
Advances in HIV Gene Therapy


Abstract
:1. Introduction
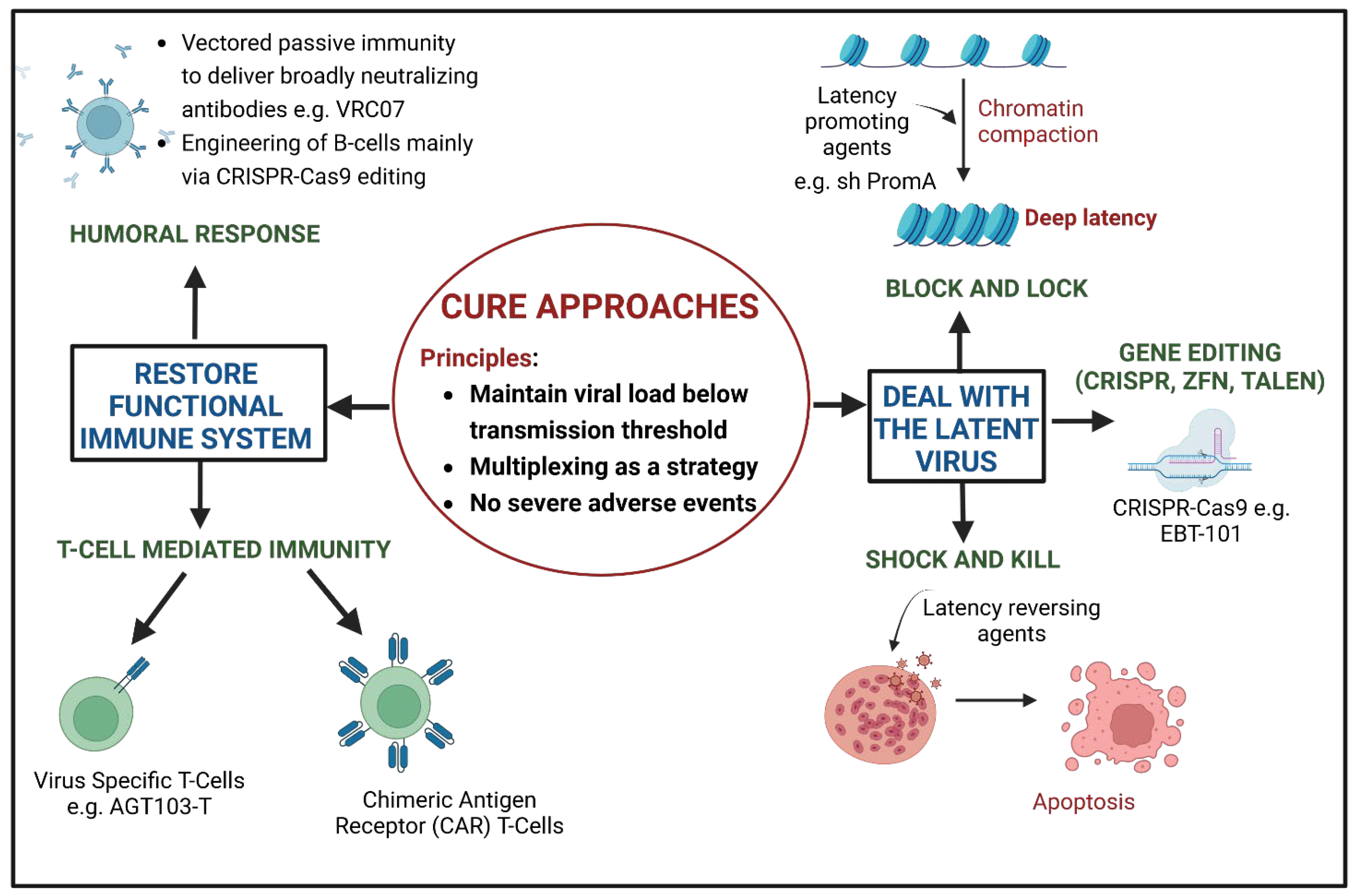
2. Cure Approaches of HIV Gene Therapy
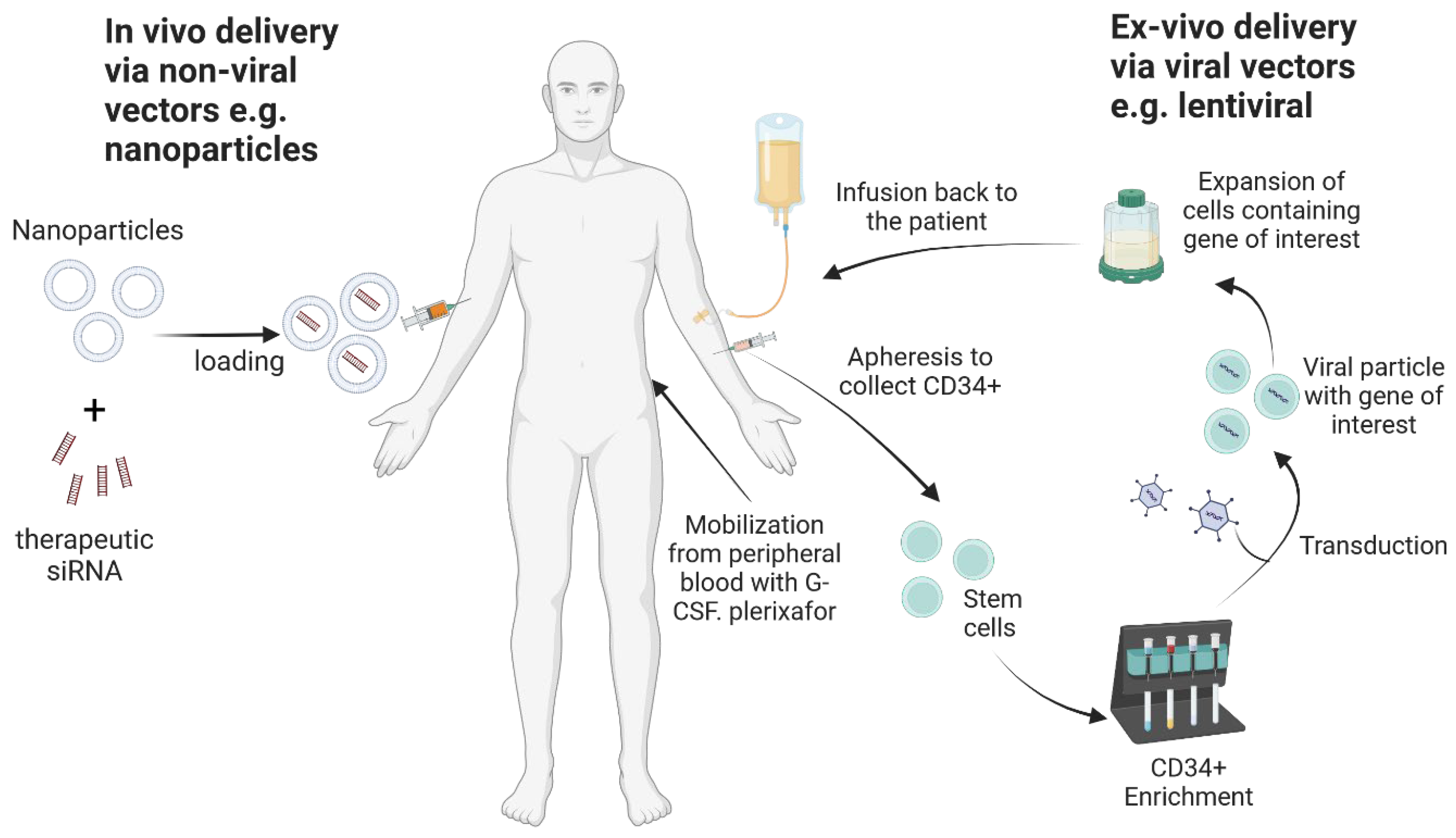
3. Ex Vivo Gene Therapy
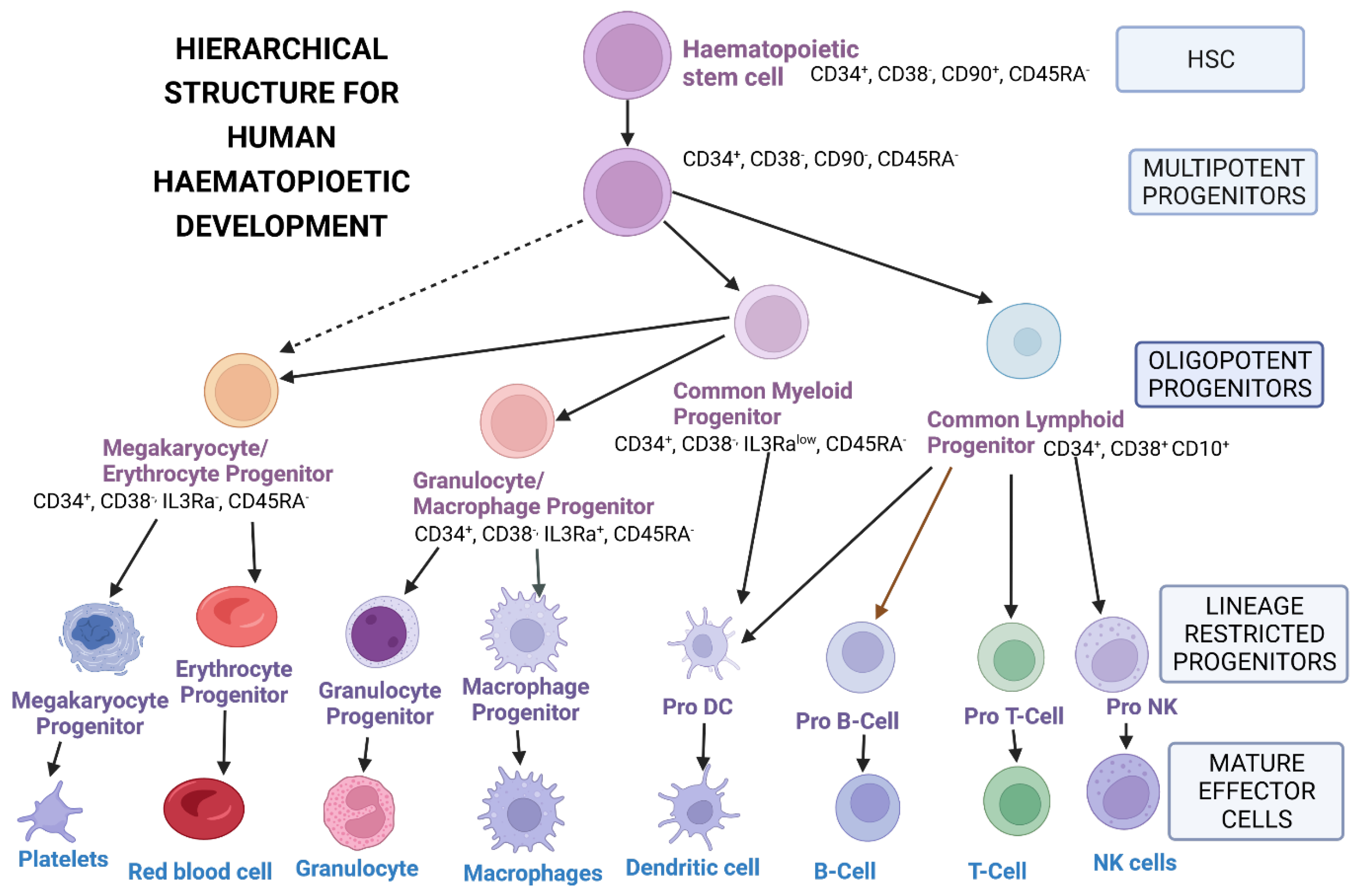
3.1. Sources of Hematopoietic Stem Cells
3.1.1. Bone Marrow-Derived Hematopoietic Stem Cells
3.1.2. Peripheral Blood-Derived Hematopoietic Stem Cells
3.1.3. Umbilical Cord-Derived Hematopoietic Stem Cells
3.1.4. Placenta-Derived Hematopoietic Stem Cells
3.2. Isolation, Purification and Enrichment of Hematopoietic Stem Cells
3.3. Modification of Hematopoietic Stem Cells with Genes of Interest
4. HIV Gene Therapy Delivery via Viral Vectors
4.1. Gamma Retroviral Vectors (γ RV) in HIV Gene Therapy: Complications and Approaches to Enhance Safety
4.2. Lentiviral Vectors for Stable Gene Expression: Challenges and Strategies to Mitigate Them
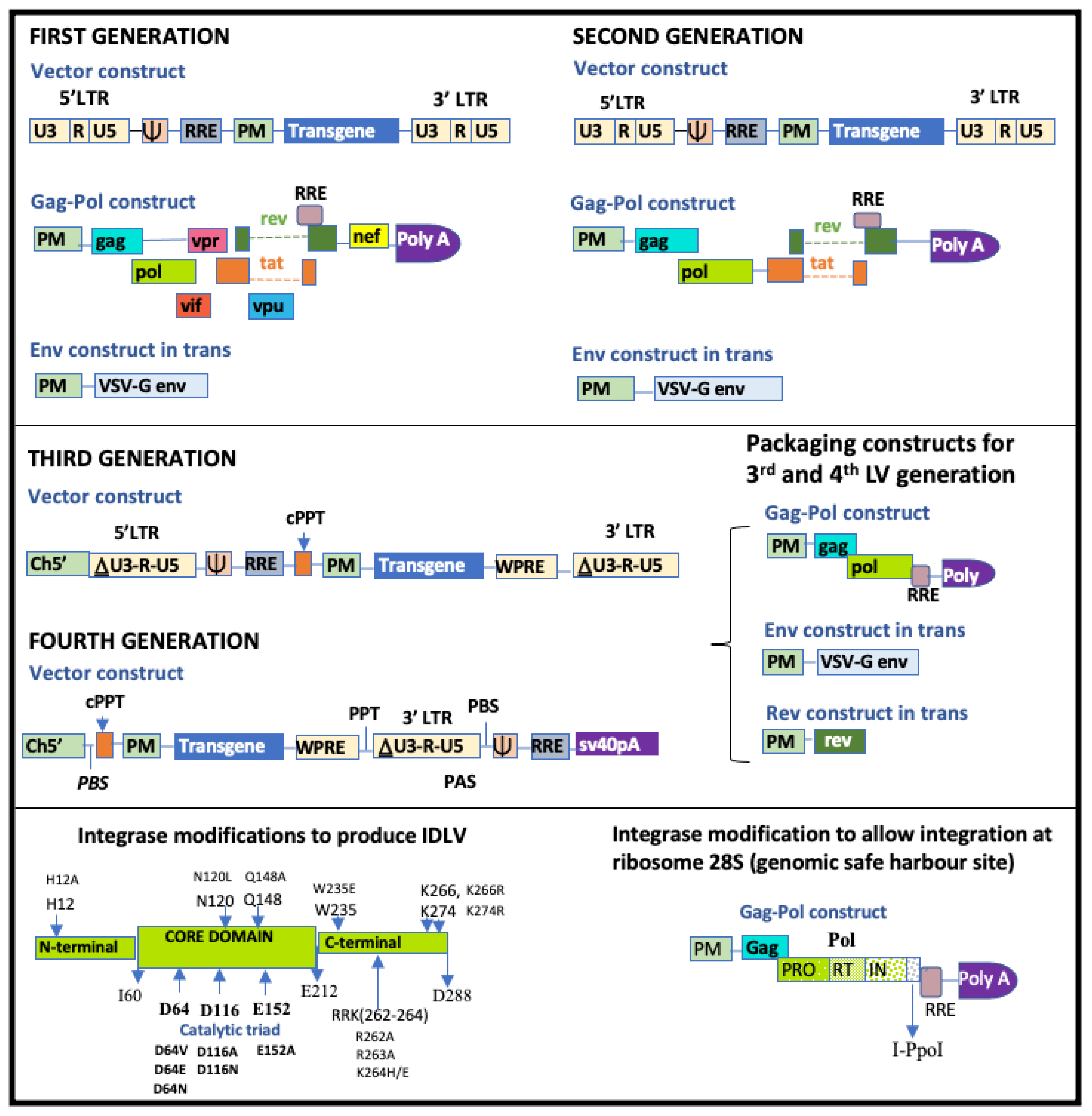
4.2.1. Generations of Lentiviral Vectors
4.2.2. Optimizing Lentiviral Transduction
4.2.3. Lentiviral Vector Silencing
4.2.4. Strategies to Reduce Insertional Mutagenesis
4.3. Adeno Virus Associated Vectors (AAVs): Challenges Limiting Their Use
4.4. Ex Vivo Cell Selection and Expansion
5. Administration of Gene-Modified Hematopoietic Stem Cells: Enhancing Preferential Engraftment of Gene-Modified Cells
5.1. Conditioning Regimen for the Clearance of the Hematopoietic Stem Cell Niche
5.2. In Vivo Selection for Gene-Corrected Hematopoietic Stem Cells
5.3. Recent Developments in Niche Clearance
6. Clinical Trials Based on Ex Vivo HIV Gene Therapy
6.1. Cell Therapy-Based HIV Clinical Trials
6.2. Clinical Trials Based on Gene Delivery via Retroviral Vectors
6.3. Lentiviral Vector-Based HIV Studies
7. HIV Gene Therapy Clinical Trials Based on In Vivo Delivery
CRISPR-Cas9 Technology for HIV Cure
8. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- National Human Genome Research Institute. The Human Genome Project Results. The Human Genome Project 2003. Available online: https://www.genome.gov/human-genome-project/results (accessed on 5 July 2022).
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 380, 45–56. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Richardson, P.G.; Riches, M.L.; Kernan, N.A.; Brochstein, J.A.; Mineishi, S.; Termuhlen, A.M.; Arai, S.; Grupp, S.A.; Guinan, E.C.; Martin, P.L.; et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 2016, 127, 1656–1665. [Google Scholar] [CrossRef]
- Kernan, N.A.; Grupp, S.; Smith, A.R.; Arai, S.; Triplett, B.; Antin, J.H.; Lehmann, L.; Shore, T.; Ho, V.T.; Bunin, N.; et al. Final results from a defibrotide treatment-IND study for patients with hepatic veno-occlusive disease/sinusoidal obstruction syndrome. Br. J. Haematol. 2018, 181, 816–827. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119–122. [Google Scholar]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T.; Feinsod, M.; Guyer, D.R. Pegaptanib for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef]
- Deev, R.V.; Bozo, I.Y.; Mzhavanadze, N.D.; Voronov, D.A.; Gavrilenko, A.V.; Chervyakov, Y.V.; Staroverov, I.N.; Kalinin, R.E.; Shvalb, P.G.; Isaev, A.A. pCMV-vegf165 Intramuscular Gene Transfer is an Effective Method of Treatment for Patients with Chronic Lower Limb Ischemia. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 473–482. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; van’t Hoff, W.G.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Alemozaffar, M.; Konety, B.R.; Shore, N.D.; Gomella, L.G.; Kamat, A.M.; Bivalacqua, T.J.; Montgomery, J.S.; Lerner, S.P.; Busby, J.E.; et al. Intravesical nadofaragene firadenovec gene therapy for BCG-unresponsive non-muscle-invasive bladder cancer: A single-arm, open-label, repeat-dose clinical trial. Lancet Oncol. 2021, 22, 107–117. [Google Scholar] [CrossRef]
- Zhang, S.; Xiao, S.; Sun, Y.; Liu, C.; Su, X.; Li, D.; Xu, G.; Zhu, G.; Xu, B. Clinical Trial of Recombinant Adenovirus-p53 (Gendicine) Combined with Radiotherapy in Nasopharyngeal Carcinoma Patients. Mol. Ther. 2006, 13, S280. [Google Scholar] [CrossRef]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef]
- Pipe, S.W.; Recht, M.; Key, N.S.; Leebeek, F.W.G.; Castaman, G.; Lattimore, S.U.; Van Der Valk, P.; Peerlinck, K.; Coppens, M.; O’Connell, N.; et al. First Data from the Phase 3 HOPE-B Gene Therapy Trial: Efficacy and Safety of Etranacogene Dezaparvovec (AAV5-Padua hFIX variant; AMT-061) in Adults with Severe or Moderate-Severe Hemophilia B Treated Irrespective of Pre-Existing Anti-Capsid Neutralizing Antibodies. Blood 2020, 136, LBA-6. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shieh, P.B.; McDonald, C.M.; Sahenk, Z.; Lehman, K.J.; Lowes, L.P.; Reash, N.F.; Iammarino, M.A.; Alfano, L.N.; Sabo, B.; et al. Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front. Cell Dev. Biol. 2023, 11, 1167762. [Google Scholar] [CrossRef]
- Chawla, S.P.; Chua, V.S.; Fernandez, L.; Quon, D.; Blackwelder, W.C.; Gordon, E.M.; Hall, F.L. Advanced Phase I/II Studies of Targeted Gene Delivery In vivo: Intravenous Rexin-G for Gemcitabine-resistant Metastatic Pancreatic Cancer. Mol. Ther. 2010, 18, 435–441. [Google Scholar] [CrossRef]
- Aiuti, A.; Roncarolo, M.G.; Naldini, L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 2017, 9, 737–740. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Shahryari, A.; Saghaeian Jazi, M.; Mohammadi, S.; Razavi Nikoo, H.; Nazari, Z.; Hosseini, E.S.; Burtscher, I.; Mowla, S.J.; Lickert, H. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 2019, 10, 868. [Google Scholar] [CrossRef]
- Naldini, L. Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 2011, 12, 301–315. [Google Scholar] [CrossRef]
- Tucci, F.; Galimberti, S.; Naldini, L.; Valsecchi, M.G.; Aiuti, A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat. Commun. 2022, 13, 1315. [Google Scholar] [CrossRef]
- Ferrari, G.; Thrasher, A.J.; Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 2021, 22, 216–234. [Google Scholar] [CrossRef]
- Jayarajan, V.; Kounatidou, E.; Qasim, W.; Di, W.-L. Ex vivo gene modification therapy for genetic skin diseases—Recent advances in gene modification technologies and delivery. Exp. Dermatol. 2021, 30, 887–896. [Google Scholar] [CrossRef]
- Gowing, G.; Svendsen, S.; Svendsen, C.N. Ex vivo gene therapy for the treatment of neurological disorders. Prog. Brain Res. 2017, 230, 99–132. [Google Scholar] [CrossRef]
- Jensen, T.L.; Gøtzsche, C.R.; Woldbye, D.P.D. Current and Future Prospects for Gene Therapy for Rare Genetic Diseases Affecting the Brain and Spinal Cord. Front. Mol. Neurosci. 2021, 14, 695937. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Orthopaedic Gene Therapy: Twenty-Five Years on. JBJS Rev. 2021, 9, e20.00220. [Google Scholar] [CrossRef] [PubMed]
- Pauza, C.D.; Huang, K.; Bordon, J. Advances in cell and gene therapy for HIV disease: It is good to be specific. Curr. Opin. HIV AIDS 2021, 16, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Alfageme-Abello, O.; Porret, R.; Perreau, M.; Perez, L.; Muller, Y.D. Chimeric antigen receptor T-cell therapy for HIV cure. Curr. Opin. HIV AIDS 2021, 16, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Lewin, S.R.; Attoye, T.; Bansbach, C.; Doehle, B.; Dubé, K.; Dybul, M.; SenGupta, D.; Jiang, A.; Johnston, R.; Lamplough, R.; et al. Multi-stakeholder consensus on a target product profile for an HIV cure. Lancet HIV 2021, 8, e42–e50. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Van Besien, K.; Glesby, M.J.; Pahwa, S.; Coletti, A.; Warshaw, M.G.; Petz, L.D.; Moore, T.B.; Chen, Y.H.; Pallikkuth, S.; et al. HIV-1 remission and possible cure in a woman after haplo-cord blood transplant. Cell 2023, 186, 1115–1126.e1118. [Google Scholar] [CrossRef]
- Klemm, V.; Mitchell, J.; Cortez-Jugo, C.; Cavalieri, F.; Symonds, G.; Caruso, F.; Kelleher, A.D.; Ahlenstiel, C. Achieving HIV-1 Control through RNA-Directed Gene Regulation. Genes 2016, 7, 119. [Google Scholar] [CrossRef]
- Chandrasekar, A.P.; Badley, A.D. Prime, shock and kill: BCL-2 inhibition for HIV cure. Front. Immunol. 2022, 13, 1033609. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Klinnert, S.; Schenkel, C.D.; Freitag, P.C.; Günthard, H.F.; Plückthun, A.; Metzner, K.J. Targeted shock-and-kill HIV-1 gene therapy approach combining CRISPR activation, suicide gene tBid and retargeted adenovirus delivery. Gene Ther. 2023. [Google Scholar] [CrossRef]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef]
- Anthony-Gonda, K.; Bardhi, A.; Ray, A.; Flerin, N.; Li, M.; Chen, W.; Ochsenbauer, C.; Kappes, J.C.; Krueger, W.; Worden, A.; et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl. Med. 2019, 11, eaav5685. [Google Scholar] [CrossRef]
- Buchholz, F.; Hauber, J. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods 2011, 53, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef]
- Shi, B.; Li, J.; Shi, X.; Jia, W.; Wen, Y.; Hu, X.; Zhuang, F.; Xi, J.; Zhang, L. TALEN-Mediated Knockout of CCR5 Confers Protection Against Infection of Human Immunodeficiency Virus. J. Acquir. Immune Defic. Syndr. 2017, 74, 229–241. [Google Scholar] [CrossRef]
- DiGiusto, D.L.; Cannon, P.M.; Holmes, M.C.; Li, L.; Rao, A.; Wang, J.; Lee, G.; Gregory, P.D.; Kim, K.A.; Hayward, S.B.; et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther. Methods Clin. Dev. 2016, 3, 16067. [Google Scholar] [CrossRef]
- Atkins, A.J.; Allen, A.G.; Dampier, W.; Haddad, E.K.; Nonnemacher, M.R.; Wigdahl, B. HIV-1 cure strategies: Why CRISPR? Expert Opin. Biol. Ther. 2021, 21, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Wolstein, O.; Boyd, M.; Millington, M.; Impey, H.; Boyer, J.; Howe, A.; Delebecque, F.; Cornetta, K.; Rothe, M.; Baum, C.; et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014, 1, 11. [Google Scholar] [CrossRef]
- Xun, J.; Zhang, X.; Guo, S.; Lu, H.; Chen, J. Editing out HIV: Application of gene editing technology to achieve functional cure. Retrovirology 2021, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Shijuuku, T.; Fukamachi, T.; Zaunders, J.; Guillemin, G.; Cooper, D.; Kelleher, A. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J. RNAi Gene Silenc. 2005, 1, 66–78. [Google Scholar]
- Suzuki, K.; Ishida, T.; Yamagishi, M.; Ahlenstiel, C.; Swaminathan, S.; Marks, K.; Murray, D.; McCartney, E.M.; Beard, M.R.; Alexander, M.; et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol. 2011, 8, 1035–1046. [Google Scholar] [CrossRef]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol. Ther. Nucleic Acids 2015, 4, e261. [Google Scholar] [CrossRef] [PubMed]
- Higaki, K.; Hirao, M.; Kawana-Tachikawa, A.; Iriguchi, S.; Kumagai, A.; Ueda, N.; Bo, W.; Kamibayashi, S.; Watanabe, A.; Nakauchi, H.; et al. Generation of HIV-Resistant Macrophages from IPSCs by Using Transcriptional Gene Silencing and Promoter-Targeted RNA. Mol. Ther. Nucleic Acids 2018, 12, 793–804. [Google Scholar] [CrossRef]
- Suzuki, K.; Hattori, S.; Marks, K.; Ahlenstiel, C.; Maeda, Y.; Ishida, T.; Millington, M.; Boyd, M.; Symonds, G.; Cooper, D.A.; et al. Promoter Targeting shRNA Suppresses HIV-1 Infection Through Transcriptional Gene Silencing. Mol. Ther. Nucleic Acids 2013, 2, e137. [Google Scholar] [CrossRef]
- Chabannon, C.; Kuball, J.; Bondanza, A.; Dazzi, F.; Pedrazzoli, P.; Toubert, A.; Ruggeri, A.; Fleischhauer, K.; Bonini, C. Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci. Transl. Med. 2018, 10, eaap9630. [Google Scholar] [CrossRef]
- Malek, A.; Bersinger, N.A. Human placental stem cells: Biomedical potential and clinical relevance. J. Stem Cells 2011, 6, 75–92. [Google Scholar] [PubMed]
- Nielsen, J.S.; McNagny, K.M. Novel functions of the CD34 family. J. Cell Sci. 2008, 121, 3683–3692. [Google Scholar] [CrossRef]
- Bryder, D.; Rossi, D.J.; Weissman, I.L. Hematopoietic stem cells: The paradigmatic tissue-specific stem cell. Am. J. Pathol. 2006, 169, 338–346. [Google Scholar] [CrossRef]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef]
- Engelhardt, M.; Lübbert, M.; Guo, Y. CD34+ or CD34−: Which is the more primitive? Leukemia 2002, 16, 1603–1608. [Google Scholar] [CrossRef]
- Taichman, R.S. Blood and bone: Two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood 2005, 105, 2631–2639. [Google Scholar] [CrossRef]
- Taichman, R.S.; Reilly, M.J.; Matthews, L.S. Human osteoblast-like cells and osteosarcoma cell lines synthesize macrophage inhibitory protein 1alpha in response to interleukin 1beta and tumour necrosis factor alpha stimulation in vitro. Br. J. Haematol. 2000, 108, 275–283. [Google Scholar] [CrossRef]
- Aiuti, A.; Tavian, M.; Cipponi, A.; Ficara, F.; Zappone, E.; Hoxie, J.; Peault, B.; Bordignon, C. Expression of CXCR4, the receptor for stromal cell-derived factor-1 on fetal and adult human lympho-hematopoietic progenitors. Eur. J. Immunol. 1999, 29, 1823–1831. [Google Scholar] [CrossRef]
- Puri, M.C.; Bernstein, A. Requirement for the TIE family of receptor tyrosine kinases in adult but not fetal hematopoiesis. Proc. Natl. Acad. Sci. USA 2003, 100, 12753–12758. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Popiołek, Ł.; Horecka, A. Characteristics of hematopoietic stem cells of umbilical cord blood. Cytotechnology 2015, 67, 387–396. [Google Scholar] [CrossRef]
- Panch, S.R.; Szymanski, J.; Savani, B.N.; Stroncek, D.F. Sources of Hematopoietic Stem and Progenitor Cells and Methods to Optimize Yields for Clinical Cell Therapy. Biol. Blood Marrow Transplant. 2017, 23, 1241–1249. [Google Scholar] [CrossRef]
- Mijovic, A.; Pamphilon, D. Harvesting, processing and inventory management of peripheral blood stem cells. Asian J. Transfus. Sci. 2007, 1, 16–23. [Google Scholar] [CrossRef]
- Hoggatt, J.; Speth, J.M.; Pelus, L.M. Concise Review: Sowing the Seeds of a Fruitful Harvest: Hematopoietic Stem Cell Mobilization. Stem Cells 2013, 31, 2599–2606. [Google Scholar] [CrossRef]
- Stroncek, D.F.; Confer, D.L.; Leitman, S.F. Peripheral blood progenitor cells for HPC transplants involving unrelated donors. Transfusion 2000, 40, 731–741. [Google Scholar] [CrossRef]
- Canarutto, D.; Tucci, F.; Gattillo, S.; Zambelli, M.; Calbi, V.; Gentner, B.; Ferrua, F.; Marktel, S.; Migliavacca, M.; Barzaghi, F.; et al. Peripheral blood stem and progenitor cell collection in pediatric candidates for ex vivo gene therapy: A 10-year series. Mol. Ther. Methods Clin. Dev. 2021, 22, 76–83. [Google Scholar] [CrossRef]
- Cencioni, C.; Capogrossi, M.C.; Napolitano, M. The SDF-1/CXCR4 axis in stem cell preconditioning. Cardiovasc. Res. 2012, 94, 400–407. [Google Scholar] [CrossRef]
- Domingues, M.J.; Nilsson, S.K.; Cao, B. New agents in HSC mobilization. Int. J. Hematol. 2017, 105, 141–152. [Google Scholar] [CrossRef]
- Rowisha, M.A.; El-Shanshory, M.R.; El-Hawary, E.E.; Ahmed, A.Y.; Altoraky, S.R.M. Impact of maternal and neonatal factors on umbilical cord CD34+ cells. Stem Cell Investig. 2020, 7, 5. [Google Scholar] [CrossRef]
- Pafumi, C.; Leanza, V.; Carbonaro, A.; Leanza, G.; Iemmola, A.; Abate, G.; Stracquadanio, M.G.; D’Agati, A. CD34+ Stem Cells from Umbilical Cord Blood. Clin. Pract. 2011, 1, e79. [Google Scholar] [CrossRef]
- Silini, A.R.; Di Pietro, R.; Lang-Olip, I.; Alviano, F.; Banerjee, A.; Basile, M.; Borutinskaite, V.; Eissner, G.; Gellhaus, A.; Giebel, B.; et al. Perinatal Derivatives: Where Do We Stand? A Roadmap of the Human Placenta and Consensus for Tissue and Cell Nomenclature. Front. Bioeng. Biotechnol. 2020, 8, 1438. [Google Scholar] [CrossRef]
- Pipino, C.; Shangaris, P.; Resca, E.; Zia, S.; Deprest, J.; Sebire, N.J.; David, A.L.; Guillot, P.V.; De Coppi, P. Placenta as a reservoir of stem cells: An underutilized resource? Br. Med. Bull. 2013, 105, 43–68. [Google Scholar] [CrossRef]
- Parolini, O.; Alviano, F.; Bagnara, G.P.; Bilic, G.; Bühring, H.J.; Evangelista, M.; Hennerbichler, S.; Liu, B.; Magatti, M.; Mao, N.; et al. Concise review: Isolation and characterization of cells from human term placenta: Outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells 2008, 26, 300–311. [Google Scholar] [CrossRef]
- Serikov, V.; Hounshell, C.; Larkin, S.; Green, W.; Ikeda, H.; Walters, M.C.; Kuypers, F.A. A BRIEF COMMUNICATION: Human Term Placenta as a Source of Hematopoietic Cells. Exp. Biol. Med. 2009, 234, 813–823. [Google Scholar] [CrossRef]
- Walenda, T.; Bork, S.; Horn, P.; Wein, F.; Saffrich, R.; Diehlmann, A.; Eckstein, V.; Ho, A.D.; Wagner, W. Co-culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J. Cell. Mol. Med. 2010, 14, 337–350. [Google Scholar] [CrossRef]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal Stem Cell: Keystone of the Hematopoietic Stem Cell Niche and a Stepping-Stone for Regenerative Medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef]
- Flower, A.; Harrison, L.; Pulsipher, M.A.; Shi, Q.; Giller, R.; Morris, E.; Klejmont, L.; Ayello, J.; Semidei-Pomales, M.; Fabricatore, S.; et al. Hematopoietic Stem Cell Transplantation with Human Placenta-Derived Stem Cells Combined with Unrelated Cord Blood for Malignant and Non-Malignant Disorders (IND 14949). Biol. Blood Marrow Transplant. 2019, 25, S214–S215. [Google Scholar] [CrossRef]
- Gramignoli, R.; Srinivasan, R.C.; Kannisto, K.; Strom, S.C. Isolation of Human Amnion Epithelial Cells According to Current Good Manufacturing Procedures. Curr. Protoc. Stem Cell Biol. 2016, 37, 1E.10.11–11E.10.13. [Google Scholar] [CrossRef]
- Aghayan, H.R.; Payab, M.; Mohamadi-Jahani, F.; Aghayan, S.S.; Larijani, B.; Arjmand, B. GMP-Compliant Production of Human Placenta-Derived Mesenchymal Stem Cells. In Stem Cells and Good Manufacturing Practices: Methods, Protocols, and Regulations; Turksen, K., Ed.; Springer: New York, NY, USA, 2021; pp. 213–225. [Google Scholar]
- Huang, Q.; Yang, Y.; Luo, C.; Wen, Y.; Liu, R.; Li, S.; Chen, T.; Sun, H.; Tang, L. An efficient protocol to generate placental chorionic plate-derived mesenchymal stem cells with superior proliferative and immunomodulatory properties. Stem Cell Res. Ther. 2019, 10, 301. [Google Scholar] [CrossRef]
- Robin, C.; Dzierzak, E. Preparation of Hematopoietic Stem and Progenitor Cells from the Human Placenta. Curr. Protoc. Stem Cell Biol. 2010, 14, 2A.9.1–2A.9.8. [Google Scholar] [CrossRef]
- Oliveira, M.S.; Barreto-Filho, J.B. Placental-derived stem cells: Culture, differentiation and challenges. World J. Stem Cells 2015, 7, 769–775. [Google Scholar] [CrossRef]
- Baldwin, K.; Urbinati, F.; Romero, Z.; Campo-Fernandez, B.; Kaufman, M.L.; Cooper, A.R.; Masiuk, K.; Hollis, R.P.; Kohn, D.B. Enrichment of human hematopoietic stem/progenitor cells facilitates transduction for stem cell gene therapy. Stem Cells 2015, 33, 1532–1542. [Google Scholar] [CrossRef]
- Radtke, S.; Pande, D.; Cui, M.; Perez, A.M.; Chan, Y.-Y.; Enstrom, M.; Schmuck, S.; Berger, A.; Eunson, T.; Adair, J.E.; et al. Purification of Human CD34+CD90+ HSCs Reduces Target Cell Population and Improves Lentiviral Transduction for Gene Therapy. Mol. Ther. Methods Clin. Dev. 2020, 18, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Kays, S.-K.; Kaufmann, K.B.; Abel, T.; Brendel, C.; Bonig, H.; Grez, M.; Buchholz, C.J.; Kneissl, S. CD105 Is a Surface Marker for Receptor-Targeted Gene Transfer into Human Long-Term Repopulating Hematopoietic Stem Cells. Stem Cells Dev. 2014, 24, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Laje, P.; Zoltick, P.W.; Flake, A.W. SLAM-enriched hematopoietic stem cells maintain long-term repopulating capacity after lentiviral transduction using an abbreviated protocol. Gene Ther. 2010, 17, 412–418. [Google Scholar] [CrossRef]
- Kallinikou, K.; Anjos-Afonso, F.; Blundell, M.P.; Ings, S.J.; Watts, M.J.; Thrasher, A.J.; Linch, D.C.; Bonnet, D.; Yong, K.L. Engraftment defect of cytokine-cultured adult human mobilized CD34+ cells is related to reduced adhesion to bone marrow niche elements. Br. J. Haematol. 2012, 158, 778–787. [Google Scholar] [CrossRef]
- Zonari, E.; Desantis, G.; Petrillo, C.; Boccalatte, F.E.; Lidonnici, M.R.; Kajaste-Rudnitski, A.; Aiuti, A.; Ferrari, G.; Naldini, L.; Gentner, B. Efficient Ex vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017, 8, 977–990. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, S.; Ding, Q.; Jiang, R.; He, J.; Wang, Q.; Jin, Y.; Huang, X.; Liu, S.; Capitano, M.L.; et al. ADGRG1 enriches for functional human hematopoietic stem cells following ex vivo expansion–induced mitochondrial oxidative stress. J. Clin. Investig. 2021, 131, e148329. [Google Scholar] [CrossRef]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef]
- Nishimura, T.; Hsu, I.; Martinez-Krams, D.C.; Nakauchi, Y.; Majeti, R.; Yamazaki, S.; Nakauchi, H.; Wilkinson, A.C. Use of polyvinyl alcohol for chimeric antigen receptor T-cell expansion. Exp. Hematol. 2019, 80, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ishida, R.; Nakauchi, H.; Yamazaki, S. Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat. Protoc. 2020, 15, 628–648. [Google Scholar] [CrossRef]
- Sudo, K.; Yamazaki, S.; Wilkinson, A.C.; Nakauchi, H.; Nakamura, Y. Polyvinyl alcohol hydrolysis rate and molecular weight influence human and murine HSC activity ex vivo. Stem Cell Res. 2021, 56, 102531. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sivanandam, V.; Wong, K., Jr. Adeno-Associated Virus and Hematopoietic Stem Cells: The Potential of Adeno-Associated Virus Hematopoietic Stem Cells in Genetic Medicines. Hum. Gene Ther. 2020, 31, 542–552. [Google Scholar] [CrossRef]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306. [Google Scholar] [CrossRef]
- Bestor, T.H. Gene silencing as a threat to the success of gene therapy. J. Clin. Investig. 2000, 105, 409–411. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Holehonnur, R.; Lella, S.K.; Ho, A.; Luong, J.A.; Ploski, J.E. The production of viral vectors designed to express large and difficult to express transgenes within neurons. Mol. Brain 2015, 8, 12. [Google Scholar] [CrossRef]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Kalidasan, V.; Ng, W.H.; Ishola, O.A.; Ravichantar, N.; Tan, J.J.; Das, K.T. A guide in lentiviral vector production for hard-to-transfect cells, using cardiac-derived c-kit expressing cells as a model system. Sci. Rep. 2021, 11, 19265. [Google Scholar] [CrossRef]
- El Ashkar, S.; De Rijck, J.; Demeulemeester, J.; Vets, S.; Madlala, P.; Cermakova, K.; Debyser, Z.; Gijsbers, R. BET-independent MLV-based Vectors Target Away From Promoters and Regulatory Elements. Mol. Ther. Nucleic Acids 2014, 3, e179. [Google Scholar] [CrossRef]
- LaFave, M.C.; Varshney, G.K.; Gildea, D.E.; Wolfsberg, T.G.; Baxevanis, A.D.; Burgess, S.M. MLV integration site selection is driven by strong enhancers and active promoters. Nucleic Acids Res. 2014, 42, 4257–4269. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D.; Emerman, M. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef]
- Yoder, K.E.; Rabe, A.J.; Fishel, R.; Larue, R.C. Strategies for Targeting Retroviral Integration for Safer Gene Therapy: Advances and Challenges. Front. Mol. Biosci. 2021, 8, 662331. [Google Scholar] [CrossRef]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The BET Family of Proteins Targets Moloney Murine Leukemia Virus Integration near Transcription Start Sites. Cell Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef]
- Van Looveren, D.; Giacomazzi, G.; Thiry, I.; Sampaolesi, M.; Gijsbers, R. Improved functionality and potency of next generation BinMLV viral vectors toward safer gene therapy. Mol. Ther. Methods Clin. Dev. 2021, 23, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Nombela, I.; Michiels, M.; Van Looveren, D.; Marcelis, L.; el Ashkar, S.; Van Belle, S.; Bruggemans, A.; Tousseyn, T.; Schwaller, J.; Christ, F.; et al. BET-Independent Murine Leukemia Virus Integration Is Retargeted In vivo and Selects Distinct Genomic Elements for Lymphomagenesis. Microbiol. Spectr. 2022, 10, e01478-01422. [Google Scholar] [CrossRef]
- El Ashkar, S.; Van Looveren, D.; Schenk, F.; Vranckx, L.S.; Demeulemeester, J.; De Rijck, J.; Debyser, Z.; Modlich, U.; Gijsbers, R. Engineering Next-Generation BET-Independent MLV Vectors for Safer Gene Therapy. Mol. Ther. Nucleic Acids 2017, 7, 231–245. [Google Scholar] [CrossRef]
- Bosticardo, M.; Ghosh, A.; Du, Y.; Jenkins, N.A.; Copeland, N.G.; Candotti, F. Self-inactivating Retroviral Vector-mediated Gene Transfer Induces Oncogene Activation and Immortalization of Primary Murine Bone Marrow Cells. Mol. Ther. 2009, 17, 1910–1918. [Google Scholar] [CrossRef]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional Transformation of Hematopoietic Cells by Self-inactivating Lentiviral and Gammaretroviral Vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle-Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P.; et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Poletti, V.; Mavilio, F. Designing Lentiviral Vectors for Gene Therapy of Genetic Diseases. Viruses 2021, 13, 1526. [Google Scholar] [CrossRef]
- Poeschla, E.M. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. CMLS 2008, 65, 1403–1424. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef]
- Gándara, C.; Affleck, V.; Stoll, E.A. Manufacture of Third-Generation Lentivirus for Preclinical Use, with Process Development Considerations for Translation to Good Manufacturing Practice. Hum. Gene Ther. Methods 2017, 29, 1–15. [Google Scholar] [CrossRef]
- Merten, O.-W.; Hebben, M.; Bovolenta, C. Production of lentiviral vectors. Mol. Ther. Methods Clin. Dev. 2016, 3, 16017. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.A.; Counsell, J.R.; Perocheau, D.P.; Karda, R.; Buckley, S.M.K.; Brugman, M.H.; Galla, M.; Schambach, A.; McKay, T.R.; Waddington, S.N.; et al. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017, 25, 1790–1804. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B. A Fourth Generation Lentiviral Vector: Simplifying Genomic Gymnastics. Mol. Ther. 2017, 25, 1741–1743. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F. Immune Evasion and Counteraction of Restriction Factors by HIV-1 and Other Primate Lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nuñez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef]
- Hofmann, H.; Logue Eric, C.; Bloch, N.; Daddacha, W.; Polsky Sylvie, B.; Schultz Megan, L.; Kim, B.; Landau Nathaniel, R. The Vpx Lentiviral Accessory Protein Targets SAMHD1 for Degradation in the Nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- McAllery, S.A.; Ahlenstiel, C.L.; Suzuki, K.; Symonds, G.P.; Kelleher, A.D.; Turville, S.G. The feasibility of incorporating Vpx into lentiviral gene therapy vectors. Mol. Ther. Methods Clin. Dev. 2016, 5, 16066. [Google Scholar] [CrossRef]
- Davis, H.E.; Morgan, J.R.; Yarmush, M.L. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys. Chem. 2002, 97, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Cornetta, K.; Anderson, W.F. Protamine sulfate as an effective alternative to polybrene in retroviral-mediated gene-transfer: Implications for human gene therapy. J. Virol. Methods 1989, 23, 187–194. [Google Scholar] [CrossRef]
- Lee, H.-J.; Lee, Y.-S.; Kim, H.-S.; Kim, Y.-K.; Kim, J.-H.; Jeon, S.-H.; Lee, H.-W.; Kim, S.; Miyoshi, H.; Chung, H.-M.; et al. Retronectin enhances lentivirus-mediated gene delivery into hematopoietic progenitor cells. Biologicals 2009, 37, 203–209. [Google Scholar] [CrossRef]
- Piovan, C.; Marin, V.; Scavullo, C.; Corna, S.; Giuliani, E.; Bossi, S.; Galy, A.; Fenard, D.; Bordignon, C.; Rizzardi, G.P.; et al. Vectofusin-1 Promotes RD114-TR-Pseudotyped Lentiviral Vector Transduction of Human HSPCs and T Lymphocytes. Mol. Ther. Methods Clin. Dev. 2017, 5, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Radek, C.; Bernadin, O.; Drechsel, K.; Cordes, N.; Pfeifer, R.; Sträßer, P.; Mormin, M.; Gutierrez-Guerrero, A.; Cosset, F.-l.; Kaiser, A.D.; et al. Vectofusin-1 Improves Transduction of Primary Human Cells with Diverse Retroviral and Lentiviral Pseudotypes, Enabling Robust, Automated Closed-System Manufacturing. Hum. Gene Ther. 2019, 30, 1477–1493. [Google Scholar] [CrossRef] [PubMed]
- Hauber, I.; Beschorner, N.; Schrödel, S.; Chemnitz, J.; Kröger, N.; Hauber, J.; Thirion, C. Improving Lentiviral Transduction of CD34+ Hematopoietic Stem and Progenitor Cells. Hum. Gene Ther. Methods 2018, 29, 104–113. [Google Scholar] [CrossRef]
- Lewis, G.; Christiansen, L.; McKenzie, J.; Luo, M.; Pasackow, E.; Smurnyy, Y.; Harrington, S.; Gregory, P.; Veres, G.; Negre, O.; et al. Staurosporine Increases Lentiviral Vector Transduction Efficiency of Human Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 313–322. [Google Scholar] [CrossRef]
- Uchida, N.; Hsieh, M.M.; Hayakawa, J.; Madison, C.; Washington, K.N.; Tisdale, J.F. Optimal conditions for lentiviral transduction of engrafting human CD34+ cells. Gene Ther. 2011, 18, 1078–1086. [Google Scholar] [CrossRef]
- Uchida, N.; Demirci, S.; Haro-Mora, J.J.; Fujita, A.; Raines, L.N.; Hsieh, M.M.; Tisdale, J.F. Serum-free Erythroid Differentiation for Efficient Genetic Modification and High-Level Adult Hemoglobin Production. Mol. Ther. Methods Clin. Dev. 2018, 9, 247–256. [Google Scholar] [CrossRef]
- Tolmachov, O.E.; Subkhankulova, T.; Tolmachova, T. Silencing of Transgene Expression: A Gene Therapy Perspective. In Gene Therapy—Tools and Potential Applications; IntechOpen, Ed.; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar] [CrossRef]
- Ramezani, A.; Hawley, R.G. Strategies to insulate lentiviral vector-expressed transgenes. Methods Mol. Biol. 2010, 614, 77–100. [Google Scholar] [CrossRef]
- Schenkwein, D.; Turkki, V.; Ahlroth, M.K.; Timonen, O.; Airenne, K.J.; Ylä-Herttuala, S. rDNA-directed integration by an HIV-1 integrase--I-PpoI fusion protein. Nucleic Acids Res. 2013, 41, e61. [Google Scholar] [CrossRef]
- Gurumoorthy, N.; Nordin, F.; Tye, G.J.; Wan Kamarul Zaman, W.S.; Ng, M.H. Non-Integrating Lentiviral Vectors in Clinical Applications: A Glance Through. Biomedicines 2022, 10, 107. [Google Scholar] [CrossRef]
- Wanisch, K.; Yáñez-Muñoz, R.J. Integration-deficient Lentiviral Vectors: A Slow Coming of Age. Mol. Ther. 2009, 17, 1316–1332. [Google Scholar] [CrossRef]
- Banasik, M.B.; McCray, P.B. Integrase-defective lentiviral vectors: Progress and applications. Gene Ther. 2010, 17, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, L.S.; Demeulemeester, J.; Debyser, Z.; Gijsbers, R. Towards a Safer, More Randomized Lentiviral Vector Integration Profile Exploring Artificial LEDGF Chimeras. PLoS ONE 2016, 11, e0164167. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Sindelar, R.D.; Meibohn, B. Pharmaceutical Biotechnology, Fundamentals and Applications, 4th ed.; Crommelin, D.J.A., Sindelar, R.D., Meibohm, B., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Kruzik, A.; Fetahagic, D.; Hartlieb, B.; Dorn, S.; Koppensteiner, H.; Horling, F.M.; Scheiflinger, F.; Reipert, B.M.; de la Rosa, M. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol. Ther. Methods Clin. Dev. 2019, 14, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.-A.; Tedesco, N.; Leborgne, C.; Ronzitti, G. Overcoming the Challenges Imposed by Humoral Immunity to AAV Vectors to Achieve Safe and Efficient Gene Transfer in Seropositive Patients. Front. Immunol. 2022, 13, 857276. [Google Scholar] [CrossRef]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.L.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Zubkova, E.S.; Beloglazova, I.B.; Ratner, E.I.; Dyikanov, D.T.; Dergilev, K.V.; Menshikov, M.Y.; Parfyonova, Y.V. Transduction of rat and human adipose-tissue derived mesenchymal stromal cells by adeno-associated viral vector serotype DJ. Biol. Open 2021, 10, bio058461. [Google Scholar] [CrossRef] [PubMed]
- Sehara, Y.; Fujimoto, K.-I.; Ikeguchi, K.; Katakai, Y.; Ono, F.; Takino, N.; Ito, M.; Ozawa, K.; Muramatsu, S.-I. Persistent Expression of Dopamine-Synthesizing Enzymes 15 Years After Gene Transfer in a Primate Model of Parkinson’s Disease. Hum. Gene Ther. Clin. Dev. 2017, 28, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.A.; Martins, M.A. Adeno-associated virus-vectored delivery of HIV biologics: The promise of a “single-shot” functional cure for HIV infection. J. Virus Erad. 2023, 9, 100316. [Google Scholar] [CrossRef] [PubMed]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Investig. 2012, 122, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Bhukhai, K.; de Dreuzy, E.; Giorgi, M.; Colomb, C.; Negre, O.; Denaro, M.; Gillet-Legrand, B.; Cheuzeville, J.; Paulard, A.; Trebeden-Negre, H.; et al. Ex vivo Selection of Transduced Hematopoietic Stem Cells for Gene Therapy of β-Hemoglobinopathies. Mol. Ther. 2018, 26, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Pawliuk, R.; Eaves, C.J.; Humphries, R.K. Sustained High-Level Reconstitution of the Hematopoietic System by Preselected Hematopoietic Cells Expressing a Transduced Cell-Surface Antigen. Hum. Gene Ther. 1997, 8, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Tajer, P.; Pike-Overzet, K.; Arias, S.; Havenga, M.; Staal, F.J.T. Ex vivo Expansion of Hematopoietic Stem Cells for Therapeutic Purposes: Lessons from Development and the Niche. Cells 2019, 8, 169. [Google Scholar] [CrossRef]
- Miller, P.H.; Knapp, D.J.H.F.; Beer, P.A.; MacAldaz, M.; Rabu, G.; Cheung, A.M.S.; Wei, L.; Eaves, C.J. Early production of human neutrophils and platelets posttransplant is severely compromised by growth factor exposure. Exp. Hematol. 2016, 44, 635–640. [Google Scholar] [CrossRef]
- Schuster, J.A.; Stupnikov, M.R.; Ma, G.; Liao, W.; Lai, R.; Ma, Y.; Aguila, J.R. Expansion of hematopoietic stem cells for transplantation: Current perspectives. Exp. Hematol. Oncol. 2012, 1, 12. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef]
- Fares, I.; Chagraoui, J.; Gareau, Y.; Gingras, S.; Ruel, R.; Mayotte, N.; Csaszar, E.; Knapp David, J.H.F.; Miller, P.; Ngom, M.; et al. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 2014, 345, 1509–1512. [Google Scholar] [CrossRef]
- Chagraoui, J.; Girard, S.; Spinella, J.-F.; Simon, L.; Bonneil, E.; Mayotte, N.; MacRae, T.; Coulombe-Huntington, J.; Bertomeu, T.; Moison, C.; et al. UM171 Preserves Epigenetic Marks that Are Reduced in Ex vivo Culture of Human HSCs via Potentiation of the CLR3-KBTBD4 Complex. Cell Stem Cell 2021, 28, 48–62.e46. [Google Scholar] [CrossRef]
- Cohen, S.; Roy, J.; Lachance, S.; Delisle, J.-S.; Marinier, A.; Busque, L.; Roy, D.-C.; Barabé, F.; Ahmad, I.; Bambace, N.; et al. Hematopoietic stem cell transplantation using single UM171-expanded cord blood: A single-arm, phase 1–2 safety and feasibility study. Lancet Haematol. 2020, 7, e134–e145. [Google Scholar] [CrossRef]
- Delaney, C.; Heimfeld, S.; Brashem-Stein, C.; Voorhies, H.; Manger, R.L.; Bernstein, I.D. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 2010, 16, 232–236. [Google Scholar] [CrossRef]
- Domashenko, A.D.; Danet-Desnoyers, G.; Aron, A.; Carroll, M.P.; Emerson, S.G. TAT-mediated transduction of NF-Ya peptide induces the ex vivo proliferation and engraftment potential of human hematopoietic progenitor cells. Blood 2010, 116, 2676–2683. [Google Scholar] [CrossRef]
- Aguila, J.R.; Liao, W.; Yang, J.; Avila, C.; Hagag, N.; Senzel, L.; Ma, Y. SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood 2011, 118, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Mann, Z.; Sengar, M.; Verma, Y.K.; Rajalingam, R.; Raghav, P.K. Hematopoietic Stem Cell Factors: Their Functional Role in Self-Renewal and Clinical Aspects. Front. Cell Dev. Biol. 2022, 10, 664261. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Pellin, D.; Scala, S.; Dionisio, F.; Basso-Ricci, L.; Leonardelli, L.; Scaramuzza, S.; Baricordi, C.; Ferrua, F.; Cicalese, M.P.; et al. In vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell 2016, 19, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Zaboikin, M.; Srinivasakumar, N.; Schuening, F. Gene therapy with drug resistance genes. Cancer Gene Ther. 2006, 13, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef]
- Abadir, E.; Bryant, C.; Larsen, S.; Clark, G.J. Targeting the niche: Depleting haemopoietic stem cells with targeted therapy. Bone Marrow Transplant. 2019, 54, 961–968. [Google Scholar] [CrossRef]
- Bacigalupo, A.; Ballen, K.; Rizzo, D.; Giralt, S.; Lazarus, H.; Ho, V.; Apperley, J.; Slavin, S.; Pasquini, M.; Sandmaier, B.M.; et al. Defining the intensity of conditioning regimens: Working definitions. Biol. Blood Marrow Transplant. 2009, 15, 1628–1633. [Google Scholar] [CrossRef]
- Pond, G.R.; Lipton, J.H.; Messner, H.A. Long-Term Survival after Blood and Marrow Transplantation: Comparison with an Age- and Gender-Matched Normative Population. Biol. Blood Marrow Transplant. 2006, 12, 422–429. [Google Scholar] [CrossRef]
- Gagelmann, N.; Kröger, N. Dose intensity for conditioning in allogeneic hematopoietic cell transplantation: Can we recommend “when and for whom” in 2021? Haematologica 2021, 106, 1794–1804. [Google Scholar] [CrossRef]
- Atilla, E.; Ataca Atilla, P.; Demirer, T. A Review of Myeloablative vs Reduced Intensity/Non-Myeloablative Regimens in Allogeneic Hematopoietic Stem Cell Transplantations. Balk. Med. J. 2017, 34, 1–9. [Google Scholar] [CrossRef]
- Uchida, N.; Patrick Weitzel, R.; Platner, C.; Ballantine, J.; Bonifacino, A.C.; Price, S.D.; Krouse, A.E.; Metzger, M.E.; Donahue, R.E.; Tisdale, J.F. 294. Myeloablative Conditioning Is Required for Efficient Engraftment of Gene-Modified Cells and Prevention of Antibody Production Against Transgene Products in a Rhesus Stem Cell Gene Therapy Model. Mol. Ther. 2015, 23, S118–S119. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Aiuti, A. The Role of Conditioning in Hematopoietic Stem-Cell Gene Therapy. Hum. Gene Ther. 2016, 27, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Gratwohl, A. The EBMT risk score. Bone Marrow Transplant. 2012, 47, 749–756. [Google Scholar] [CrossRef]
- Lachmann, N.; Brennig, S.; Phaltane, R.; Flasshove, M.; Dilloo, D.; Moritz, T. Myeloprotection by Cytidine Deaminase Gene Transfer in Antileukemic Therapy. Neoplasia 2013, 15, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, R.; Baturin, D.; Fosmire, S.; Freed, B.; Porter, C.C. Knockdown of HPRT for Selection of Genetically Modified Human Hematopoietic Progenitor Cells. PLoS ONE 2013, 8, e59594. [Google Scholar] [CrossRef]
- Beard, B.C.; Trobridge, G.D.; Ironside, C.; McCune, J.S.; Adair, J.E.; Kiem, H.-P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Investig. 2010, 120, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Glassner, B.J.; Weeda, G.; Allan, J.M.; Broekhof, J.L.M.; Carls, N.H.E.; Donker, I.; Engelward, B.P.; Hampson, R.J.; Hersmus, R.; Hickman, M.J.; et al. DNA repair methyltransferase (Mgmt) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis 1999, 14, 339–347. [Google Scholar] [CrossRef]
- Hacke, K.; Treger, J.A.; Bogan, B.T.; Schiestl, R.H.; Kasahara, N. Genetic modification of mouse bone marrow by lentiviral vector-mediated delivery of hypoxanthine-Guanine phosphoribosyltransferase short hairpin RNA confers chemoprotection against 6-thioguanine cytotoxicity. Transplant. Proc. 2013, 45, 2040–2044. [Google Scholar] [CrossRef]
- Czechowicz, A.; Palchaudhuri, R.; Scheck, A.; Hu, Y.; Hoggatt, J.; Saez, B.; Pang, W.W.; Mansour, M.K.; Tate, T.A.; Chan, Y.Y.; et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 2019, 10, 617. [Google Scholar] [CrossRef] [PubMed]
- Palchaudhuri, R.; Saez, B.; Hoggatt, J.; Schajnovitz, A.; Sykes, D.B.; Tate, T.A.; Czechowicz, A.; Kfoury, Y.; Ruchika, F.; Rossi, D.J.; et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 2016, 34, 738–745. [Google Scholar] [CrossRef]
- Czechowicz, A.; Kraft, D.; Weissman Irving, L.; Bhattacharya, D. Efficient Transplantation via Antibody-Based Clearance of Hematopoietic Stem Cell Niches. Science 2007, 318, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-S.; Logan, A.C.; Chhabra, A.; Pang, W.W.; Czechowicz, A.; Tate, K.; Le, A.; Poyser, J.; Hollis, R.; Kelly, B.V.; et al. Anti-human CD117 antibody-mediated bone marrow niche clearance in nonhuman primates and humanized NSG mice. Blood 2019, 133, 2104–2108. [Google Scholar] [CrossRef]
- Lanieri, L.; Lamothe, T.L.; Miske, O.; McDonough, S.M.; Sarma, G.N.; Bhattarai, P.; Latimer, K.; Dushime, J.; Jain, N.; Palchaudhuri, R.; et al. A Single Dose of a Novel Anti-Human CD117-Amanitin Antibody Drug Conjugate (ADC) Engineered for a Short Half-Life Provides Dual Conditioning and Anti-Leukemic Activity and Extends Survival Compared to Standard of Care in Multiple Preclinical Models of Acute Myeloid Leukemia (AML). Blood 2020, 136 (Suppl. S1), 47–48. [Google Scholar] [CrossRef]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A Phase II Randomized Study of HIV-Specific T-Cell Gene Therapy in Subjects with Undetectable Plasma Viremia on Combination Antiretroviral Therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef]
- Muvarak, N.; Li, H.; Lahusen, T.; Galvin, J.A.; Kumar, P.N.; Pauza, C.D.; Bordon, J. Safety and durability of AGT103-T autologous T cell therapy for HIV infection in a Phase 1 trial. Front. Med. 2022, 9, 1044713. [Google Scholar] [CrossRef]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.L.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Walker, R.E.; Carter, C.S.; Muul, L.; Natarajan, V.; Herpin, B.R.; Leitman, S.F.; Klein, H.G.; Mullen, C.A.; Metcalf, J.A.; Baseler, M.; et al. Peripheral expansion of pre-existing mature T cells is an important means of CD4+ T-cell regeneration HIV-infected adults. Nat. Med. 1998, 4, 852–856. [Google Scholar] [CrossRef]
- VandenDriessche, T.; Chuah, M.K.; Chiang, L.; Chang, H.K.; Ensoli, B.; Morgan, R.A. Inhibition of clinical human immunodeficiency virus (HIV) type 1 isolates in primary CD4+ T lymphocytes by retroviral vectors expressing anti-HIV genes. J. Virol. 1995, 69, 4045–4052. [Google Scholar] [CrossRef]
- Morgan, R.A.; Walker, R.; Carter, C.S.; Natarajan, V.; Tavel, J.A.; Bechtel, C.; Herpin, B.; Muul, L.; Zheng, Z.; Jagannatha, S.; et al. Preferential Survival of CD4+ T Lymphocytes Engineered with Anti-Human Immunodeficiency Virus (HIV) Genes in HIV-Infected Individuals. Hum. Gene Ther. 2005, 16, 1065–1074. [Google Scholar] [CrossRef]
- Jacobson, J.M.; Jadlowsky, J.K.; Lacey, S.F.; Fraietta, J.A.; Plesa, G.; Chono, H.; Lee, D.H.; Kulikovskaya, I.; Bartoszek, C.; Chen, F.; et al. Autologous CD4 T Lymphocytes Modified with a Tat-Dependent, Virus-Specific Endoribonuclease Gene in HIV-Infected Individuals. Mol. Ther. 2021, 29, 626–635. [Google Scholar] [CrossRef]
- Macpherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Zack, J.A.; Macpherson, J.L.; Symonds, G.P. Phase I/II Clinical Trials Using Gene-Modified Adult Hematopoietic Stem Cells for HIV: Lessons Learnt. Stem Cells Int. 2011, 2011, 393698. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Binder-Scholl, G.; Mukherjee, R.; Brady, T.; Rebello, T.; Humeau, L.; Kalos, M.; Papasavvas, E.; Montaner, L.J.; et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 2013, 121, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Javien, J.; Nolta, J.A.; Bauer, G. Preintegration HIV-1 Inhibition by a Combination Lentiviral Vector Containing a Chimeric TRIM5α Protein, a CCR5 shRNA, and a TAR Decoy. Mol. Ther. 2009, 17, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34 + cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [PubMed]
- Ledger, S.; Howe, A.; Turville, S.; Aggarwal, A.; Savkovic, B.; Ong, A.; Wolstein, O.; Boyd, M.; Millington, M.; Gorry, P.R.; et al. Analysis and dissociation of anti-HIV effects of shRNA to CCR5 and the fusion inhibitor C46. J. Gene Med. 2018, 20, e3006. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.W.; Haworth, K.G.; Burke, B.P.; Polacino, P.; Norman, K.K.; Adair, J.E.; Hu, S.-L.; Bartlett, J.S.; Symonds, G.P.; Kiem, H.-P. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol. Ther. Methods Clin. Dev. 2016, 3, 16007. [Google Scholar] [CrossRef] [PubMed]
- Méndez, C.; Ledger, S.; Petoumenos, K.; Ahlenstiel, C.; Kelleher, A.D. RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology 2018, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Kariya, R.; Marks, K.; Hattori, S.; Ahlenstiel, C.; Symonds, G.; Okada, S.; Kelleher, A. Transcriptional gene silencing limits CXCR4-associated depletion of bone marrow CD34+ cells in HIV-1 infection: Erratum. AIDS 2018, 32, 2857–2858. [Google Scholar] [CrossRef] [PubMed]
- Dhody, K.; Kazempour, K.; Pourhassan, N.; Paul, J.M. PRO 140 (leronlimab) SC: Long-acting single-agent maintenance therapy (SAMT) for HIV-1 infection. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, WA, USA, 4–7 March 2019. [Google Scholar]
- Bhowmik, R.; Chaubey, B. CRISPR/Cas9: A tool to eradicate HIV-1. AIDS Res. Ther. 2022, 19, 58. [Google Scholar] [CrossRef]
- Xiao, Q.; Guo, D.; Chen, S. Application of CRISPR/Cas9-Based Gene Editing in HIV-1/AIDS Therapy. Front. Cell. Infect. Microbiol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vector | Type | Insert Size Kilobase (kb) | Reference |
|---|---|---|---|
| Adenoviral | First | 4.5 | [113] |
| Second | 10.5 | ||
| Third | 36 | ||
| Adeno-Virus Associated Vectors (AAVs) | 5 | [113,114] | |
| Bi-directional vectors | Dual gene cassettes—10 | [113] | |
| Self-complementary AAVs (double stranded genome packaged) | Approx. 2.5 | [113] | |
| Gamma retroviral | 5.5 optimal, but up to 10 | [115] | |
| Lentiviral | First | Up to 10 | [116] |
| Second | Up to 10 | ||
| Third | Up to 10 | ||
| Fourth | Up to 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitawi, R.; Ledger, S.; Kelleher, A.D.; Ahlenstiel, C.L. Advances in HIV Gene Therapy. Int. J. Mol. Sci. 2024, 25, 2771. https://doi.org/10.3390/ijms25052771
Kitawi R, Ledger S, Kelleher AD, Ahlenstiel CL. Advances in HIV Gene Therapy. International Journal of Molecular Sciences. 2024; 25(5):2771. https://doi.org/10.3390/ijms25052771
Chicago/Turabian StyleKitawi, Rose, Scott Ledger, Anthony D. Kelleher, and Chantelle L. Ahlenstiel. 2024. "Advances in HIV Gene Therapy" International Journal of Molecular Sciences 25, no. 5: 2771. https://doi.org/10.3390/ijms25052771
APA StyleKitawi, R., Ledger, S., Kelleher, A. D., & Ahlenstiel, C. L. (2024). Advances in HIV Gene Therapy. International Journal of Molecular Sciences, 25(5), 2771. https://doi.org/10.3390/ijms25052771