
γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae



Abstract
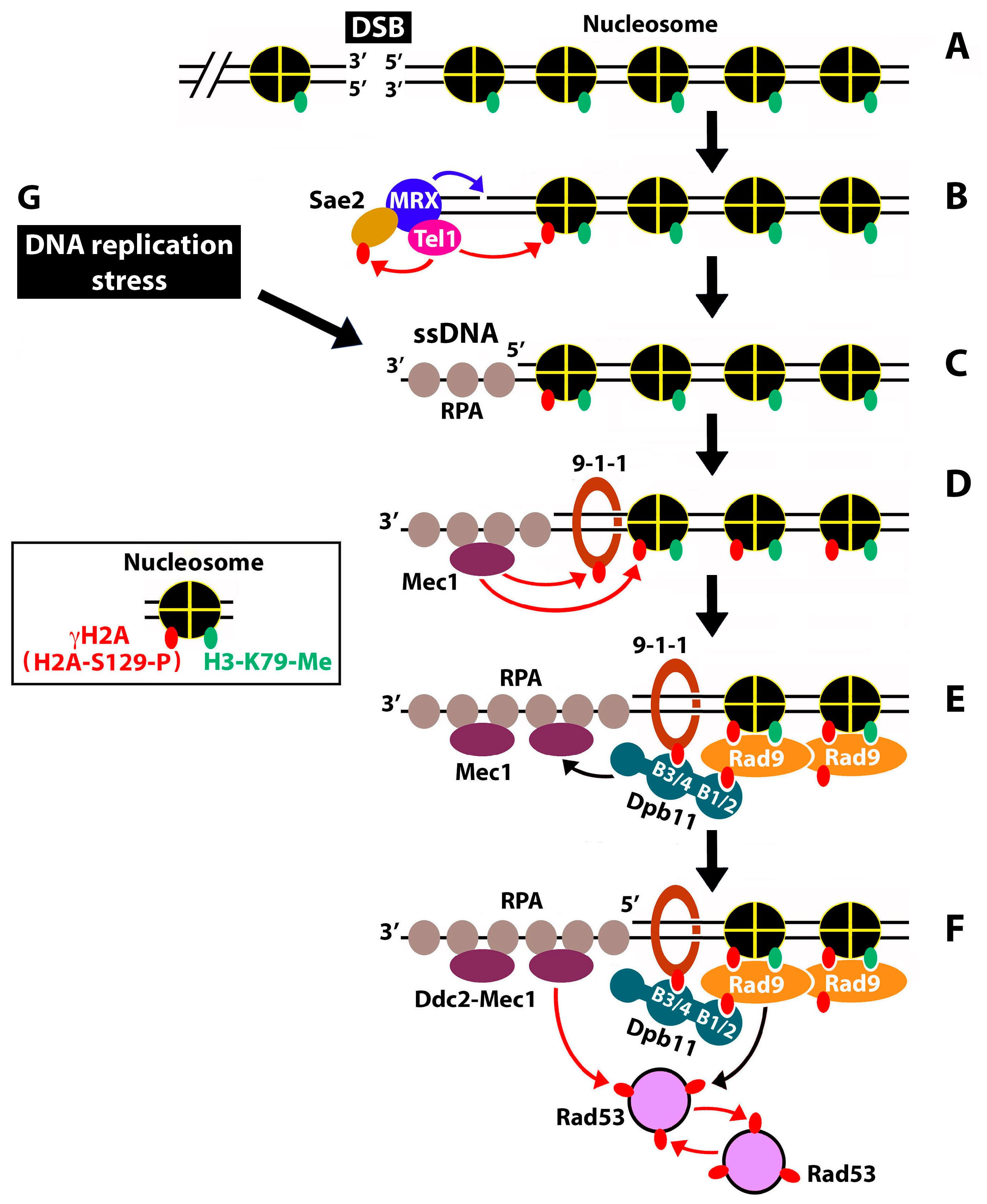
:1. Introduction
2. Results
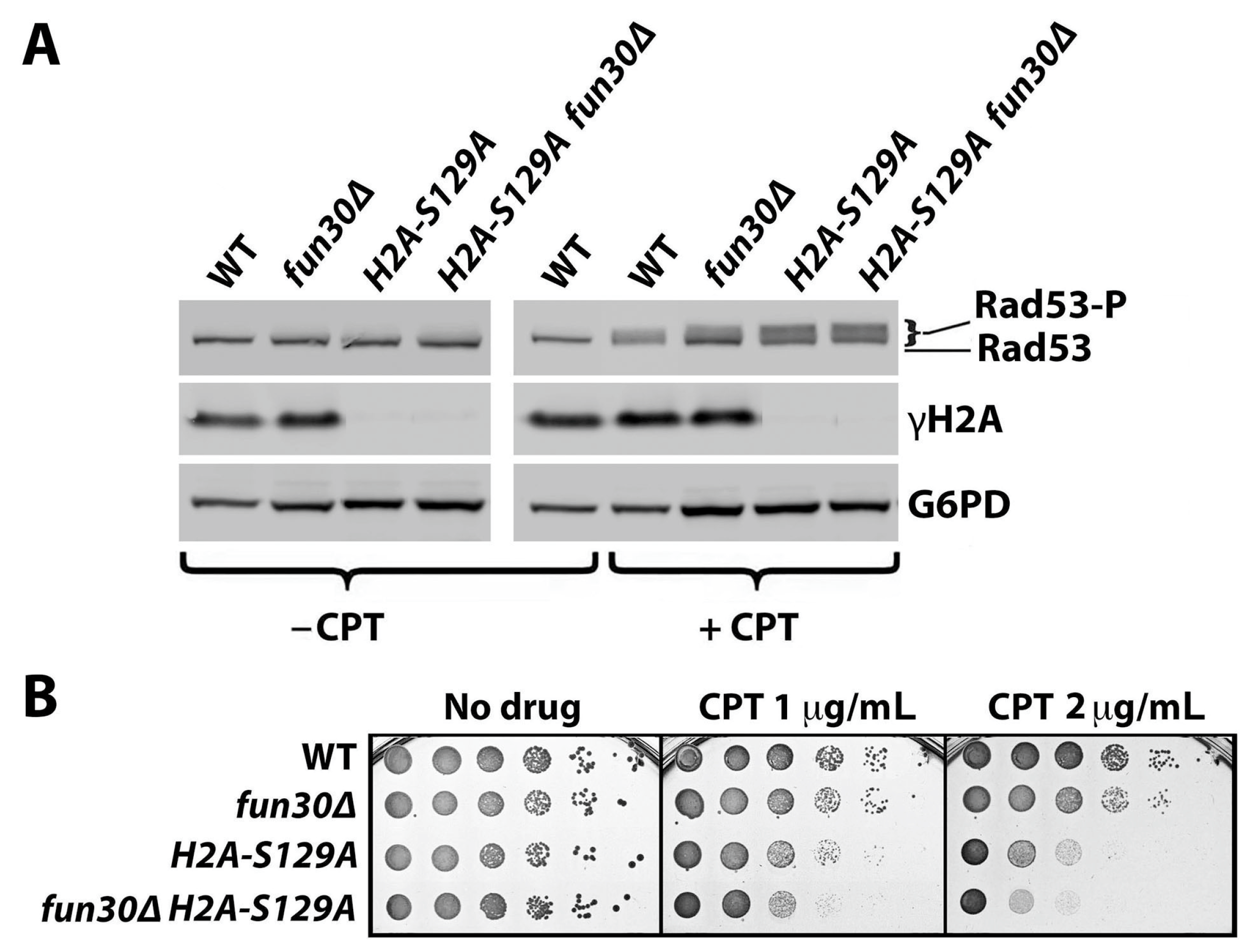
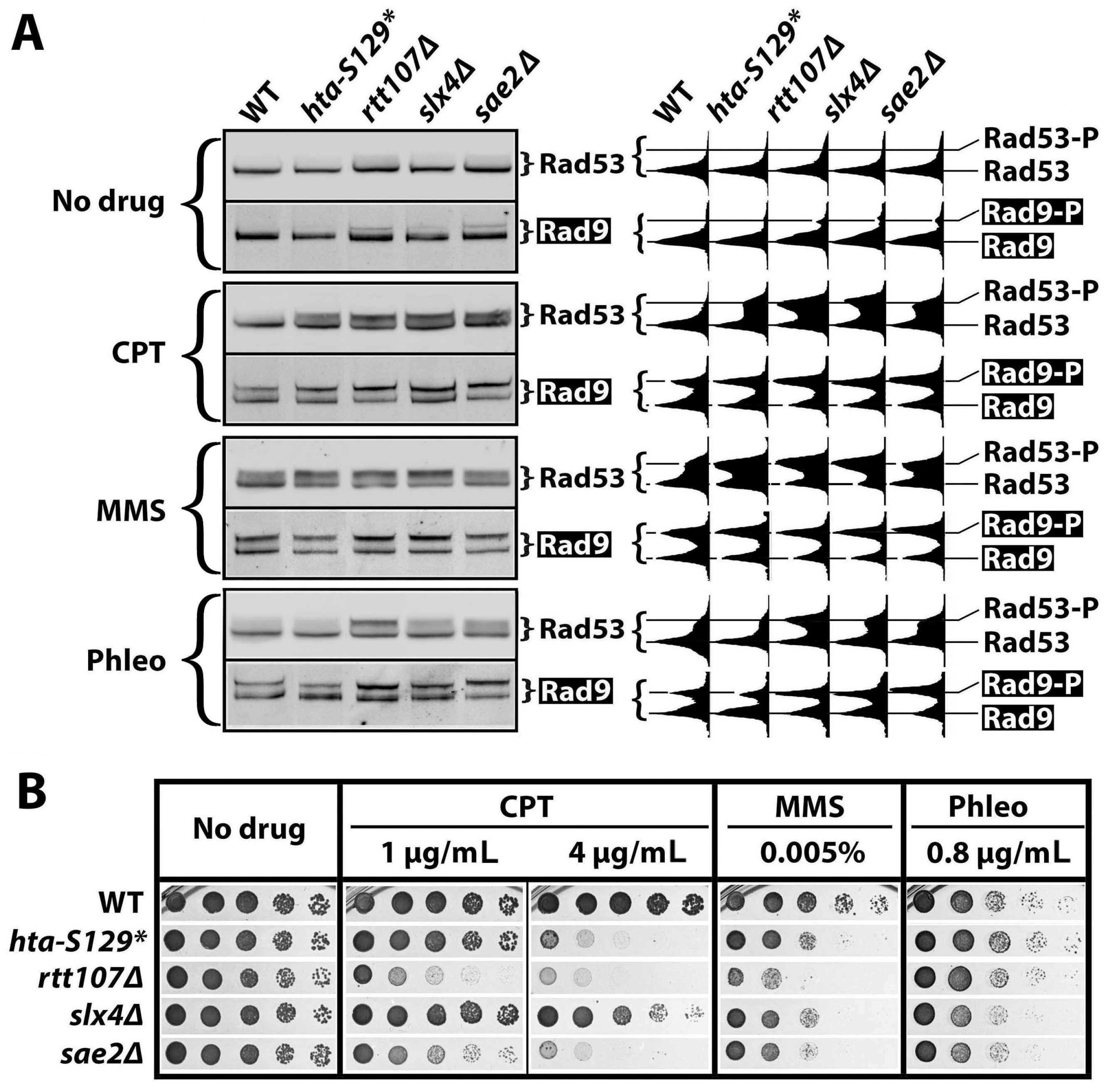
2.1. γH2A Negatively Regulates DNA Damage Signaling
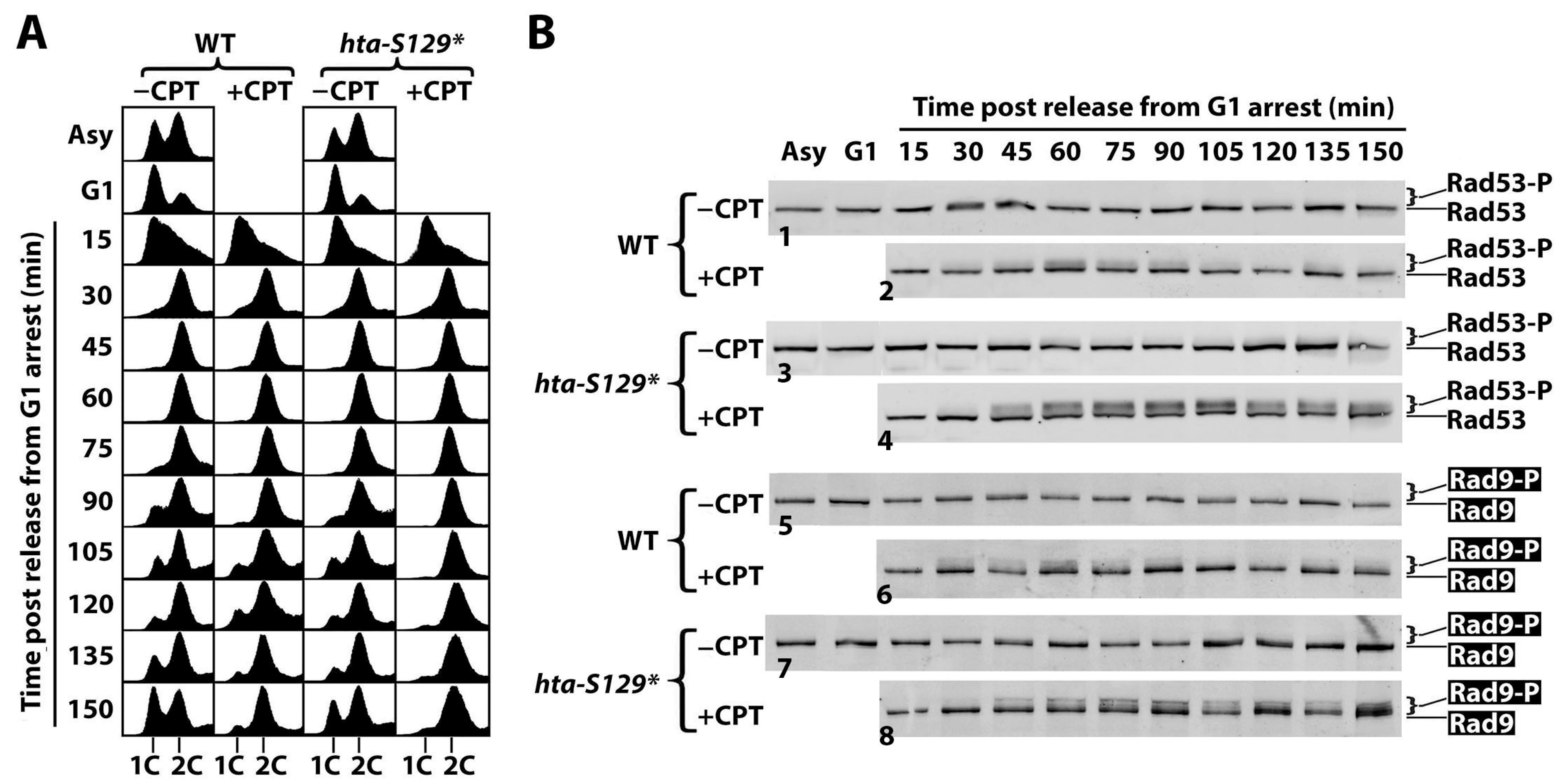
2.2. γH2A Controls CPT-Induced G2/M Checkpoint
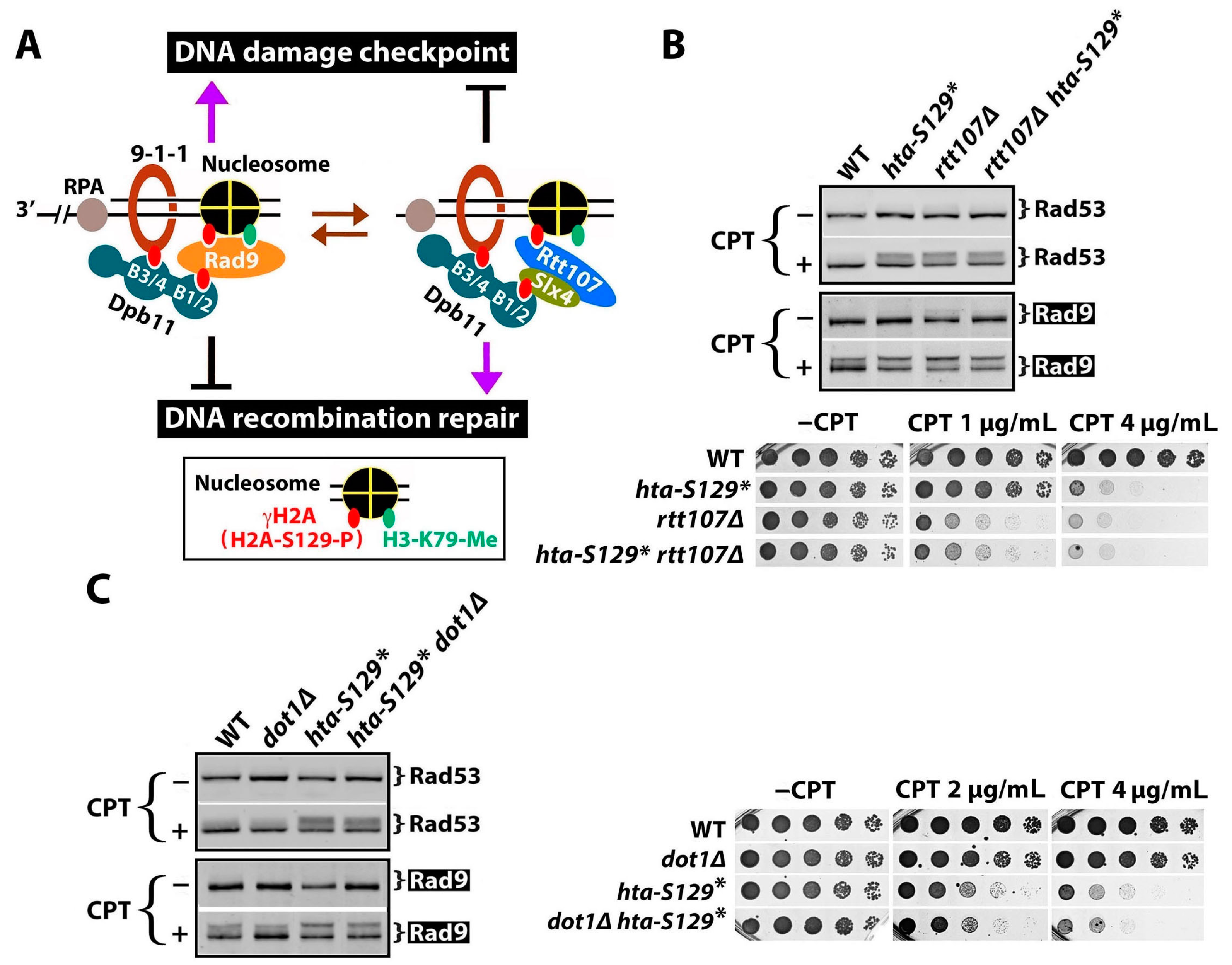
2.3. γH2A May Downregulate DDCs by Hindering Rad9 Recruitment to DNA Lesions
2.4. The Effect of γH2A on Checkpoint Signaling Is DNA Damage-Dependent
3. Discussion
4. Materials and Methods
4.1. Yeast Strains
4.2. Yeast Growth Phenotype Test
4.3. SDS-PAGE and Western Blotting
4.4. Fluorescence Activated Cell Sorting (FACS)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Pizzul, P.; Casari, E.; Gnugnoli, M.; Rinaldi, C.; Corallo, F.; Longhese, M.P. The DNA damage checkpoint: A tale from budding yeast. Front. Genet. 2022, 13, 995163. [Google Scholar] [CrossRef] [PubMed]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.M.; Tagle, D.A.; Shiloh, Y.; Collins, F.S.; Hieter, P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 1995, 82, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Desany, B.A.; Jones, W.J.; Liu, Q.; Wang, B.; Elledge, S.J. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 1996, 271, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Baldo, V.; Mantiero, D.; Lottersberger, F.; Lucchini, G.; Longhese, M.P. A Tel1/MRX-dependent checkpoint inhibits the metaphase-to-anaphase transition after UV irradiation in the absence of Mec1. Mol. Cell. Biol. 2004, 24, 10126–10144. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; Cassani, C.; Gobbini, E.; Bonetti, D.; Longhese, M.P. Coupling end resection with the checkpoint response at DNA double-strand breaks. Cell Mol. Life Sci. 2016, 73, 3655–3663. [Google Scholar] [CrossRef]
- Hustedt, N.; Gasser, S.M.; Shimada, K. Replication checkpoint: Tuning and coordination of replication forks in s phase. Genes 2013, 4, 388–434. [Google Scholar] [CrossRef] [PubMed]
- Gobbini, E.; Cassani, C.; Villa, M.; Bonetti, D.; Longhese, M.P. Functions and regulation of the MRX complex at DNA double-strand breaks. Microb. Cell 2016, 3, 329–337. [Google Scholar] [CrossRef]
- Fukunaga, K.; Kwon, Y.; Sung, P.; Sugimoto, K. Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol. Cell. Biol. 2011, 31, 1959–1971. [Google Scholar] [CrossRef]
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 2013, 12, 791–799. [Google Scholar] [CrossRef]
- Downs, J.A.; Lowndes, N.F.; Jackson, S.P. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000, 408, 1001–1004. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef]
- Foster, E.R.; Downs, J.A. Histone H2A phosphorylation in DNA double-strand break repair. FEBS J. 2005, 272, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Nagase, Y.; Tsubouchi, H.; Murakami-Murofushi, K.; Shibata, T.; Ohta, K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 1998, 17, 6412–6425. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Shim, E.Y.; Chung, W.H.; Nicolette, M.L.; Zhang, Y.; Davis, M.; Zhu, Z.; Paull, T.T.; Ira, G.; Lee, S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010, 29, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef]
- Paciotti, V.; Lucchini, G.; Plevani, P.; Longhese, M.P. Mec1p is essential for phosphorylation of the yeast DNA damage checkpoint protein Ddc1p, which physically interacts with Mec3p. EMBO J. 1998, 17, 4199–4209. [Google Scholar] [CrossRef]
- Mordes, D.A.; Nam, E.A.; Cortez, D. Dpb11 activates the Mec1-Ddc2 complex. Proc. Natl. Acad. Sci. USA 2008, 105, 18730–18734. [Google Scholar] [CrossRef] [PubMed]
- Cussiol, J.R.; Dibitetto, D.; Pellicioli, A.; Smolka, M.B. Slx4 scaffolding in homologous recombination and checkpoint control: Lessons from yeast. Chromosoma 2017, 126, 45–58. [Google Scholar] [CrossRef]
- Bantele, S.C.; Ferreira, P.; Gritenaite, D.; Boos, D.; Pfander, B. Targeting of the Fun30 nucleosome remodeller by the Dpb11 scaffold facilitates cell cycle-regulated DNA end resection. eLife 2017, 6, e21687. [Google Scholar] [CrossRef] [PubMed]
- Navadgi-Patil, V.M.; Burgers, P.M. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J. Biol. Chem. 2008, 283, 35853–35859. [Google Scholar] [CrossRef] [PubMed]
- Pfander, B.; Diffley, J.F. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 2011, 30, 4897–4907. [Google Scholar] [CrossRef] [PubMed]
- Hammet, A.; Magill, C.; Heierhorst, J.; Jackson, S.P. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007, 8, 851–857. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef]
- Vialard, J.E.; Gilbert, C.S.; Green, C.M.; Lowndes, N.F. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998, 17, 5679–5688. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Weng, S.; Zhou, H. Multiple phosphorylation of Rad9 by CDK is required for DNA damage checkpoint activation. Cell Cycle 2012, 11, 3792–3800. [Google Scholar] [CrossRef]
- Schwartz, M.F.; Duong, J.K.; Sun, Z.; Morrow, J.S.; Pradhan, D.; Stern, D.F. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol. Cell 2002, 9, 1055–1065. [Google Scholar] [CrossRef]
- Sweeney, F.D.; Yang, F.; Chi, A.; Shabanowitz, J.; Hunt, D.F.; Durocher, D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr. Biol. 2005, 15, 1364–1375. [Google Scholar] [CrossRef]
- Pellicioli, A.; Foiani, M. Signal transduction: How rad53 kinase is activated. Curr. Biol. 2005, 15, R769–R771. [Google Scholar] [CrossRef]
- Sanchez, Y.; Bachant, J.; Wang, H.; Hu, F.; Liu, D.; Tetzlaff, M.; Elledge, S.J. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999, 286, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Crabbé, L.; Pasero, P. Signaling pathways of replication stress in yeast. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Almeida, B.S.; Smolka, M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 2010, 39, 300–306. [Google Scholar] [CrossRef]
- Cussiol, J.R.; Jablonowski, C.M.; Yimit, A.; Brown, G.W.; Smolka, M.B. Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J. 2015, 34, 1704–1717. [Google Scholar] [CrossRef]
- Rozenzhak, S.; Mejía-Ramírez, E.; Williams, J.S.; Schaffer, L.; Hammond, J.A.; Head, S.R.; Russell, P. Rad3ATR decorates critical chromosomal domains with γH2A to protect genome integrity during S-phase in fission yeast. PLoS Genet. 2010, 6, e1001032. [Google Scholar] [CrossRef]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Liu, Y.; Ma, C.J.; Smolka, M.B. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 2013, 493, 120–124. [Google Scholar] [CrossRef]
- Siler, J.; Xia, B.; Wong, C.; Kath, M.; Bi, X. Cell cycle-dependent positive and negative functions of Fun30 chromatin remodeler in DNA damage response. DNA Repair 2017, 50, 61–70. [Google Scholar] [CrossRef]
- Avemann, K.; Knippers, R.; Koller, T.; Sogo, J.M. Camptothecin, a specific inhibitor of type I DNA topoisomerase, induces DNA breakage at replication forks. Mol. Cell. Biol. 1988, 8, 3026–3034. [Google Scholar]
- Sleigh, M.J. The mechanism of DNA breakage by phleomycin in vitro. Nucleic Acids Res. 1976, 3, 891–901. [Google Scholar] [CrossRef]
- Eapen, V.V.; Sugawara, N.; Tsabar, M.; Wu, W.H.; Haber, J.E. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell. Biol. 2012, 32, 4727–4740. [Google Scholar] [CrossRef]
- Javaheri, A.; Wysocki, R.; Jobin-Robitaille, O.; Altaf, M.; Côté, J.; Kron, S.J. Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc. Natl. Acad. Sci. USA 2006, 103, 13771–13776. [Google Scholar] [CrossRef]
- Princz, L.N.; Gritenaite, D.; Pfander, B. The Slx4-Dpb11 scaffold complex: Coordinating the response to replication fork stalling in S-phase and the subsequent mitosis. Cell Cycle 2015, 14, 488–4894. [Google Scholar] [CrossRef]
- di Cicco, G.; Bantele, S.C.S.; Reusswig, K.U.; Pfander, B. A cell cycle-independent mode of the Rad9-Dpb11 interaction is induced by DNA damage. Sci. Rep. 2017, 7, 11650. [Google Scholar] [CrossRef]
- Vidanes, G.M.; Bonilla, C.Y.; Toczyski, D.P. Complicated tails: Histone modifications and the DNA damage response. Cell 2005, 121, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, R.; Javaheri, A.; Allard, S.; Sha, F.; Côté, J.; Kron, S.J. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell. Biol. 2005, 25, 8430–8443. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 2006, 7, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.; Pilch, D.R.; Rogakou, E.P.; Orr, A.H.; Lowndes, N.F.; Bonner, W.M. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003, 4, 678–684. [Google Scholar] [CrossRef]
- Bi, X.; Yu, Q.; Siler, J.; Li, C.; Khan, A. Functions of Fun30 chromatin remodeler in regulating cellular resistance to genotoxic stress. PLoS ONE 2015, 10, e0121341. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Jackson, S.P. The Mre11 complex: At the crossroads of dna repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef]
- Haase, S.B.; Reed, S.I. Improved flow cytometric analysis of the budding yeast cell cycle. Cell Cycle 2002, 1, 132–136. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Name | Genotype | Source/Reference |
|---|---|---|---|
| 1 | W303-1A | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 rad5-535 | Ref. [44] |
| 2 | YXB1812-15 | W303-1A, fun30Δ::NatMX | This work |
| 3 | SKY2939 | W303-1A, hta1S129A::his3MX6 hta2S129A::TRP1 | Ref. [44] |
| 4 | YXB1812-18 | SKY2939, fun30Δ::NatMX | This work |
| 5 | QY364 | JKM139, RAD9-HA-KanMX6 | Ref. [44] |
| 6 | QY375 | QY364, hta1-S129*, hta2-S129* | Ref. [44] |
| 7 | YXB1812-24 | QY364, bar1Δ::TRP1 | This work |
| 8 | YXB1812-25 | QY375, bar1Δ::TRP1 | This work |
| 9 | YXB1812-36 | QY364, rtt107Δ::NatMX | This work |
| 10 | YXB1812-37 | QY375, rtt107Δ::NatMX | This work |
| 11 | YXB1812-38 | QY364, do1Δ::NatMX | This work |
| 12 | YXB1812-39 | QY375, dot1Δ::NatMX | This work |
| 13 | YXB1812-41 | QY364, slx4Δ::NatMX | This work |
| 14 | YXB1812-42 | QY364, sae2Δ::NatMX | This work |
| 15 | JKM139 | MATa hoΔhml::ADE1 hmr::ADE1 ade leu2–3,112 trp1::hisG lys5 ura3–52 ade3::GAL::HO1–100 | Ref. [43] |
| 16 | R726 | JKM139, hta1-S129A hta2-S129A | Ref. [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siler, J.; Guo, N.; Liu, Z.; Qin, Y.; Bi, X. γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2024, 25, 2462. https://doi.org/10.3390/ijms25052462
Siler J, Guo N, Liu Z, Qin Y, Bi X. γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2024; 25(5):2462. https://doi.org/10.3390/ijms25052462
Chicago/Turabian StyleSiler, Jasmine, Na Guo, Zhengfeng Liu, Yuhua Qin, and Xin Bi. 2024. "γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae" International Journal of Molecular Sciences 25, no. 5: 2462. https://doi.org/10.3390/ijms25052462
APA StyleSiler, J., Guo, N., Liu, Z., Qin, Y., & Bi, X. (2024). γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae. International Journal of Molecular Sciences, 25(5), 2462. https://doi.org/10.3390/ijms25052462