, O.; Kilic, G.; Zoodsma, M.; Liu, Z.; Oldenburg, M.; Rüchel, N.; Zhang, B.; Xu, C.-J.; et al. Induction of Trained Immunity by Influenza Vaccination-Impact on COVID-19. PLoS Pathog. 2021, 17, e1009928. [Google Scholar] [CrossRef] [PubMed]
- Murugathasan, M.; Jafari, A.; Amandeep, A.; Hassan, S.A.; Chihata, M.; Abdul-Sater, A.A. Moderate Exercise Induces Trained Immunity in Macrophages. Am. J. Physiol.-Cell Physiol. 2023, 325, C429–C442. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.N.; Caffrey, D.R.; de Souza Silva, L.; Kurt-Jones, E.A.; Dobbs, K.; Dent, A.; Fitzgerald, K.A.; Golenbock, D.T. Lymphocyte Crosstalk Is Required for Monocyte-Intrinsic Trained Immunity to Plasmodium falciparum. J. Clin. Investig. 2022, 132, e139298. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Braun, M.; Kranig, S.A.; Frommhold, D.; Pöschl, J.; Hudalla, H. LPS Induces Opposing Memory-like Inflammatory Responses in Mouse Bone Marrow Neutrophils. Int. J. Mol. Sci. 2021, 22, 9803. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Quintin, J.; Joosten, L.A.B.; van der Meer, J.W.M.; Netea, M.G.; Riksen, N.P. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Crișan, T.O.; Cleophas, M.C.P.; Oosting, M.; Lemmers, H.; Toenhake-Dijkstra, H.; Netea, M.G.; Jansen, T.L.; Joosten, L.A.B. Soluble Uric Acid Primes TLR-Induced Proinflammatory Cytokine Production by Human Primary Cells via Inhibition of IL-1Ra. Ann. Rheum. Dis. 2016, 75, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Novakovic, B.; ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.-C.; Wang, S.-Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Arts, R.J.W.; Carvalho, A.; La Rocca, C.; Palma, C.; Rodrigues, F.; Silvestre, R.; Kleinnijenhuis, J.; Lachmandas, E.; Gonçalves, L.G.; Belinha, A.; et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016, 17, 2562–2571. [Google Scholar] [CrossRef]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e9. [Google Scholar] [CrossRef]
- van der Heijden, C.D.C.C.; Keating, S.T.; Groh, L.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Aldosterone Induces Trained Immunity: The Role of Fatty Acid Synthesis. Cardiovasc. Res. 2020, 116, 317–328. [Google Scholar] [CrossRef]
- van der Heijden, C.D.C.C.; Groh, L.; Keating, S.T.; Kaffa, C.; Noz, M.P.; Kersten, S.; van Herwaarden, A.E.; Hoischen, A.; Joosten, L.A.B.; Timmers, H.J.L.M.; et al. Catecholamines Induce Trained Immunity in Monocytes In Vitro and In Vivo. Circ. Res. 2020, 127, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Blok, B.A.; Joosten, L.A.B.; Riksen, N.P.; van Crevel, R.; Netea, M.G. In Vitro Experimental Model of Trained Innate Immunity in Human Primary Monocytes. Clin. Vaccine Immunol. 2016, 23, 926–933. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.-C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Dandapat, S.; Manikandan, R.; Dinesh, M.; Subbaiyan, A.; Mani, P.; Dhawan, M.; Tiwari, R.; Bilal, M.; Emran, T.B.; et al. Prophylactic and Therapeutic Insights into Trained Immunity: A Renewed Concept of Innate Immune Memory. Hum. Vaccines Immunother. 2022, 18, 2040238. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, J.W.M.; Joosten, L.A.B.; Riksen, N.; Netea, M.G. Trained Immunity: A Smart Way to Enhance Innate Immune Defence. Mol. Immunol. 2015, 68, 40–44. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α–Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed]
- Ciarlo, E.; Heinonen, T.; Théroude, C.; Asgari, F.; Le Roy, D.; Netea, M.G.; Roger, T. Trained Immunity Confers Broad-Spectrum Protection Against Bacterial Infections. J. Infect. Dis. 2020, 222, 1869–1881. [Google Scholar] [CrossRef]
- Dagenais, A.; Villalba-Guerrero, C.; Olivier, M. Trained Immunity: A “New” Weapon in the Fight against Infectious Diseases. Front. Immunol. 2023, 14, 1147476. [Google Scholar] [CrossRef]
- Mitroulis, I.; Hajishengallis, G.; Chavakis, T. Bone Marrow Inflammatory Memory in Cardiometabolic Disease and Inflammatory Comorbidities. Cardiovasc. Res. 2023, cvad003. [Google Scholar] [CrossRef]
- Mitroulis, I.; Hajishengallis, G.; Chavakis, T. Trained Immunity and Cardiometabolic Disease: The Role of Bone Marrow. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 48–54. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2020, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Schloss, M.J.; Swirski, F.K.; Nahrendorf, M. Modifiable Cardiovascular Risk, Hematopoiesis, and Innate Immunity. Circ. Res. 2020, 126, 1242–1259. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Dimmeler, S.; Schunk, S.J.; Fliser, D.; Ridker, P.M. Targeting Innate Immunity-Driven Inflammation in CKD and Cardiovascular Disease. Nat. Rev. Nephrol. 2022, 18, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, Y.; Lagache, S.M.M.; Schnack, L.; Godfrey, R.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. mTOR-Dependent Oxidative Stress Regulates oxLDL-Induced Trained Innate Immunity in Human Monocytes. Front. Immunol. 2019, 9, 3155. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The Glycolytic Enzyme PKM2 Bridges Metabolic and Inflammatory Dysfunction in Coronary Artery Disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, L.; Vavassori, C.; Colombo, G.I. Trained Immunity in Perivascular Adipose Tissue of Abdominal Aortic Aneurysm—A Novel Concept for a Still Elusive Disease. Front. Cell Dev. Biol. 2022, 10, 886086. [Google Scholar] [CrossRef] [PubMed]
- Groh, L.A.; Verel, D.E.; van der Heijden, C.D.C.C.; Matzaraki, V.; Moorlag, S.J.C.F.M.; de Bree, L.C.; Koeken, V.A.C.M.; Mourits, V.P.; Keating, S.T.; van Puffelen, J.H.; et al. Immune Modulatory Effects of Progesterone on oxLDL-Induced Trained Immunity in Monocytes. J. Leukoc. Biol. 2022, 112, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.M.; Mills, K.H.G.; Basdeo, S.A. The Effects of Trained Innate Immunity on T Cell Responses; Clinical Implications and Knowledge Gaps for Future Research. Front. Immunol. 2021, 12, 706583. [Google Scholar] [CrossRef]
- Jeljeli, M.; Riccio, L.G.C.; Doridot, L.; Chêne, C.; Nicco, C.; Chouzenoux, S.; Deletang, Q.; Allanore, Y.; Kavian, N.; Batteux, F. Trained Immunity Modulates Inflammation-Induced Fibrosis. Nat. Commun. 2019, 10, 5670. [Google Scholar] [CrossRef]
- Yao, Y.; Jeyanathan, M.; Haddadi, S.; Barra, N.G.; Vaseghi-Shanjani, M.; Damjanovic, D.; Lai, R.; Afkhami, S.; Chen, Y.; Dvorkin-Gheva, A.; et al. Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell 2018, 175, 1634–1650.e17. [Google Scholar] [CrossRef]
- Lin, J.; Huang, L.; Li, Y.; Zhang, P.; Yu, Q.; Yang, Q. Bacillus Subtilis Spore-Trained Dendritic Cells Enhance the Generation of Memory T Cells via ICAM1. Cells 2021, 10, 2267. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Jiang, D.; Chen, W.-R.; Lin, J.S.; Zhang, X.-Y.; Chen, C.-H.; Hsu, C.-L.; Lai, L.-C.; Chen, P.-H.; Yang, K.-C.; et al. Trained Immunity Induced by High-Salt Diet Impedes Stroke Recovery. EMBO Rep. 2023, 24, e57164. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium Chloride Drives Autoimmune Disease by the Induction of Pathogenic TH17 Cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Côrte-Real, B.F.; Hamad, I.; Arroyo Hornero, R.; Geisberger, S.; Roels, J.; Van Zeebroeck, L.; Dyczko, A.; van Gisbergen, M.W.; Kurniawan, H.; Wagner, A.; et al. Sodium Perturbs Mitochondrial Respiration and Induces Dysfunctional Tregs. Cell Metab. 2023, 35, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Itani, H.A.; Xiao, L.; Saleh, M.A.; Wu, J.; Pilkinton, M.A.; Dale, B.L.; Barbaro, N.R.; Foss, J.D.; Kirabo, A.; Montaniel, K.R.; et al. CD70 Exacerbates Blood Pressure Elevation and Renal Damage in Response to Repeated Hypertensive Stimuli. Circ. Res. 2016, 118, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Li, J.; Cheng, X. Regulatory T Cells and Cardiovascular Diseases. Chin. Med. J. 2023, 136, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Hinkley, H.; Counts, D.A.; VonCanon, E.; Lacy, M. T Cells in Atherosclerosis: Key Players in the Pathogenesis of Vascular Disease. Cells 2023, 12, 2152. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Mann, D.L. The Emerging Role of B Lymphocytes in Cardiovascular Disease. Annu. Rev. Immunol. 2020, 38, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Pedicino, D.; Giglio, A.F.; Ruggio, A.; Massaro, G.; D’Aiello, A.; Trotta, F.; Lucci, C.; Graziani, F.; Biasucci, L.M.; Crea, F.; et al. Inflammasome, T Lymphocytes and Innate-Adaptive Immunity Crosstalk: Role in Cardiovascular Disease and Therapeutic Perspectives. Thromb. Haemost. 2018, 118, 1352–1369. [Google Scholar] [CrossRef]
- Roy, P.; Orecchioni, M.; Ley, K. How the Immune System Shapes Atherosclerosis: Roles of Innate and Adaptive Immunity. Nat. Rev. Immunol. 2022, 22, 251–265. [Google Scholar] [CrossRef]
- Seijkens, T.; Hoeksema, M.A.; Beckers, L.; Smeets, E.; Meiler, S.; Levels, J.; Tjwa, M.; de Winther, M.P.J.; Lutgens, E. Hypercholesterolemia-Induced Priming of Hematopoietic Stem and Progenitor Cells Aggravates Atherosclerosis. FASEB J. 2014, 28, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [PubMed]
- Edgar, L.; Akbar, N.; Braithwaite, A.T.; Krausgruber, T.; Gallart-Ayala, H.; Bailey, J.; Corbin, A.L.; Khoyratty, T.E.; Chai, J.T.; Alkhalil, M.; et al. Hyperglycemia Induces Trained Immunity in Macrophages and Their Precursors and Promotes Atherosclerosis. Circulation 2021, 144, 961–982. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Chen, K.; Yuan, R.; Peng, L.; Maitra, U.; Diao, N.; Chen, C.; Zhang, Y.; Hu, Y.; Qi, C.-F.; et al. The Persistence of Low-Grade Inflammatory Monocytes Contributes to Aggravated Atherosclerosis. Nat. Commun. 2016, 7, 13436. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Sun, Y.; Saaoud, F.; Shao, Y.; Xu, K.; Jiang, X.; Wu, S.; Yu, J.; Snyder, N.W.; Yang, L.; et al. ER Stress Mediates Angiotensin II-Augmented Innate Immunity Memory and Facilitates Distinct Susceptibilities of Thoracic from Abdominal Aorta to Aneurysm Development. Front. Immunol. 2023, 14, 1268916. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Sun, Y.; Xu, K.; Saaoud, F.; Shao, Y.; Drummer, C.; Wu, S.; Hu, W.; Yu, J.; Kunapuli, S.P.; et al. Aorta in Pathologies May Function as an Immune Organ by Upregulating Secretomes for Immune and Vascular Cell Activation, Differentiation and Trans-Differentiation-Early Secretomes May Serve as Drivers for Trained Immunity. Front. Immunol. 2022, 13, 858256. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial Infarction Accelerates Breast Cancer via Innate Immune Reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Mohmmad-Rezaei, M.; Arefnezhad, R.; Ahmadi, R.; Abdollahpour-Alitappeh, M.; Mirzaei, Y.; Arjmand, M.-H.; Ferns, G.A.; Bashash, D.; Bagheri, N. An Overview of the Innate and Adaptive Immune System in Atherosclerosis. IUBMB Life 2021, 73, 64–91. [Google Scholar] [CrossRef]
- Soehnlein, O.; Swirski, F.K. Hypercholesterolemia Links Hematopoiesis with Atherosclerosis. Trends Endocrinol. Metab. 2013, 24, 129–136. [Google Scholar] [CrossRef]
- Feng, Y.; Schouteden, S.; Geenens, R.; Duppen, V.V.; Herijgers, P.; Holvoet, P.; Veldhoven, P.P.V.; Verfaillie, C.M. Hematopoietic Stem/Progenitor Cell Proliferation and Differentiation Is Differentially Regulated by High-Density and Low-Density Lipoproteins in Mice. PLoS ONE 2012, 7, e47286. [Google Scholar] [CrossRef]
- Itabe, H.; Obama, T. The Oxidized Lipoproteins In Vivo: Its Diversity and Behavior in the Human Circulation. Int. J. Mol. Sci. 2023, 24, 5747. [Google Scholar] [CrossRef]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL Oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed]
- Jerala, R. Structural Biology of the LPS Recognition. Int. J. Med. Microbiol. 2007, 297, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Munford, R.S. Endotoxemia—Menace, Marker, or Mistake? J. Leukoc. Biol. 2016, 100, 687–698. [Google Scholar] [CrossRef]
- Novakovic, B.; Habibi, E.; Wang, S.-Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; LeMaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic Aneurysms and Dissections Series. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef] [PubMed]
- Raffort, J.; Lareyre, F.; Clément, M.; Hassen-Khodja, R.; Chinetti, G.; Mallat, Z. Monocytes and Macrophages in Abdominal Aortic Aneurysm. Nat. Rev. Cardiol. 2017, 14, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.Y.; Jeong, Y.J. Sodium Intake, Blood Pressure and Cardiovascular Disease. Korean Circ. J. 2020, 50, 555–571. [Google Scholar] [CrossRef]
- Li, X.; Alu, A.; Wei, Y.; Wei, X.; Luo, M. The Modulatory Effect of High Salt on Immune Cells and Related Diseases. Cell Prolif. 2022, 55, e13250. [Google Scholar] [CrossRef]
- Müller, D.N.; Wilck, N.; Haase, S.; Kleinewietfeld, M.; Linker, R.A. Sodium in the Microenvironment Regulates Immune Responses and Tissue Homeostasis. Nat. Rev. Immunol. 2019, 19, 243–254. [Google Scholar] [CrossRef]
- Wenstedt, E.F.E.; Verberk, S.G.S.; Kroon, J.; Neele, A.E.; Baardman, J.; Claessen, N.; Pasaoglu, Ö.T.; Rademaker, E.; Schrooten, E.M.; Wouda, R.D.; et al. Salt Increases Monocyte CCR2 Expression and Inflammatory Responses in Humans. Available online: https://insight.jci.org/articles/view/130508/sd/1 (accessed on 6 July 2022).
- Yi, B.; Titze, J.; Rykova, M.; Feuerecker, M.; Vassilieva, G.; Nichiporuk, I.; Schelling, G.; Morukov, B.; Choukèr, A. Effects of Dietary Salt Levels on Monocytic Cells and Immune Responses in Healthy Human Subjects: A Longitudinal Study. Transl. Res. 2015, 166, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri Barbaro, N.; Van Beusecum, J.; Xiao, L.; do Carmo, L.; Pitzer, A.; Loperena, R.; Foss, J.D.; Elijovich, F.; Laffer, C.L.; Montaniel, K.R.; et al. Sodium Activates Human Monocytes via the NADPH Oxidase and Isolevuglandin Formation. Cardiovasc. Res. 2021, 117, 1358–1371. [Google Scholar] [CrossRef] [PubMed]
- Hucke, S.; Eschborn, M.; Liebmann, M.; Herold, M.; Freise, N.; Engbers, A.; Ehling, P.; Meuth, S.G.; Roth, J.; Kuhlmann, T.; et al. Sodium Chloride Promotes Pro-Inflammatory Macrophage Polarization Thereby Aggravating CNS Autoimmunity. J. Autoimmun. 2016, 67, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-Responsive Gut Commensal Modulates TH17 Axis and Disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Dasari, R.; Bonsack, F.; Sukumari-Ramesh, S. Brain Injury and Repair after Intracerebral Hemorrhage: The Role of Microglia and Brain-Infiltrating Macrophages. Neurochem. Int. 2021, 142, 104923. [Google Scholar] [CrossRef] [PubMed]
- Herring, J.A.; Elison, W.S.; Tessem, J.S. Function of Nr4a Orphan Nuclear Receptors in Proliferation, Apoptosis and Fuel Utilization Across Tissues. Cells 2019, 8, 1373. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.P.; Crean, D. NR4A1-3 Nuclear Receptor Activity and Immune Cell Dysregulation in Rheumatic Diseases. Front. Med. 2022, 9, 874182. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.F.; Lei, X.; Haas, A.; Baylis, R.A.; Gao, H.; Luo, L.; Giordano, S.H.; Wehner, M.R.; Nead, K.T.; Leeper, N.J. Risk of Cancer After Diagnosis of Cardiovascular Disease. JACC CardioOncology 2023, 5, 431–440. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Fang, L.; Moore, X.-L.; Dart, A.M.; Wang, L.-M. Systemic Inflammatory Response Following Acute Myocardial Infarction. J. Geriatr. Cardiol. JGC 2015, 12, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Attalla, S.; Taifour, T.; Bui, T.; Muller, W. Insights from Transgenic Mouse Models of PyMT-Induced Breast Cancer: Recapitulating Human Breast Cancer Progression in Vivo. Oncogene 2021, 40, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Bouvain, P.; Ding, Z.; Kadir, S.; Kleimann, P.; Kluge, N.; Tiren, Z.-B.; Steckel, B.; Flocke, V.; Zalfen, R.; Petzsch, P.; et al. Non-Invasive Mapping of Systemic Neutrophil Dynamics upon Cardiovascular Injury. Nat. Cardiovasc. Res. 2023, 2, 126–143. [Google Scholar] [CrossRef]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct Vascular Channels Connect Skull Bone Marrow and the Brain Surface Enabling Myeloid Cell Migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef]
- Lee, C.Z.W.; Ginhoux, F. Biology of Resident Tissue Macrophages. Development 2022, 149, dev200270. [Google Scholar] [CrossRef] [PubMed]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate Immune Memory in the Brain Shapes Neurological Disease Hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, O.; Biesiekierska, M.; Panthu, B.; Vialichka, V.; Pirola, L.; Balcerczyk, A. The Epigenetic Profile of Tumor Endothelial Cells. Effects of Combined Therapy with Antiangiogenic and Epigenetic Drugs on Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2606. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Bhonde, R. Genetic and Epigenetic Stability of Stem Cells: Epigenetic Modifiers Modulate the Fate of Mesenchymal Stem Cells. Genomics 2020, 112, 3615–3623. [Google Scholar] [CrossRef]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef]
- Zurek, M.; Aavik, E.; Mallick, R.; Ylä-Herttuala, S. Epigenetic Regulation of Vascular Smooth Muscle Cell Phenotype Switching in Atherosclerotic Artery Remodeling: A Mini-Review. Front. Genet. 2021, 12, 719456. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient High Glucose Causes Persistent Epigenetic Changes and Altered Gene Expression during Subsequent Normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Schnack, L.; Sohrabi, Y.; Lagache, S.M.M.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. Mechanisms of Trained Innate Immunity in oxLDL Primed Human Coronary Smooth Muscle Cells. Front. Immunol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Joyner, M.J.; Green, D.J. Exercise Protects the Cardiovascular System: Effects beyond Traditional Risk Factors. J. Physiol. 2009, 587, 5551–5558. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, T.; Ren, J.; Xia, Y.; Onuma, A.; Wang, Y.; He, J.; Wu, J.; Wang, H.; Hamad, A.; et al. Pre-Operative Exercise Therapy Triggers Anti-Inflammatory Trained Immunity of Kupffer Cells through Metabolic Reprogramming. Nat. Metab. 2021, 3, 843–858. [Google Scholar] [CrossRef] [PubMed]


| Species | Cell Type | 1st Stimulus | 2nd Stimulus | Biological Effect | References |
|---|---|---|---|---|---|
| Human | Monocytes | oxLDL | TLR2 or TLR4 ligands |
| [35] |
| Human | PBMCs | Uric acid | TLR2 or TLR4 ligands |
| [36] |
| Human | Monocytes | Methylfumarate | LPS |
| [37] |
| Human | Monocytes | BCG | LPS |
| [38] |
| Human | Monocytes | Mevalonate | LPS |
| [39] |
| Human | Monocytes | Aldosterone | TLR2 or TLR4 ligands |
| [40] |
| Human | Monocytes | (Nor)adrenaline | LPS |
| [41] |
| Human | Monocytes | Plasmodium falciparum-infected red blood cells | LPS |
| [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleimann, P.; Irschfeld, L.-M.; Grandoch, M.; Flögel, U.; Temme, S. Trained Innate Immunity in Animal Models of Cardiovascular Diseases. Int. J. Mol. Sci. 2024, 25, 2312. https://doi.org/10.3390/ijms25042312
Kleimann P, Irschfeld L-M, Grandoch M, Flögel U, Temme S. Trained Innate Immunity in Animal Models of Cardiovascular Diseases. International Journal of Molecular Sciences. 2024; 25(4):2312. https://doi.org/10.3390/ijms25042312
Chicago/Turabian StyleKleimann, Patricia, Lisa-Marie Irschfeld, Maria Grandoch, Ulrich Flögel, and Sebastian Temme. 2024. "Trained Innate Immunity in Animal Models of Cardiovascular Diseases" International Journal of Molecular Sciences 25, no. 4: 2312. https://doi.org/10.3390/ijms25042312
APA StyleKleimann, P., Irschfeld, L. -M., Grandoch, M., Flögel, U., & Temme, S. (2024). Trained Innate Immunity in Animal Models of Cardiovascular Diseases. International Journal of Molecular Sciences, 25(4), 2312. https://doi.org/10.3390/ijms25042312