1. Introduction
Tumor cells have a high metabolic demand due to their increased ability to proliferate. Otto Warburg discovered that this metabolic demand is met by anaerobic glycolysis despite the availability of oxygen, in a process known as the Warburg effect [
1]. The increased use of anaerobic glycolysis allows tumor cells to produce energy rapidly. However, this also leads to increased production of lactate, resulting in an acidic tumor microenvironment. To prevent cellular acidification, tumor cells upregulate the expression of the lactate transporter MCT4, allowing them to recycle lactate as their main energy source [
2]. Because anaerobic glycolysis is an inefficient method of meeting total energy requirements, tumor cells increase their rate of glycolysis 20- to 30-fold compared to healthy cells [
3]. This is accompanied by an upregulation of glucose transporters (GLUT) and thus increased glucose uptake. In tumor cells, the expression of GLUT-1 and GLUT-3 in particular is upregulated [
2,
4]. In addition, increased glucose consumption by the tumor cells leads to nutrient deprivation within the tumor microenvironment (TME). The available energy sources in the TME in turn attenuate the functionality of immune cells such as natural killer (NK) cells [
5].
NK cells are lymphocytes belonging to the innate immune system and account for 5–15% of peripheral blood mononuclear cells. They play an important role in immune responses against infections and cancer by killing infected or transformed cells [
6]. NK cell cytotoxicity is mediated by activating NK cell receptors such as NKp30, NKp46, NKG2D, 2B4, and CD16 [
7]. NK cell effector functions are energy-consuming processes, so cellular metabolism has a major influence on their activity. Two metabolic pathways play important roles in the activation and proliferation of NK cells: oxidative phosphorylation (OxPhos) and glycolysis [
8]. Previous studies have shown that NK cells primarily use OxPhos in the resting state, whereas upon activation they increase glycolysis and upregulate OxPhos [
5]. This increases energy production and the synthesis of building blocks required for biosynthetic processes that support proliferation and effector molecule production in NK cells [
9,
10]. Thus, stimulation of NK cells by cytokines such as IL-15 and IL-12 leads to their proliferation and upregulation of OxPhos and glycolysis [
5]. In line with this, Keppel et al. have shown that inhibition of OxPhos impairs receptor-mediated IFN-γ production [
11]. While glucose is an important source of energy, glutamine and fatty acid metabolism are also important for several cellular functions [
12]. Thus, glutamine is known to exert an important influence on NK cell metabolism and effector functions by contributing to the maintenance of the transcription factor cMyc [
13]. In contrast, the role of fatty acids in the metabolism of NK cells has not yet been thoroughly investigated.
Given the high glucose dependence of tumor cells, glucose transporters are potential targets for cancer therapy. On one hand, GLUT inhibition could reduce the proliferation of tumor cells, and on the other hand, the TME would have enough glucose for the functional capabilities of immune cells. Based on this, we have recently developed the GLUT inhibitors Glutor [
14] and Glupin [
15], which are potent inhibitors of glucose uptake. We recently showed that Glutor, an inhibitor of GLUT1, 2, and 3, blocks NK cell proliferation, inhibits IFN-γ secretion, and reduces degranulation [
16], which shows that GLUT function is also important for NK cell activities. Here, we were interested in investigating the effects of Glupin, which is a slightly less potent inhibitor of GLUT1 and 3. We treated primary human NK cells with Glupin in short-term and long-term experiments and analyzed their functions, phenotype, and proliferation. Our results show that short-term as well as long-term treatment with Glupin has no negative effect on NK cell activities, except for decreased IFN-γ secretion after long-term treatment. Interestingly, long-term treatment even results in an increase in the serial killing capacity of NK cells.
3. Discussion
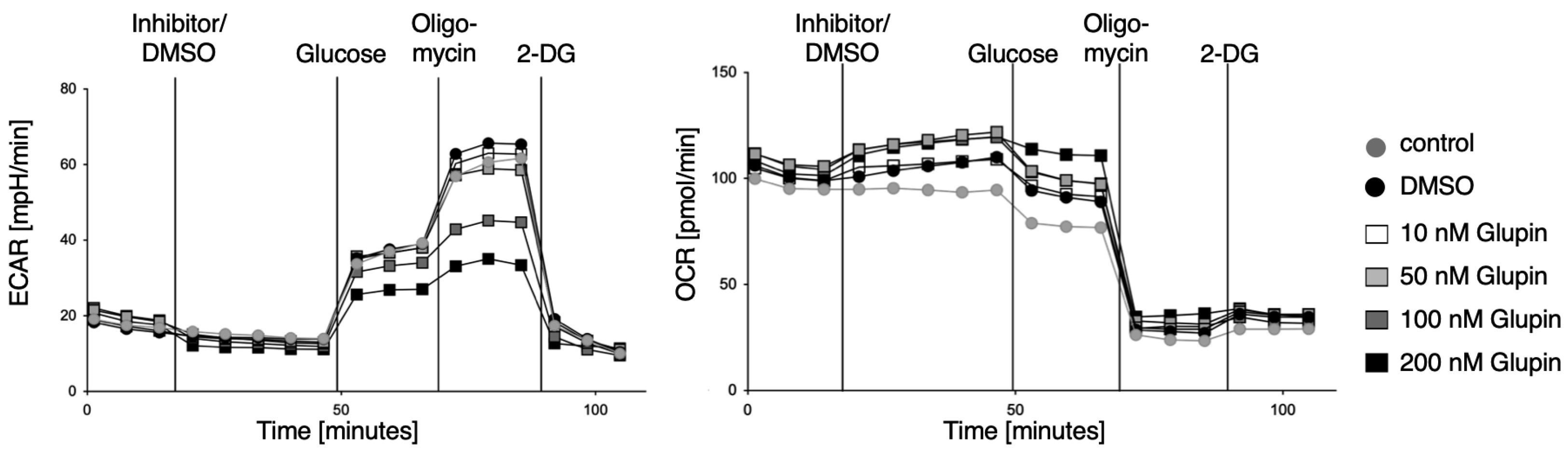
Here we characterized the effect of the GLUT inhibitor Glupin on the functional activities of human NK cells. Upon the blocking of glucose uptake by Glupin, NK cells immediately showed reduced glycolysis and increased OxPhos, demonstrating their fuel flexibility. However, these acute changes in their metabolism did not affect their functions, as NK cell cytotoxicity and IFN-γ production were not altered upon acute Glupin treatment. This demonstrates that NK cell activities are very robust and are not affected by short-term changes in metabolism and energy production, which is consistent with our previous findings [
16]. This could be due to the intracellular storage of glucose and the use of alternative energy sources such as glutamine or fatty acids. Additionally, NK cell cytotoxicity relies mostly on pre-formed effector proteins that are stored within the lytic granules and that just need to be mobilized upon NK cell activation [
21]. Therefore, little energy is needed for their immediate effector functions, and it has been described that the low basal metabolic rate of resting NK cells is sufficient to promote acute NK cell effector responses [
11].
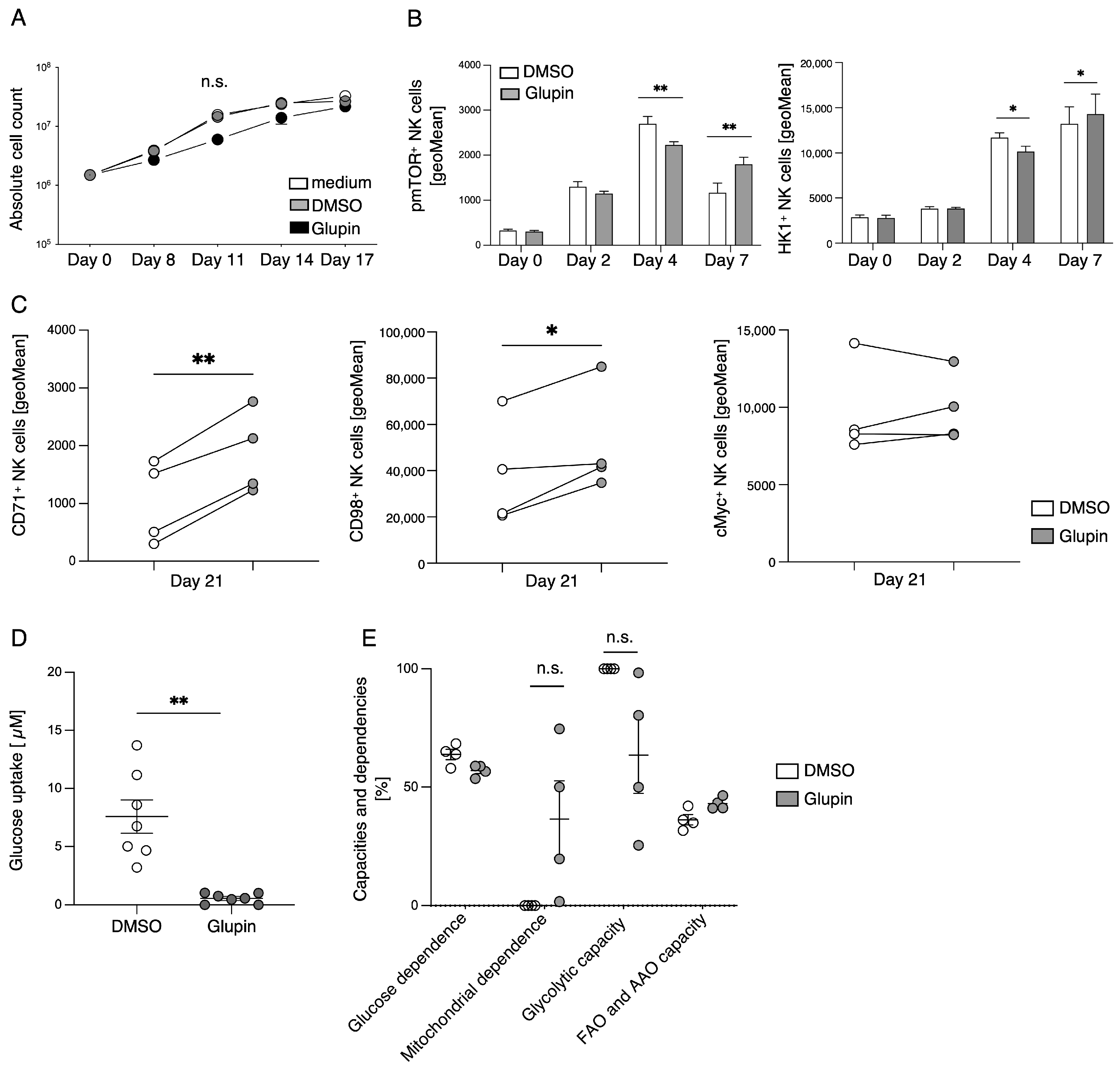
The stimulation of resting NK cells with feeder cells and cytokines induces NK cell proliferation and activation. This is accompanied by a strong increase in glycolytic activity to provide sufficient energy and precursors for anabolic processes [
9,
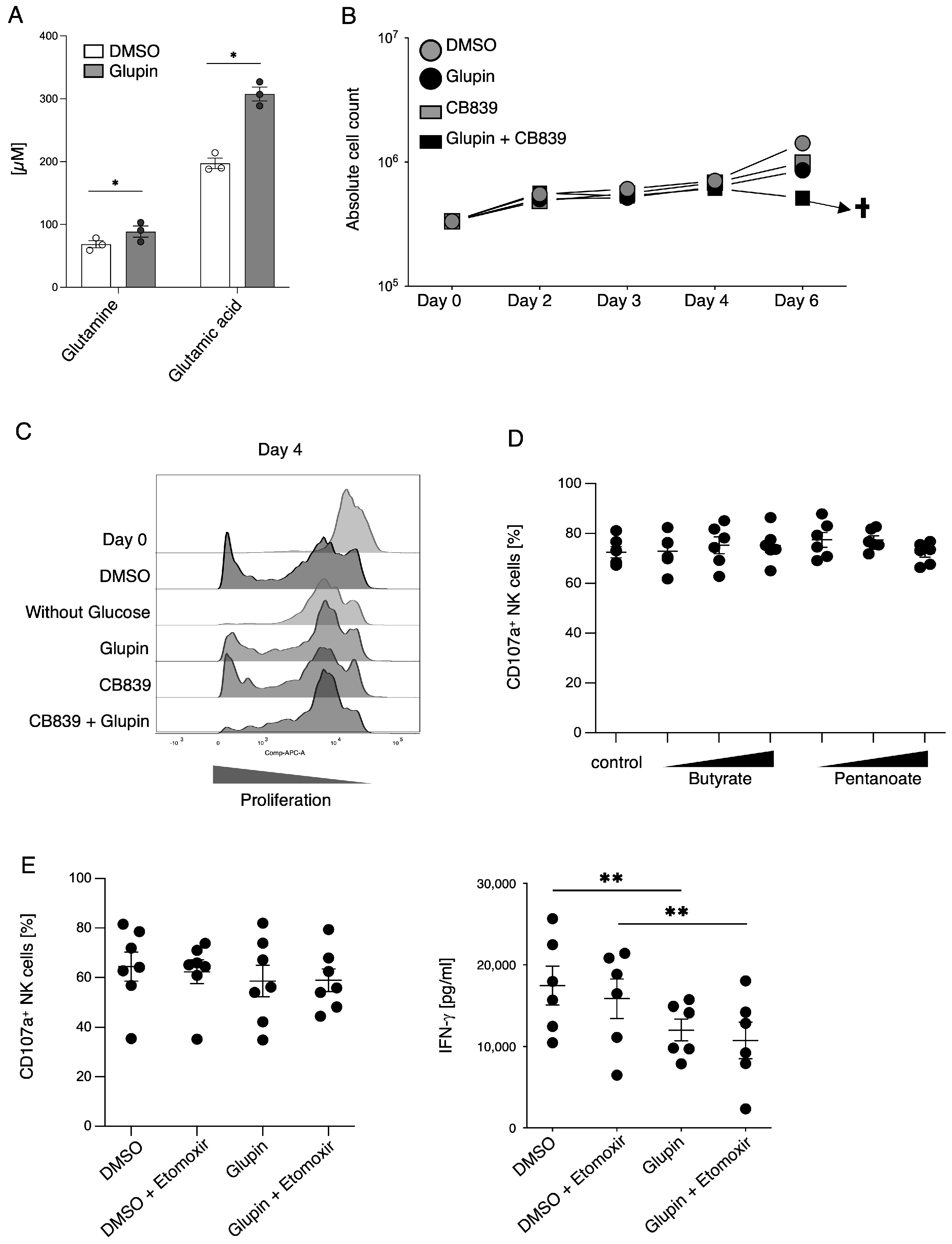
10]. In the presence of Glupin, glucose uptake and therefore glycolysis were severely reduced. Surprisingly, Glupin-treatment only resulted in decreased proliferation for the first 11 days of cultivation. Thereafter, NK cells showed a normal proliferation, suggesting that the Glupin-treated NK cells have to adapt to alternative fuels. Our metabolic profiling found increased concentrations of glutamine and glutamic acid in the Glupin-cultured NK cells. Glutamine is an important fuel for NK cell proliferation and activation [
13]. Blocking glutamine metabolism using CB839 in addition to blocking GLUT via Glupin completely blocked NK cell proliferation and resulted in cell death. Therefore, glutamine metabolism is necessary for the continued proliferation and survival of Glupin-treated NK cells. Interestingly, when we previously used the more potent GLUT inhibitor Glutor, NK cell proliferation was completely inhibited [
16]. Similarly, NK cell proliferation was much more inhibited in glucose-free medium compared to Glupin treatment (
Figure 5C). This suggests that while Glupin severely limits glucose uptake, it does not completely block it. This limited glucose uptake may be necessary to allow the NK cells to switch to alternative energy sources for their proliferation and to retain their functional activity.
Cytokines such as IL-2, IL-15, and IL-21 are known to relay their signals via mTOR, and mTOR plays an important role in the maturation, activation, and metabolism of NK cells [
22,
23]. Glupin-treated NK cells displayed a delayed increase in the phosphorylation of mTOR in comparison to the control NK cells. This could indicate counter-regulation due to decreased glucose availability. CD98 (SLC3A2) is an amino acid transporter which is upregulated on proliferating and activated lymphocytes [
24]. Cytokine stimulation of NK cells leads to an upregulation of their nutrient receptors, such as CD98 and CD71, to fulfil their biosynthetic requirements [
25]. Therefore, the increased expression of CD98 and CD71 on Glupin-treated NK cells is a further indication of their metabolic changes in response to a lack of glucose. This is supported by our metabolic SCENITH analysis, which showed that Glupin-treated NK cells had reduced flexibility to use glucose as fuel, and that these NK cells instead strongly depend on mitochondrial OxPhos, showing higher mitochondrial dependency than control NK cells.
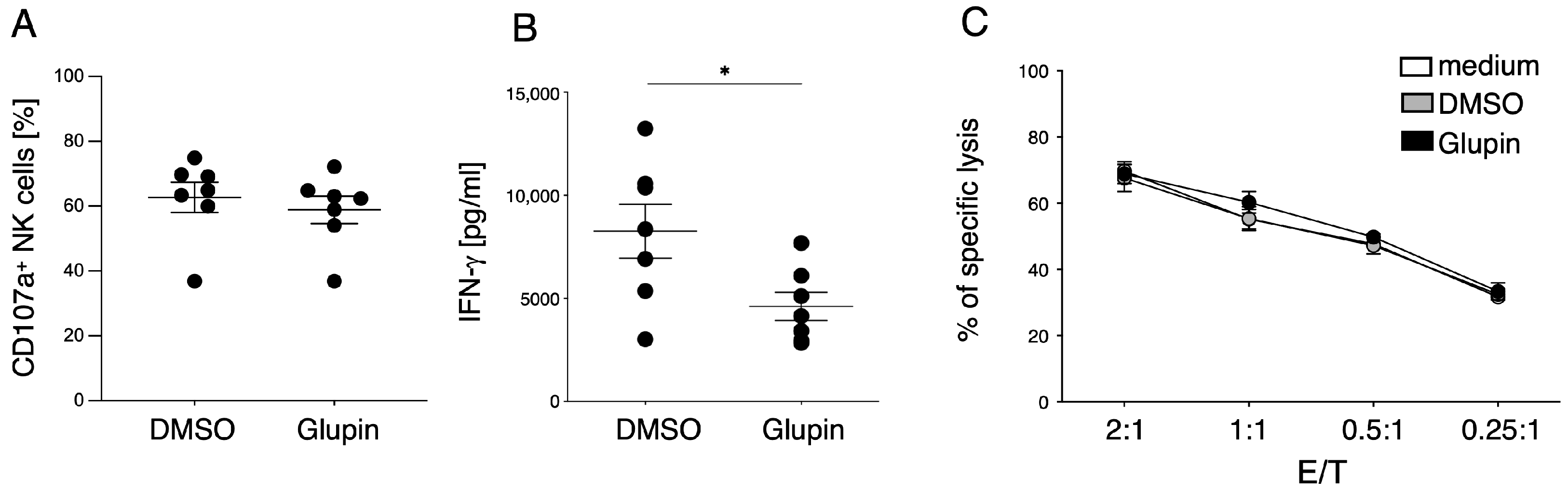
Although long term Glupin-treated NK cells had strongly reduced glucose uptake and clear metabolic changes, their degranulation and cytotoxic activity were not impaired. This contrasts with NK cells treated with Glutor, a more potent GLUT inhibitor, which showed reduced degranulation and a block in proliferation [
16]. Therefore, the energy obtained via glutamine metabolism and other metabolic adaptations seems not only important for proliferation, but also for the maintenance of NK cell cytotoxic activities. In contrast, IFN-γ production by Glupin-treated NK cells was clearly reduced, demonstrating different metabolic requirements for NK cell cytotoxicity and cytokine production [
11].
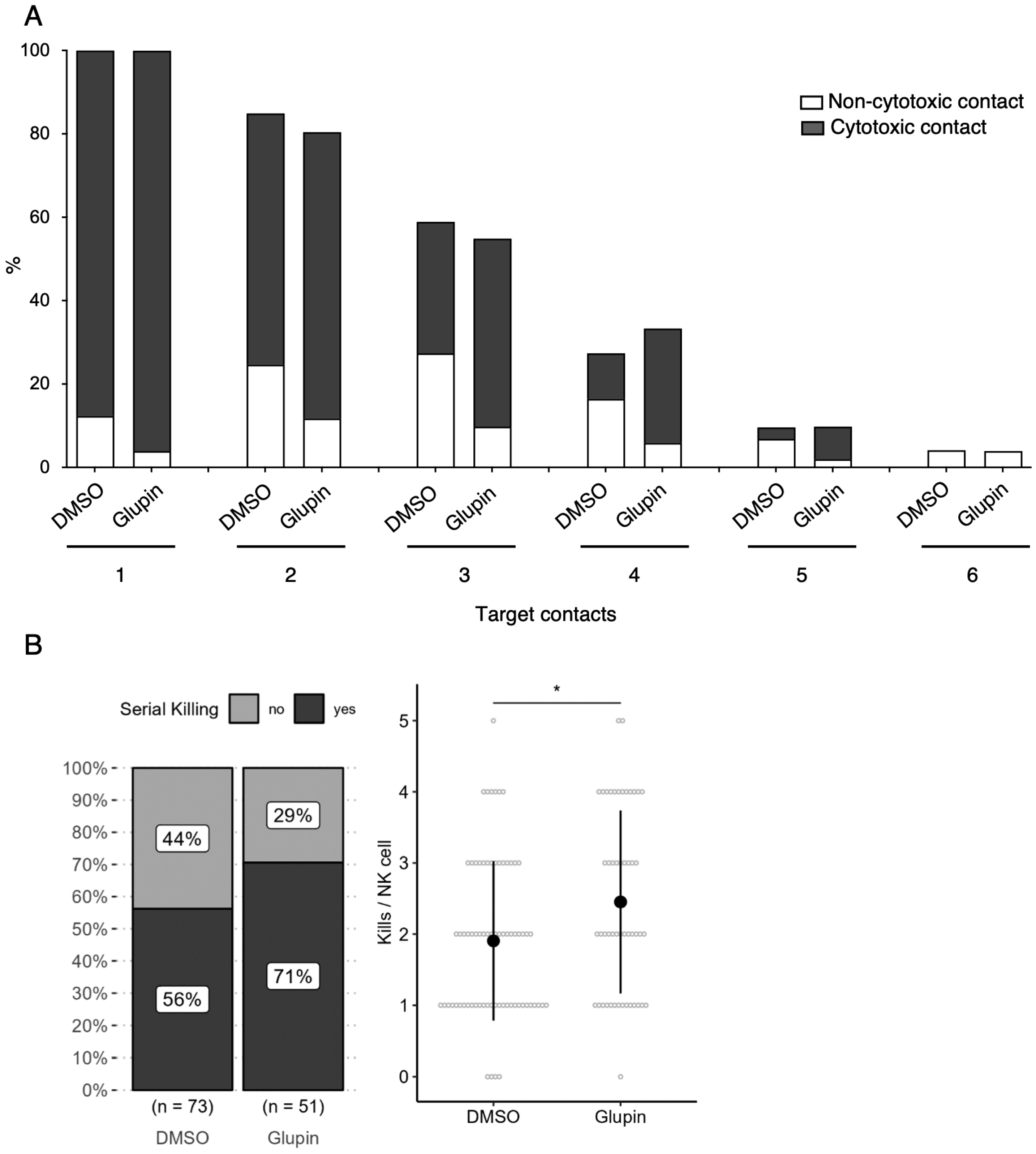
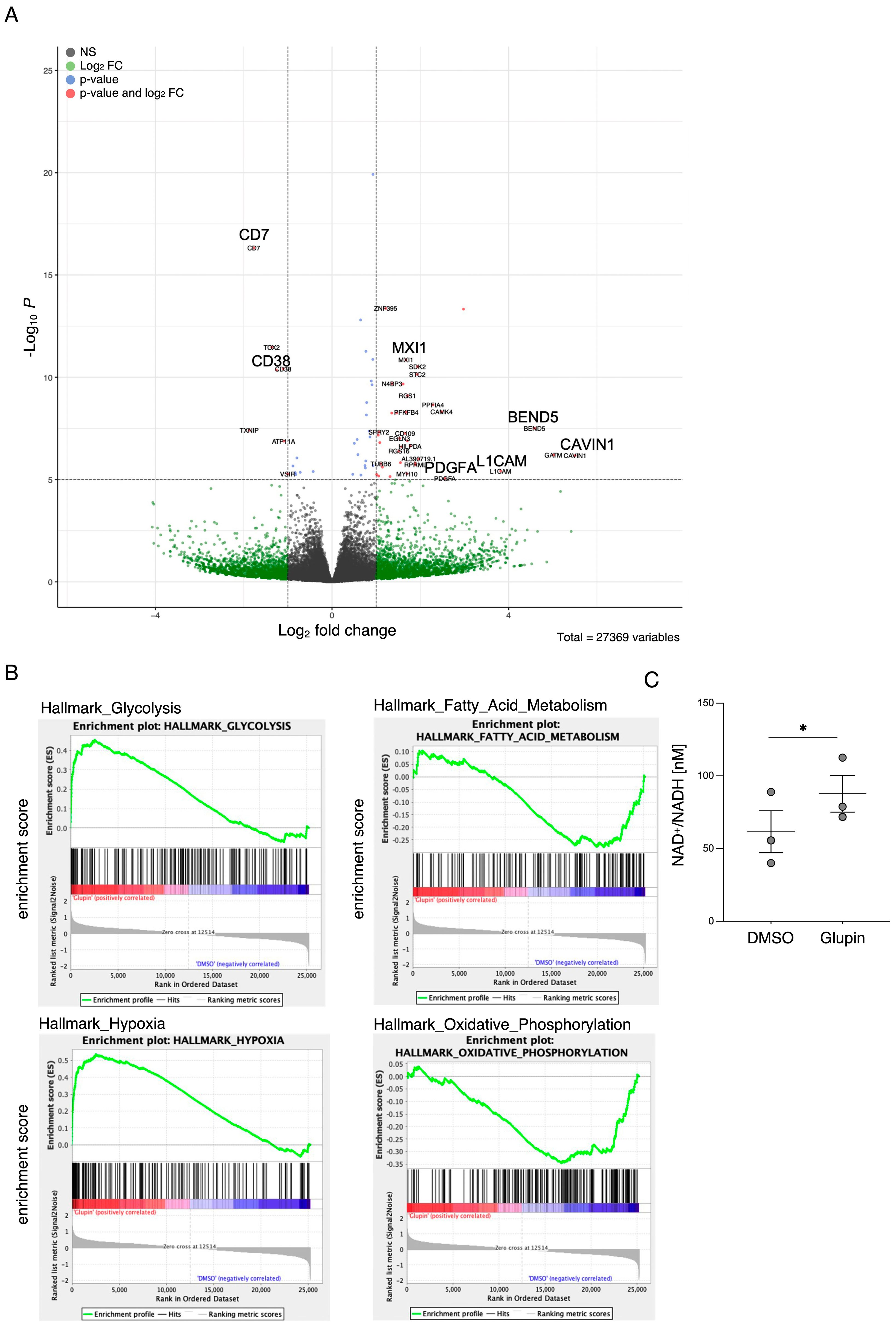
Interestingly, long-term Glupin-treated NK cells showed increased serial killing activity; we found a slightly higher percentage of serial killers and more kills per NK cell. Our data excluded alternative energy sources such as glutamine or fatty acids as solely responsible for this effect. This is consistent with a previous study, which showed a negative effect of fatty acid metabolism on NK cell function [
26]. However, we found increased levels of granzyme B in Glupin-treated NK cells. Additionally, a down-regulation of the NADase CD38 was revealed in our RNAseq and in our flow cytometry analysis. This was consistent with an increase in NAD
+ levels in Glupin-treated NK cells. Interestingly, antibody-mediated degradation of CD38 was shown to increase NK cell cytotoxic activity [
27], and CD38 negative NK cells possess higher metabolic fitness [
28]. However, CRISPR/Cas9-mediated deletion of CD38 in primary human NK cells did not result in increased serial killing activity. Therefore, a complete absence of CD38 alone cannot recapitulate our phenotype. In T cells, a reduced expression of CD38 was shown to be associated with higher NAD+, enhanced oxidative phosphorylation, higher glutaminolysis, and altered mitochondrial dynamics that improved their tumor control [
29]. This is reminiscent of many of the changes we found in Glupin-treated NK cells and suggests that the reduced CD38 levels, in combination with the increased granzyme B expression and the altered metabolism, may be responsible for their increased serial killing activities.
In summary, reducing glucose uptake via Glupin has only a minor impact on NK cell effector functions and can increase their serial killing capacity while additionally inhibiting the proliferation of tumor cells. This demonstrates that cellular metabolism can be an attractive target for immune-mediated tumor therapies.
4. Materials and Methods
4.1. Isolation of NK Cells and Cell Culture
Human NK cells were isolated from peripheral blood mononuclear cells of healthy donors with the Dynabeads® UntouchedTM Human NK Cell-Kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Darmstadt, Germany). The purity of the NK cells was routinely 95–97% CD56+, CD3−, 7AAD− as determined by flow cytometry. The study was approved by the Ethics Committee of the Leibniz Research Center (#213) and all blood donors gave informed consent. For experiments with resting NK cells, isolated NK cells were rested in medium overnight and then used for experiments. To generate pre-activated NK cells, isolated NK cells were seeded in 96-well round-bottom plates with irradiated feeder cells (K562-mbIL15-41BBL) with 200 U/mL IL-2 (National Institutes of Health Cytokine Repository, Frederick, MD, USA) and 100 ng/mL IL-21 (Miltenyi Biotec, Bergisch Gladbach, Germany). On day 8, NK cells were re-stimulated with fresh feeder cells and then cultured with 100 U/mL IL-2. On day 14, 2.5 ng/mL recombinant IL-15 (PAN Biotech, Aidenbach, Germany) was added, and after three weeks NK cells were used for experiments. In cases of long-term treatment, Glupin (100 nM) or DMSO (0.1%) were added to the culture every 72 h. For inhibition of CPT1, long-term treated NK cells were treated with 1 µM of the CPT1-inhibitor Etomoxir (Sigma-Aldrich, St. Louis, MO, USA) for 24 h and analyzed for degranulation or IFN-γ secretion as described below. For cultivation with short-chain fatty acids, expanded NK cells were cultivated with 0.1% DMSO, Pentanoate (Sigma-Aldrich; 2.5 mM, 5 mM, 7.5 mM), or Butyrate (Sigma-Aldrich; 0.5 µM, 0.75 µM, 1 µM) for 72 h, followed by a degranulation-assay against K562 targets. K562 cells were cultured in DMEM with 10% FCS and 1% penicillin/streptomycin (all from Thermo Fisher Scientific). MCF7 cells were cultured in DMEM with 10% FCS and 1% penicillin/streptomycin, 1% Sodium Pyruvate, 0.1% NeAA, and 0.1% Insulin (all from Thermo Fisher Scientific). HepG2 cells were cultured in DMEM with 10% FCS and 1% penicillin/streptomycin.
4.2. RTCA Analysis
3 × 104 MCF7 cells or HepG2 cells were seeded in e-plates and placed into the xCELLigence device (OMNI Life Science, Bremen, Germany). After 16 h of measurement, 3.75 × 103 NK cells in combination with DMSO (0.1%) or Glupin (100 nM) were added. Cell index was measured every 10 min for 72 h.
4.3. 51Chromium-Release-Assay
Cytotoxicity was analyzed using a standard 4 h chromium release assay, as already described [
30]. In short, target cells were labeled for 1 h at 37 °C and 5% CO
2 on a Rotator with
51Cr (Hartmann Analytic, Braunschweig, Germany). Afterwards, target cells were washed twice and added to the NK cells in a 96 v-well plate. NK cells were washed to remove any residual inhibitor and serially diluted (1:2) starting at an E:T 2:1. After 4 h incubation time, supernatant was collected and analyzed with the Wizard
2 (PerkinElmer, Waltham, MA, USA) gamma counter. The percentage of specific lysis was determined as follows:
4.4. Flow Cytometry
To measure degranulation, resting or pre-activated NK cells were treated with Glupin (100 nM) or DMSO (0.1%) for 30 min and then stimulated via plate-bound CD16-mAb (2 µg/mL), NKp30 (2 µg/mL), NKG2D + 2B4 (2 µg/mL), or control IgG (2 µg/mL) in the presence of anti-CD107a PE-Cy5 for 2 h. CD107a-expression was then measured by flow cytometry. For intracellular staining, NK cells were stained for live/dead (Zombie NIR; BioLegend, San Diego, CA, USA) and CD56 using anti-CD56 BV421 (Clone:NCAM16.2; BD Biosciences, San Jose, CA, USA) for 15 min at RT. Intracellular staining was performed using 2% paraformaldehyde for fixation, Permeabilizing Solution 2 (BD Bioscience) for permeabilization and anti-GLUT1 (PE; Clone:202915; R&D Systems, Minneapolis, MN, USA), anti-pmTOR (PE-Cy7; Clone:MRRBY; Thermo Fisher Scientific), and anti-HK1 (AF647; Clone:EPR10134(B); abcam, Cambridge, UK) or Granzyme B (AF647; Clone:GB11; BioLegend) and Perforin (FITC; Clone:dG9; BioLegend). Surface staining was performed using two different panels. Panel 1: Zombie NIR (BioLegend), CD56 (BUV805; Clone:B159; BD Biosciences), CD3 (BUV563; Clone:UCHT1; BD Biosciences), NKp46 (BV421; Clone:9E2; BioLegend), 2B4 (FITC; Clone:C1.7; BioLegend), NKp44 (PerCP-Cy5.5; Clone:P44-8; BioLegend), DNAM-1 (AF647; Clone:DX11; BD Biosciences), NKp30 (APC-Fire750; Clone:P30-15; BioLegend), CD16 (PE-Dazzle; Clone:3G8; BioLegend), and NKG2D (AF700; Clone:FAB139N; R&D Systems). Panel 2: CD38 (BUV395; Clone:HB7; BD Biosciences), NKG2C (BUV496; Clone:134591; BD Biosciences), CD3 (BUV563; Clone:UCHT1; BD Biosciences), KLRB1 (BUV661; Clone:NKR-3G10; BD Biosciences), CD56 (BUV805; Clone:B159; BD Biosciences), 4-1BB (BV421; Clone:4B4-1; BioLegend), KLRG1 (BV510; Clone:2F1; BioLegend), HLA-DR (BV605; Clone:L243; BioLegend), FasL (BV650; Clone:NOK-1, BD Biosciences), TIGIT (BV786; Clone:741182; BD Biosciences), CD11a (AF488; Clone:HI111; BioLegend), CD8 (AF532; Clone:RPA-T8; Thermo Fisher Scientific), CD27 (PerCP-Cy5.5; Clone:M-T271; BD Biosciences), NKG2A (PE; Clone:Z199; Beckmann Coulter, Brea, CA, USA), CTLA-4 (PE-Dazzle; Clone:BNI3; BioLegend), TRAIL (PE-Cy7; Clone:N2B2; BioLegend), PD-1 (AF647; Clone:EH12.1; BD Biosciences), CD18 (AF700; Clone:TS1/18; BioLegend), Tim-3 (APC-Fire 750; Clone:F38-2E2; BioLegend), and Zombie NIR (BioLegend). Cells were measured on a Cytek Aurora spectral flow cytometer and analyzed with FlowJo (Version 10.5.3, FlowJo, LLC, Ashland, OH, USA). Gating strategy: 1. Lymphocytes according to FSC/SSC. 2. Single cells according to FSC-H/FSC-A. 3. Live cells according to Zombie NIR−. 4. NK cells according to CD56+, CD3−.
4.5. SCENITH
Assays were performed as described in [
16]. In brief, NK cells were incubated in the presence or absence of Oligomycin (1 µM), 2-DG (100 nM) or Oligomycin + 2-DG for 30 min. Next, Puromycin (10 µg/mL) was added for 20 min followed by a wash using ice cold PBS. After an Fc-Block for 5 min, surface staining was performed using Zombie NIR (BioLegend), anti-CD56 BV421 (Clone:NCAM16.2; BD Biosciences), and anti-CD16 PE-Dazzle (Clone:3G8; BioLegend). Cells were then washed, fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) followed by intracellular staining using anti-Puromycin AF488 (Clone:12D10; Sigma-Aldrich) and anti-pmTOR PE-Cy7 (Clone:MRRBY; Thermo Fisher Scientific) and analysis using a Cytek Aurora flow cytometer. Glycolytic dependency, mitochondrial dependency, glycolytic capacity, and FAO (fatty acid oxidation) and AAO (amino acid oxidation) capacity were calculated using the formula as already described by Argüello et al. [
18].
4.6. ELISA
Resting or pre-activated NK cells were treated with Glupin (100 nM) or DMSO (0.1%) for 30 min followed by stimulation via plate-bound CD16-mAb (2 µg/mL), NKp30 (2 µg/mL), NKG2D + 2B4 (2 µg/mL), or control IgG (2 µg/mL), or via IL-12 (0.25 ng/µL) + IL-18 (1.25 ng/µL). as previously described [
31]. After 16 h, supernatants were collected and IFN-γ was analyzed using the ELISA MAX™ Deluxe Set Human (BioLegend).
4.7. Glucose-Uptake Assay
Glucose uptake-GloTM Assay (Promega, Fitchburg, WI, USA) was performed according to the manufacturer’s instructions. Luminescence was recorded using a GloMaxR instrument (Promega).
4.8. NAD/NADH-Assay
NK cells were analyzed for NAD+/NADH concentrations using NAD/NADH-Glo™ Assay (Promega), according to the manufacturer’s instructions. Measurement was done with a GloMaxR instrument.
4.9. Serial Killing Assays
Assays were performed as described in [
19]. In brief, microchips were seeded with MCF7 cells to obtain 60–80 tumor cells/well. After attachment of tumor cells, medium containing the dead cell stain SYTOX Blue (1:1000) was added and the microchip was placed into the incubation chamber of the ApoTome System with an Axio Observer 7 microscope (Zeiss, Jena, Germany) equipped, using a 20×/0.8 Plan-Apochromat objective and an incubation chamber with environmental control (37 °C, 5% CO
2, humidity device PM S1, PeCon GmbH, Erbach, Germany). NK cells were washed to remove any residual inhibitor and 10–20 NK cells per well were added. Time-lapse microscopy was started. For a duration of 16 h, a picture was taken every 3 min using an Axiocam 506 mono camera. SYTOX Blue was excited using the Colibri 7 LED-module 475 (filter set 38 HE LED) and brightfield was acquired using the TL LED module (both Zeiss, Jena, Germany).
4.10. Seahorse Analysis
The energetic phenotype of pre-activated NK cells was analyzed using the glycolysis stress test kit (Agilent, Santa Clara, CA, USA). In brief, culture plates were coated with poly-l-lysine, followed by the addition of pre-activated NK cells. Injection ports were loaded with 10 × concentrated substance (final concentration after injection: Port A: 100 nM Glupin/0.1% DMSO/medium; Port B: 10 mM Glucose; Port C: Oligomycin; Port D: 50 mM 2-DG) and the experiment was performed according to the manufacturer’s instructions (Agilent).
4.11. Metabolomics
For metabolomics, 10 × 106 long-term treated NK cells were lysed and analyzed by the Metabolomics Innovation Centre (TMIC Canada) using a combination of direct injection mass spectrometry with a reverse-phase LC-MS/MS custom assay for the targeted identification and quantification of up to 143 different endogenous metabolites including amino acids, acylcarnitines, biogenic amines and derivatives, glycerophospholipids, sphingolipids, and sugars. Isotope-labeled internal standards and other internal standards were used for metabolite quantification. Data analysis was done using Analyst 1.6.2.
4.12. RNA-Seq
10 × 106 long-term treated NK cells were used for RNA isolation using an RNAeasy Mini Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer’s instructions, with an additional step of DNAse treatment. RNA-sequencing were performed at the genomics and transcriptomics facility (GTF) at the University Hospital Essen. In brief, the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD was used for library preparation. Sequencing was performed on the Illumina NextSeq500. Reads were processed with standard Illumina base calling and trimmed with Trim Galore v0.6.0 using standard settings. Alignment was performed with hisat2 v2.2.1 to grch38 and standard settings. Raw counts were summed up using Summarize Overlaps from R-Package Genomic Alignments v1.8.4. RNA-Seq data were stored in the Gene expression omnibus archive (accession: GSE216156).
4.13. Statistics
Statistical analyses were performed using GraphPad Prism version 9 (GraphPad, La Jolla, CA, USA). The statistical tests used for the analyses are mentioned in the figure legends.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





