
Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Cationic Materials
2.1.1. (11-Hydroxyundecyl) Triphenyl Phosphonium Bromide (1)
2.1.2. (Z/E)-11-Tetradecen-1-ol (2)
2.1.3. (Z/E)-11-tetradecenyl-1-Mesylate (3)
2.1.4. N,N-Dimethyl-2,3-bis(tetradec-11-enyloxy)propylamine (4)
2.1.5. N,N,N-Trimethyl-2,3-bis(tetradec-11-enyloxy)propylammonium Iodide (5)
2.1.6. N,N,N-Trimethyl-2,3-bis(tetradec-11-enyloxy)propylammonium Chloride (6)
2.2. Non-Aqueous Potentiometric Titration of 1 and 6
2.3. Antibacterial Properties
2.3.1. Antibacterial Activity of 1 and 6 by Determination of MIC Values (MICs)
2.3.2. Time-Killing Curves
2.4. Cytotoxicity Experiments on Human Liver Cells
Concentration-Dependent Cytotoxicity Test
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. Preparation of a Butyl Lithium (BuLi) Solution in Et2O
3.3. (11-Hydroxyundecyl) tri-Phenyl Phosphonium Bromide (1)
3.4. (Z/E)-11-Tetradecen-1-ol (2)
3.5. (Z/E)-11-Tetradecenyl-1-mesylate (3)
3.6. N,N-Dimethyl-2,3-bis(tetradec-11-enyloxy)propylamine (4)
3.7. N,N,N-Trimethyl-2,3-bis(tetradec-11-enyloxy)propylammonium Iodide (5)
3.8. N,N,N-Trimethyl-2,3-bis(tetradec-11-enyloxy)propylammonium Chloride (6)
3.9. Potentiometric Titrations of 1 and 6
Preparation of a 0.1 M Perchloric Acid Volumetric Solution
3.10. Microbiology
3.10.1. Microorganisms
3.10.2. Determination of the MICs
3.10.3. Killing Curves
3.11. MTT Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- My Personal Trailer. Available online: https://www.my-personaltrainer.it/farmaci/citrosil.html (accessed on 15 April 2021).
- Zhou, Z.; Zhou, S.; Zhang, X.; Zeng, S.; Xu, Y.; Nie, W.; Zhou, Y.; Xu, T.; Chen, P. Quaternary Ammonium Salts: Insights into Synthesis and New Directions in Antibacterial Applications. Bioconjug. Chem. 2023, 34, 302–325. [Google Scholar] [CrossRef]
- Lei, Y.; Zhou, S.; Dong, C.; Zhang, A.; Lin, Y. PDMS Tri-Block Copolymers Bearing Quaternary Ammonium Salts for Epidermal Antimicrobial Agents: Synthesis, Surface Adsorption and Non-Skin-Penetration. React. Funct. Polym. 2018, 124, 20–28. [Google Scholar] [CrossRef]
- Alfei, S. Shifting from Ammonium to Phosphonium Salts: A Promising Strategy to Develop Next-Generation Weapons against Biofilms. Pharmaceutics 2024, 16, 80. [Google Scholar] [CrossRef]
- Alfei, S. Cationic Materials for Gene Therapy: A Look Back to the Birth and Development of 2,2-Bis-(Hydroxymethyl)Propanoic Acid-Based Dendrimer Scaffolds. Int. J. Mol. Sci. 2023, 24, 16006. [Google Scholar] [CrossRef]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Alfei, S. Broad-Spectrum Bactericidal Activity of a Synthetic Random Copolymer Based on 2-Methoxy-6-(4-Vinylbenzyloxy)-Benzylammonium Hydrochloride. Int. J. Mol. Sci. 2021, 22, 5021. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.C.; Minbiole, K.P.C.; Wuest, W.M. Quaternary Ammonium Compounds: An Antimicrobial Mainstay and Platform for Innovation to Address Bacterial Resistance. ACS Infect. Dis. 2015, 1, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Niu, L.; Ma, S.; Li, J.; Tay, F.R.; Chen, J. Quaternary Ammonium-Based Biomedical Materials: State-of-the-Art, Toxicological Aspects and Antimicrobial Resistance. Prog. Polym. Sci. 2017, 71, 53–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cui, F.; Zeng, G.; Jiang, M.; Yang, Z.; Yu, Z.; Zhu, M.; Shen, L. Quaternary Ammonium Compounds (QACs): A Review on Occurrence, Fate and Toxicity in the Environment. Sci. Total Environ. 2015, 518–519, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Conejo-García, A.; Pisani, L.; del Carmen Núñez, M.; Catto, M.; Nicolotti, O.; Leonetti, F.; Campos, J.M.; Gallo, M.A.; Espinosa, A.; Carotti, A. Homodimeric Bis-Quaternary Heterocyclic Ammonium Salts as Potent Acetyl- and Butyrylcholinesterase Inhibitors: A Systematic Investigation of the Influence of Linker and Cationic Heads over Affinity and Selectivity. J. Med. Chem. 2011, 54, 2627–2645. [Google Scholar] [CrossRef] [PubMed]
- Kwaśniewska, D.; Chen, Y.-L.; Wieczorek, D. Biological Activity of Quaternary Ammonium Salts and Their Derivatives. Pathogens 2020, 9, 459. [Google Scholar] [CrossRef]
- Ye, X.; Qin, X.; Yan, X.; Guo, J.; Huang, L.; Chen, D.; Wu, T.; Shi, Q.; Tan, S.; Cai, X. π–π Conjugations Improve the Long-Term Antibacterial Properties of Graphene Oxide/Quaternary Ammonium Salt Nanocomposites. Chem. Eng. J. 2016, 304, 873–881. [Google Scholar] [CrossRef]
- Lin, W.; Niu, B.; Yi, J.; Deng, Z.; Song, J.; Chen, Q. Toxicity and Metal Corrosion of Glutaraldehyde-Didecyldimethylammonium Bromide as a Disinfectant Agent. Biomed. Res. Int. 2018, 2018, 9814209. [Google Scholar] [CrossRef] [PubMed]
- Jantafong, T.; Ruenphet, S.; Punyadarsaniya, D.; Takehara, K. The Study of Effect of Didecyl Dimethyl Ammonium Bromide on Bacterial and Viral Decontamination for Biosecurity in the Animal Farm. Vet. World 2018, 11, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Zheng, X.; Li, Z.; Wang, M.; Luo, K.; Zhang, C.; Xia, X.; Wang, Y.; Shi, C. Didecyldimethylammonium Bromide: Application to Control Biofilms of Staphylococcus Aureus and Pseudomonas Aeruginosa Alone and in Combination with Slightly Acidic Electrolyzed Water. Food Res. Int. 2022, 157, 111236. [Google Scholar] [CrossRef] [PubMed]
- Seferyan, M.A.; Saverina, E.A.; Frolov, N.A.; Detusheva, E.V.; Kamanina, O.A.; Arlyapov, V.A.; Ostashevskaya, I.I.; Ananikov, V.P.; Vereshchagin, A.N. Multicationic Quaternary Ammonium Compounds: A Framework for Combating Bacterial Resistance. ACS Infect. Dis. 2023, 9, 1206–1220. [Google Scholar] [CrossRef]
- Jennings, M.C.; Buttaro, B.A.; Minbiole, K.P.C.; Wuest, W.M. Bioorganic Investigation of Multicationic Antimicrobials to Combat QAC-Resistant Staphylococcus aureus. ACS Infect. Dis. 2015, 1, 304–309. [Google Scholar] [CrossRef]
- Al-Khalifa, S.E.; Jennings, M.C.; Wuest, W.M.; Minbiole, K.P.C. The Development of Next-Generation Pyridinium-Based MultiQAC Antiseptics. ChemMedChem 2017, 12, 280–283. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Minaeva, A.P.; Egorov, M.P. Synthesis and Antibacterial Activity of New Tetrakisquaternary Ammonium Compounds Based on Pentaerythritol and 3-Hydroxypyridine. Russ. Chem. Bull. 2021, 70, 545–548. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Frolov, N.A.; Minaeva, A.P.; Detusheva, E.V.; Derkach, Y.V.; Egorov, M.P. Synthesis and Biological Evaluation of Novel Cyanuric Acid-Tethered Tris-Pyridinium Derivatives. Mendeleev Commun. 2021, 31, 368–369. [Google Scholar] [CrossRef]
- Pugachev, M.V.; Shtyrlin, N.V.; Sysoeva, L.P.; Nikitina, E.V.; Abdullin, T.I.; Iksanova, A.G.; Ilaeva, A.A.; Musin, R.Z.; Berdnikov, E.A.; Shtyrlin, Y.G. Synthesis and Antibacterial Activity of Novel Phosphonium Salts on the Basis of Pyridoxine. Bioorg. Med. Chem. 2013, 21, 4388–4395. [Google Scholar] [CrossRef]
- Cieniecka-Rosłonkiewicz, A.; Pernak, J.; Kubis-Feder, J.; Ramani, A.; Robertson, A.J.; Seddon, K.R. Synthesis, Anti-Microbial Activities and Anti-Electrostatic Properties of Phosphonium-Based Ionic Liquids. Green. Chem. 2005, 7, 855. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M. Positively Charged Polymers as Promising Devices against Multidrug Resistant Gram-Negative Bacteria: A Review. Polymers 2020, 12, 1195. [Google Scholar] [CrossRef] [PubMed]
- Ermolaev, V.V.; Arkhipova, D.M.; Miluykov, V.A.; Lyubina, A.P.; Amerhanova, S.K.; Kulik, N.V.; Voloshina, A.D.; Ananikov, V.P. Sterically Hindered Quaternary Phosphonium Salts (QPSs): Antimicrobial Activity and Hemolytic and Cytotoxic Properties. Int. J. Mol. Sci. 2021, 23, 86. [Google Scholar] [CrossRef] [PubMed]
- Castellaro, S. Materiali per Terapia Genica. Doctoral Thesis, Università di Genova, Genova, Italy, 2013. [Google Scholar]
- Irvine, S.A.; Meng, Q.-H.; Afzal, F.; Ho, J.; Wong, J.B.; Hailes, H.C.; Tabor, A.B.; McEwan, J.R.; Hart, S.L. Receptor-Targeted Nanocomplexes Optimized for Gene Transfer to Primary Vascular Cells and Explant Cultures of Rabbit Aorta. Mol. Ther. 2008, 16, 508–515. [Google Scholar] [CrossRef]
- Kudsiova, L.; Ho, J.; Fridrich, B.; Harvey, R.; Keppler, M.; Ng, T.; Hart, S.L.; Tabor, A.B.; Hailes, H.C.; Lawrence, M.J. Lipid Chain Geometry of C14 Glycerol-Based Lipids: Effect on Lipoplex Structure and Transfection. Mol. BioSyst. 2011, 7, 422–436. [Google Scholar] [CrossRef]
- Avalos, F.; Ortiz, J.; Zitzumbo, R.; Lopezmanchado, M.; Verdejo, R.; Arroyo, M. Phosphonium Salt Intercalated Montmorillonites. Appl. Clay Sci. 2009, 43, 27–32. [Google Scholar] [CrossRef]
- Li, Q.; Hu, Y.; Zhang, B. Phosphonium-Based Ionic Liquids as Antifungal Agents for Conservation of Heritage Sandstone. RSC Adv. 2022, 12, 1922–1931. [Google Scholar] [CrossRef]
- Li, J.-M.; Yong, J.-P.; Aisa, H.A. Synthesis of (Z/E)-11-Tetradecen-1-Ol, a Component of Ostrinia Nubilalis Sex Pheromone. Chem. Nat. Compd. 2008, 44, 224–226. [Google Scholar] [CrossRef]
- Roosjen, A.; Šmisterová, J.; Driessen, C.; Anders, J.T.; Wagenaar, A.; Hoekstra, D.; Hulst, R.; Engberts, J.B.F.N. Synthesis and Characteristics of Biodegradable Pyridinium Amphiphiles Used for in Vitro DNA Delivery. Eur. J. Org. Chem. 2002, 2002, 1271–1277. [Google Scholar] [CrossRef]
- Hurley, C.A.; Wong, J.B.; Hailes, H.C.; Tabor, A.B. Asymmetric Synthesis of Dialkyloxy-3-Alkylammonium Cationic Lipids. J. Org. Chem. 2004, 69, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Kratochvil, B. Titrations in Nonaqueous Solvents. Anal. Chem. 1982, 54, 105–121. [Google Scholar] [CrossRef]
- Seher, A. Dr. I. Gyenes, C. Sc. (Chim.), Titrationen in Nichtwäßrigen Medien, 3. Neubearb. u. Erg. Aufl., 701 S., 206 Abb., 108 Tab., Gln., Ferdinand Enke Verlag, Stuttgart 1970, Preis: 84.—DM. Fette Seifen Anstrichm. 1973, 75, 232. [Google Scholar] [CrossRef]
- Šafařík, L.; Stránský, Z.; Svehla, G.; Burns, D.T. Titrimetric Analysis in Organic Solvents (Comprehensive Analytical Chemistry, Vol. XXII). Anal. Chim. Acta 1987, 201, 367. [Google Scholar] [CrossRef]
- Pifer, C.W.; Wollish, E.G. Potentiometric Titration of Salts of Organic Bases in Acetic Acid. Anal. Chem. 1952, 24, 300–306. [Google Scholar] [CrossRef]
- Mascellani, G.; Casalini, C. Use of Mercuric Acetate in Potentiometric Titrations in a Nonaqueous Medium. Anal. Chem. 1975, 47, 2468–2470. [Google Scholar] [CrossRef]
- Alfei, S.; Castellaro, S.; Taptue, G.B. Synthesis and NMR Characterization of Dendrimers Based on 2, 2-Bis-(Hydroxymethyl)-Propanoic Acid (Bis-HMPA) Containing Peripheral Amino Acid Residues for Gene Transfection. Org. Commun. 2017, 10, 144–177. [Google Scholar] [CrossRef]
- Alfei, S.; Castellaro, S. Synthesis and Characterization of Polyester-Based Dendrimers Containing Peripheral Arginine or Mixed Amino Acids as Potential Vectors for Gene and Drug Delivery. Macromol. Res. 2017, 25, 1172–1186. [Google Scholar] [CrossRef]
- Alfei, S.; Taptue, G.B.; Catena, S.; Bisio, A. Synthesis of Water-Soluble, Polyester-Based Dendrimer Prodrugs for Exploiting Therapeutic Properties of Two Triterpenoid Acids. Chin. J. Polym. Sci. 2018, 36, 999–1010. [Google Scholar] [CrossRef]
- Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles. Nanomaterials 2022, 12, 2743. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic Resistance of Bacterial Biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef]
- Shi, L.-W.; Zhuang, Q.-Q.; Wang, T.-Q.; Jiang, X.-D.; Liu, Y.; Deng, J.-W.; Sun, H.-H.; Li, Y.; Li, H.-H.; Liu, T.-B.; et al. Synthetic Antibacterial Quaternary Phosphorus Salts Promote Methicillin-Resistant Staphylococcus Aureus-Infected Wound Healing. Int. J. Nanomed. 2023, 18, 1145–1158. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, L.; Hou, P.; Liu, J.; Fu, S. Design, Synthesis, Antibacterial, and Antitumor Activity of Linear Polyisocyanide Quaternary Ammonium Salts with Different Structures and Chain Lengths. Molecules 2021, 26, 5686. [Google Scholar] [CrossRef]
- Gurusamy, K.S.; Koti, R.; Toon, C.D.; Wilson, P.; Davidson, B.R. Antibiotic Therapy for the Treatment of Methicillin-Resistant Staphylococcus Aureus (MRSA) in Non Surgical Wounds. In Cochrane Database of Systematic Reviews; Gurusamy, K.S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2013. [Google Scholar]
- Rosa, T.F.; Coelho, S.S.; Foletto, V.S.; Bottega, A.; Serafin, M.B.; Machado, C.d.S.; Franco, L.N.; Paula, B.R.; Hörner, R. Alternatives for the Treatment of Infections Caused by ESKAPE Pathogens. J. Clin. Pharm. Ther. 2020, 45, 863–873. [Google Scholar] [CrossRef]
- Alfei, S.; Brullo, C.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, A.M. Pyrazole-Based Water-Soluble Dendrimer Nanoparticles as a Potential New Agent against Staphylococci. Biomedicines 2021, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Bozdogan, B. Antibacterial Susceptibility of a Vancomycin-Resistant Staphylococcus Aureus Strain Isolated at the Hershey Medical Center. J. Antimicrob. Chemother. 2003, 52, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Tsiodras, S.; Gold, H.S.; Sakoulas, G.; Eliopoulos, G.M.; Wennersten, C.; Venkataraman, L.; Moellering, R.C.; Ferraro, M.J. Linezolid Resistance in a Clinical Isolate of Staphylococcus Aureus. Lancet 2001, 358, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin-Resistant Staphylococcus Aureus Infections in Adults and Children. Clin. Infect. Dis. 2011, 52, e18–e55. [Google Scholar] [CrossRef] [PubMed]
- Nour El-Din, H.T.; Yassin, A.S.; Ragab, Y.M.; Hashem, A.M. Phenotype-Genotype Characterization and Antibiotic-Resistance Correlations Among Colonizing and Infectious Methicillin-Resistant Staphylococcus Aureus Recovered from Intensive Care Units. Infect. Drug Resist. 2021, 14, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Tolosa, L.; Gómez-Lechón, M.J. Culture and Functional Characterization of Human Hepatoma HepG2 Cells. Methods Mol. Biol. 2015, 1250, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Gómez-Lechón, M.J.; López, S.; Guzmán, C.; Castell, J.V.; Donato, M.T.; Jover, R. Human Upcyte Hepatocytes: Characterization of the Hepatic Phenotype and Evaluation for Acute and Long-Term Hepatotoxicity Routine Testing. Toxicol. Sci. 2016, 152, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Peña-Morán, O.; Villarreal, M.; Álvarez-Berber, L.; Meneses-Acosta, A.; Rodríguez-López, V. Cytotoxicity, Post-Treatment Recovery, and Selectivity Analysis of Naturally Occurring Podophyllotoxins from Bursera Fagaroides Var. Fagaroides on Breast Cancer Cell Lines. Molecules 2016, 21, 1013. [Google Scholar] [CrossRef] [PubMed]
- Awouafack, M.D.; McGaw, L.J.; Gottfried, S.; Mbouangouere, R.; Tane, P.; Spiteller, M.; Eloff, J.N. Antimicrobial Activity and Cytotoxicity of the Ethanol Extract, Fractions and Eight Compounds Isolated from Eriosema Robustum (Fabaceae). BMC Complement. Altern. Med. 2013, 13, 289. [Google Scholar] [CrossRef] [PubMed]
- Weerapreeyakul, N.; Nonpunya, A.; Barusrux, S.; Thitimetharoch, T.; Sripanidkulchai, B. Evaluation of the Anticancer Potential of Six Herbs against a Hepatoma Cell Line. Chin. Med. 2012, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Adamu, M.; Naidoo, V.; Eloff, J.N. Efficacy and Toxicity of Thirteen Plant Leaf Acetone Extracts Used in Ethnoveterinary Medicine in South Africa on Egg Hatching and Larval Development of Haemonchus Contortus. BMC Vet. Res. 2013, 9, 38. [Google Scholar] [CrossRef]
- Adamu, M.; Naidoo, V.; Eloff, J.N. The Antibacterial Activity, Antioxidant Activity and Selectivity Index of Leaf Extracts of Thirteen South African Tree Species Used in Ethnoveterinary Medicine to Treat Helminth Infections. BMC Vet. Res. 2014, 10, 52. [Google Scholar] [CrossRef]
- Famuyide, I.M.; Aro, A.O.; Fasina, F.O.; Eloff, J.N.; McGaw, L.J. Antibacterial and Antibiofilm Activity of Acetone Leaf Extracts of Nine Under-Investigated South African Eugenia and Syzygium (Myrtaceae) Species and Their Selectivity Indices. BMC Complement. Altern. Med. 2019, 19, 141. [Google Scholar] [CrossRef]
- Nogueira, F.; do Rosário, V.E. Methods for Assessment of Antimalarial Activity in the Different Phases of the Plasmodium Life Cycle. Rev. Panamazonica Saude 2010, 1, 109–124. [Google Scholar] [CrossRef]
- EUCAST. European Committee on Antimicrobial Susceptibility Testing. Available online: https://www.eucast.org/ast_of_bacteria/ (accessed on 20 January 2024).
- Schito, A.M.; Piatti, G.; Stauder, M.; Bisio, A.; Giacomelli, E.; Romussi, G.; Pruzzo, C. Effects of Demethylfruticuline A and Fruticuline A from Salvia Corrugata Vahl. on Biofilm Production in Vitro by Multiresistant Strains of Staphylococcus aureus, Staphylococcus epidermidis and Enterococcus faecalis. Int. J. Antimicrob. Agents 2011, 37, 129–134. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
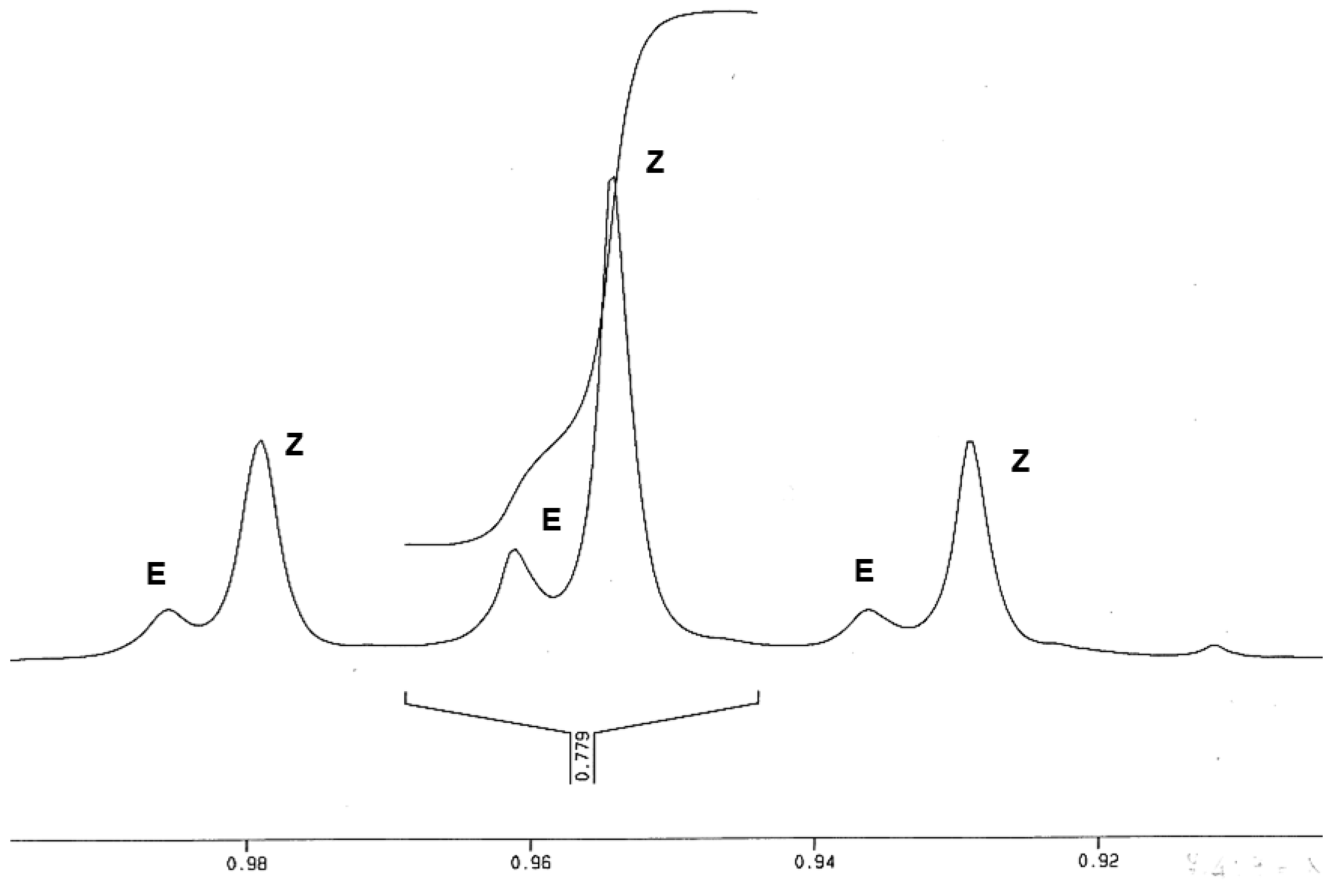{kind=link}
{kind=link}
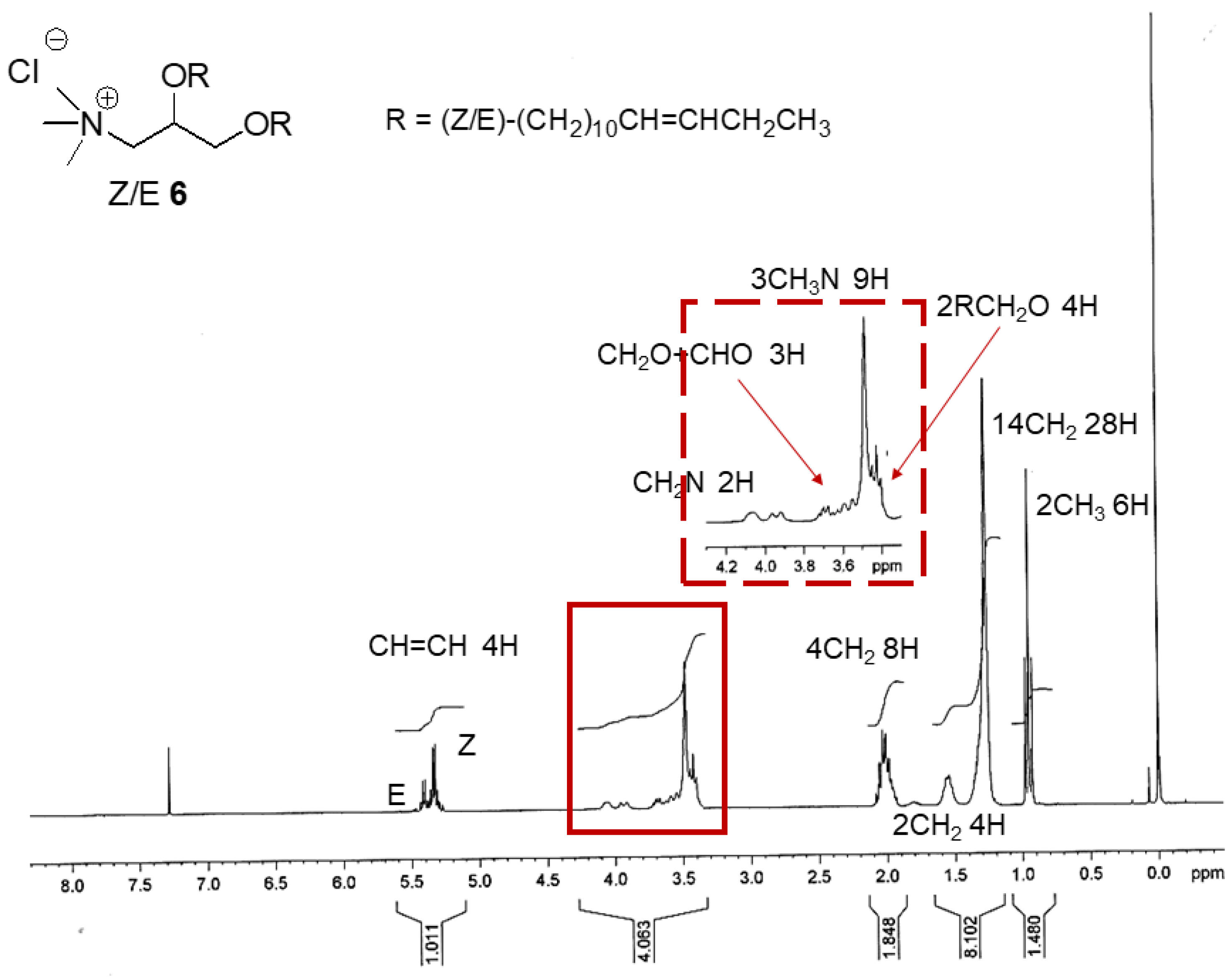{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Weight (mg) | * MW | * NH3+ (mmol) | 0.1 N HClO4 (mL) | ** NH3+ (mmol) | ** MW | *** Residuals | Error (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 280.0 | 513.5 | 0.5453 | 5.45 | 0.5450 | 513.8 | 0.3 | 0.06 |
| 6 | 245.5 | 558.4 | 0.4396 | 4.44 | 0.4390 | 559.2 | 0.8 | 0.14 |
| Strains | 1 (513.5) 1 | 6 (558.376) 1 |
|---|---|---|
| MIC µg/mL | MIC µg/mL | |
| E. faecalis * | 8 | 16 |
| E. faecium * | 8 | 4 |
| S. aureus ** | 4 | 32 |
| S. epidermidis *** | 4 | 32 |
| E. coli | >128 | >128 |
| P. Aeruginosa | >128 | >128 |
| Strains | 1 (513.5) 1 | 6 (558.376) 1 | Reference Antibiotics |
|---|---|---|---|
| MIC µg/mL | MIC µg/mL | MIC µg/mL | |
| E. faecalis 1 VRE * | 16 | 32 | 256 2 |
| E. faecalis 356 VRE * | 16 | 128 | 256 2 |
| E. faecalis 425 VRE | 8 | 32 | 128 2 |
| E. faecalis 432 VRE | 16 | 16 | 128 2 |
| E. faecium 152 VRE | 8 | 8 | 128 2 |
| E. faecium 182 VRE | 8 | 8 | 256 2 |
| E. faecium 364 VRE | 8 | 4 | 256 2 |
| E. faecium 479 VRE | 16 | 32 | 256 2 |
| S. aureus 17 MRSA | 4 | 64 | 256 3 |
| S. aureus 18 MRSA | 4 | 32 | 256 3 |
| S. aureus 187 MRSA | 8 | 64 | 256 3 |
| S. aureus 195 MRSA | 4 | 64 | 256 3 |
| S. epidermidis 22 MRSE | 4 | 32 | 256 3 |
| S. epidermidis 180 MRSE | 8 | 32 | 128 3 |
| S. epidermidis 274 MRSE | 8 | 32 | 256 3 |
| S. epidermidis 275 MRSE | 8 | 64 | 256 3 |
| Strains | 1 (27.95 µg/mL) 1 | SI | 6 (30.80 µg/mL) 1 | SI |
|---|---|---|---|---|
| MIC µg/mL | MIC µg/mL | |||
| E. faecalis 1 VRE * | 16 | 1.75 | 32 | 0.96 |
| E. faecalis 356 VRE * | 16 | 1.75 | 128 | 0.24 |
| E. faecalis 425 VRE | 8 | 3.50 | 32 | 0.96 |
| E. faecalis 432 VRE | 16 | 1.75 | 16 | 1.93 |
| E. faecium 152 VRE | 8 | 3.50 | 8 | 3.85 |
| E. faecium 182 VRE | 8 | 3.50 | 8 | 3.85 |
| E. faecium 364 VRE | 8 | 3.50 | 4 | 7.7 |
| E. faecium 479 VRE | 16 | 1.75 | 32 | 0.96 |
| S. aureus 17 MRSA | 4 | 7.00 | 64 | 0.48 |
| S. aureus 18 MRSA | 4 | 7.00 | 32 | 0.96 |
| S. aureus 187 MRSA | 8 | 3.50 | 64 | 0.48 |
| S. aureus 195 MRSA | 4 | 7.00 | 64 | 0.48 |
| S. epidermidis 22 MRSE | 4 | 7.00 | 32 | 0.96 |
| S. epidermidis 180 MRSE | 8 | 3.50 | 32 | 0.96 |
| S. epidermidis 274 MRSE | 8 | 3.50 | 32 | 0.96 |
| S. epidermidis 275 MRSE | 8 | 3.50 | 64 | 0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacchetti, F.; Schito, A.M.; Milanese, M.; Castellaro, S.; Alfei, S. Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt. Int. J. Mol. Sci. 2024, 25, 2761. https://doi.org/10.3390/ijms25052761
Bacchetti F, Schito AM, Milanese M, Castellaro S, Alfei S. Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt. International Journal of Molecular Sciences. 2024; 25(5):2761. https://doi.org/10.3390/ijms25052761
Chicago/Turabian StyleBacchetti, Francesca, Anna Maria Schito, Marco Milanese, Sara Castellaro, and Silvana Alfei. 2024. "Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt" International Journal of Molecular Sciences 25, no. 5: 2761. https://doi.org/10.3390/ijms25052761
APA StyleBacchetti, F., Schito, A. M., Milanese, M., Castellaro, S., & Alfei, S. (2024). Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt. International Journal of Molecular Sciences, 25(5), 2761. https://doi.org/10.3390/ijms25052761