
Comprehensive Evolutionary Analysis of the SMXL Gene Family in Rosaceae: Further Insights into Its Origin, Expansion, Diversification, and Role in Regulating Pear Branching

{kind=link}
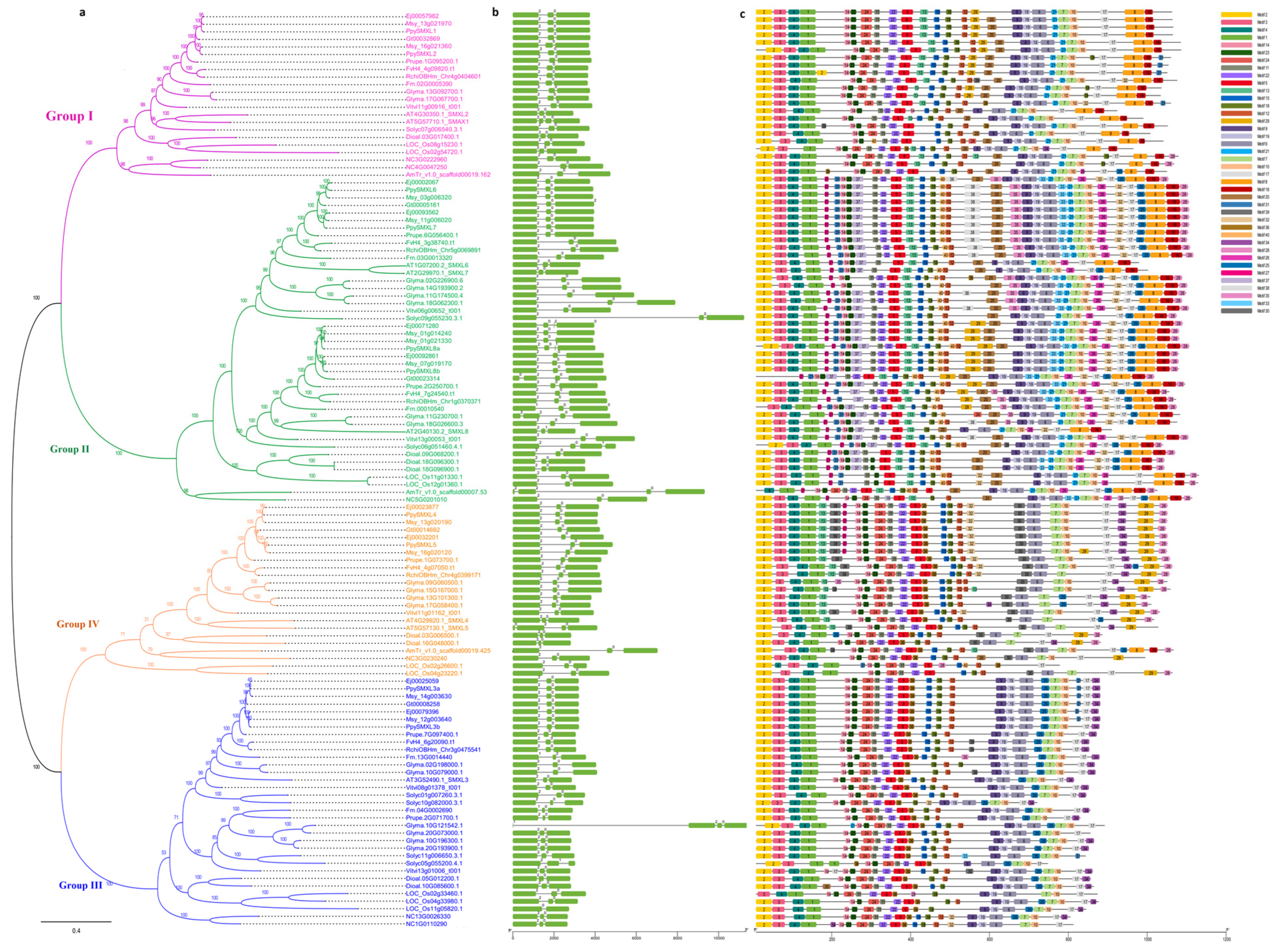{kind=link}
{kind=link}
{kind=link}
{kind=link}
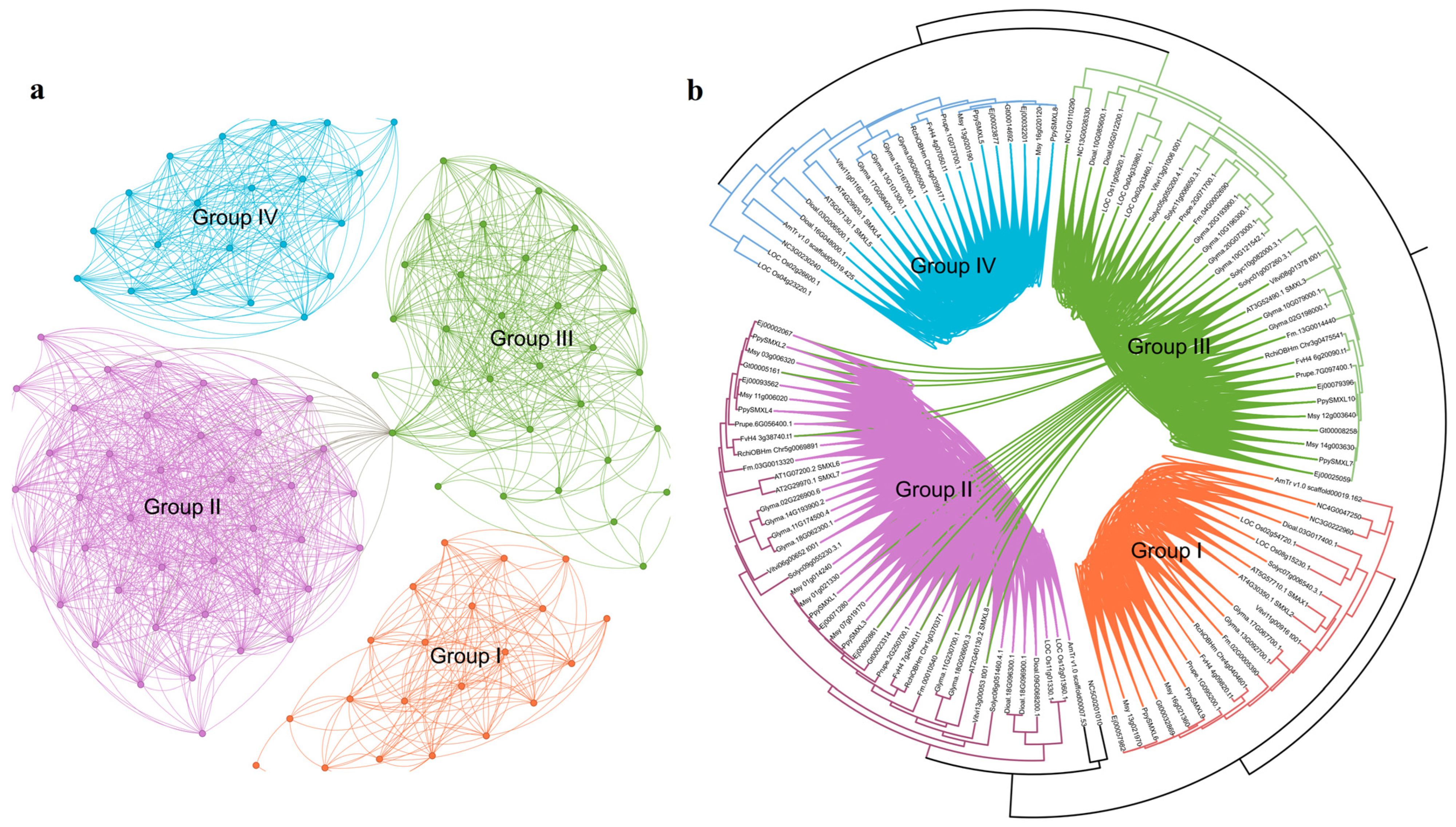{kind=link}
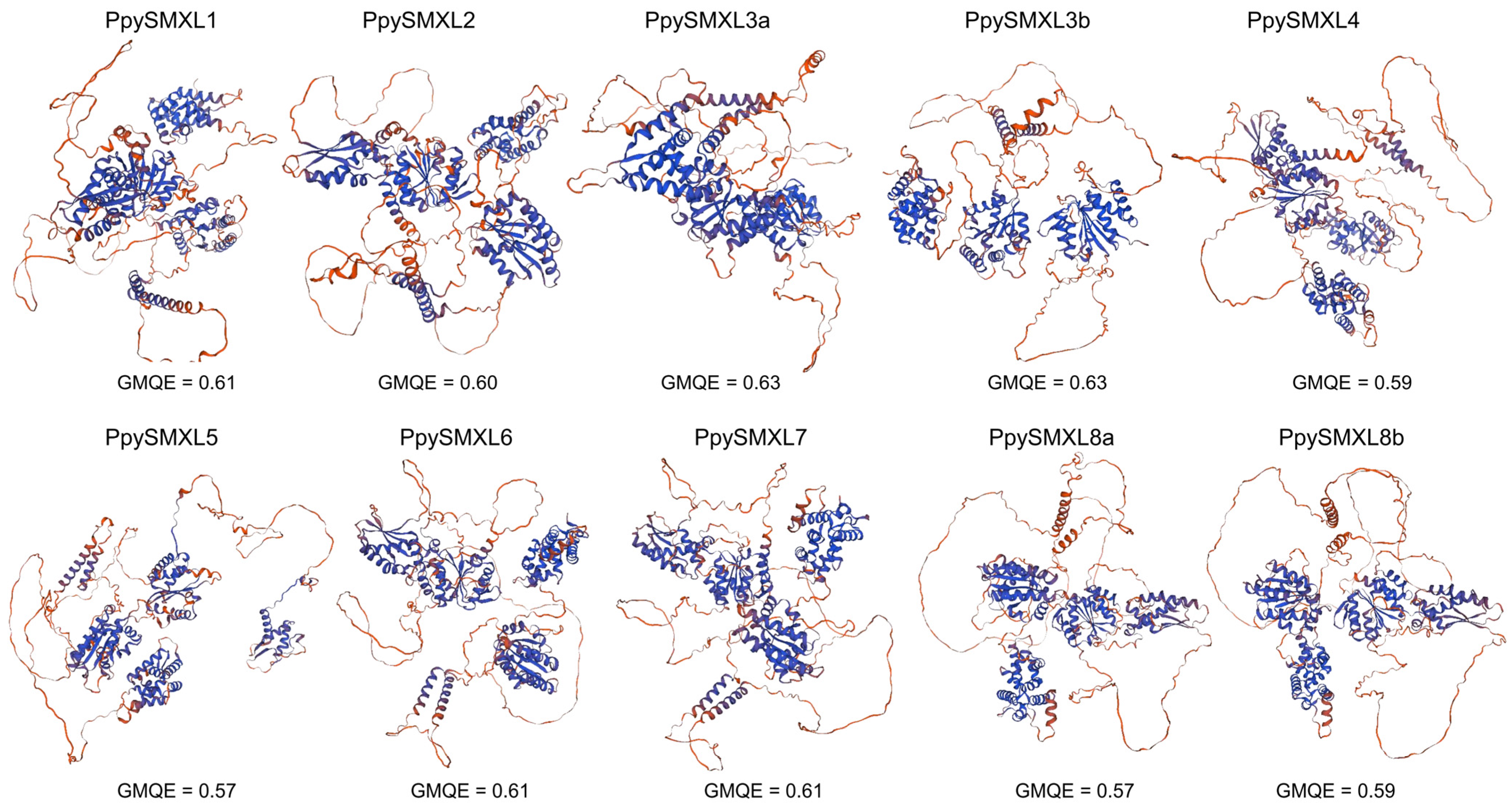{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification and Classification of SMXL Genes
2.2. Gene Structures and Conserved Motif Composition of SMXL Genes
2.3. Synteny Analysis and Evolution of SMXL Genes
2.4. Secondary Structure Analysis and Prediction of Three-Dimensional Structure of SMXL Proteins
2.5. Expression Patterns of SMXL Genes in Regulating Pear Branching
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. SMXL Gene Identification and Classification
4.3. Phylogenetic Tree, Gene, and Three-Dimensional Protein Structures, Conserved Domains, and Motifs
4.4. Genome Synteny and Gene Duplication Analysis
4.5. Plant Materials and Treatment
4.6. Expression Analysis of Pear SMXL Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, B.; Smith, S.M.; Li, J. Genetic Regulation of Shoot Architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef]
- Xu, D.; Qi, X.; Li, J.; Han, X.; Wang, J.; Jiang, Y.; Tian, Y.; Wang, Y. PzTAC and PzLAZY from a narrow-crown poplar contribute to regulation of branch angles. Plant Physiol. Biochem. 2017, 118, 571–578. [Google Scholar] [CrossRef]
- Bevacqua, D.; Melià, P.; Cividini, M.; Mattioli, F.; Lescourret, F.; Génard, M.; Casagrandi, R. A parsimonious mechanistic model of reproductive and vegetative growth in fruit trees predicts consequences of fruit thinning and branch pruning. Tree Physiol. 2021, 41, 1794–1807. [Google Scholar] [CrossRef] [PubMed]
- Umehara, M.; Hanada, A.; Yoshida, S.; Akiyama, K.; Arite, T.; Takeda-Kamiya, N.; Magome, H.; Kamiya, Y.; Shirasu, K.; Yoneyama, K.; et al. Inhibition of shoot branching by new terpenoid plant hormones. Nature 2008, 455, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pages, V.; Dun, E.A.; Pillot, J.P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.C.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Yoneyama, K.; Yoneyama, K. The strigolactone story. Annu. Rev. Phytopathol. 2010, 48, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Ruyter-Spira, C.; Al-Babili, S.; van der Krol, S.; Bouwmeester, H. The biology of strigolactones. Trends Plant Sci. 2013, 18, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Delaux, P.M.; Xie, X.; Timme, R.E.; Puech-Pages, V.; Dunand, C.; Lecompte, E.; Delwiche, C.F.; Yoneyama, K.; Bécard, G.; Séjalon-Delmas, N. Origin of strigolactones in the green lineage. N. Phytol. 2012, 195, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.E.; Whichard, L.P.; Turner, B.; Wall, M.E.; Egley, G.H. Germination of witchweed (Striga lutea Lour.): Isolation and properties of a potent stimulant. Science 1966, 154, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.T.; Gutjahr, C.; Bennett, T.; Nelson, D.C. Strigolactone signaling and evolution. Annu. Rev. Plant Biol. 2017, 68, 291–322. [Google Scholar] [CrossRef]
- Akiyama, K.; Matsuzaki, K.; Hayashi, H. Plant sesquiterpenes induce hyphal branching in arbuscular mycorrhizal fungi. Nature 2005, 435, 824–827. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Xiong, G.; Liu, H.; Chen, F.; Wang, L.; Meng, X.; Liu, G.; Yu, H.; Yuan, Y.; et al. DWARF 53 acts as a repressor of strigolactone signalling in rice. Nature 2013, 504, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lin, Q.; Zhu, L.; Ren, Y.; Zhou, K.; Shabek, N.; Wu, F.; Mao, H.; Dong, W.; Gan, L.; et al. D14-SCF (D3)-dependent degradation of D53 regulates strigolactone signalling. Nature 2013, 504, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Zhou, X.E.; Wu, Z.S.; Yi, W.; Xu, Y.; Li, S.L.; Xu, T.H.; Liu, Y.; Chen, R.Z.; Kovach, A.; et al. Crystal structures of two phytohormone signal-transducing α/β hydrolases: Karrikin-signaling KAI2 and strigolactone-signaling DWARF14. Cell Res. 2013, 23, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Ming, Z.; Yan, L.; Li, S.; Wang, F.; Ma, S.; Yu, C.; Yang, M.; Chen, L.; Chen, L.; et al. DWARF14 is a non-canonical hormone receptor for strigolactone. Nature 2016, 536, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Seto, Y.; Yasui, R.; Kameoka, H.; Tamiru, M.; Cao, M.M.; Terauchi, R.; Sakurada, A.; Hirano, R.; Kisugi, T.; Hanada, A.; et al. Strigolactone perception and deactivation by a hydrolase receptor DWARF14. Nat. Commun. 2019, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Moturu, T.R.; Thula, S.; Singh, R.K.; Nodzynski, T.; Varekova, R.S.; Friml, J.; Simon, S. Molecular evolution and diversification of the SMXL gene family. J. Exp. Bot. 2018, 69, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Temmerman, A.; Guillory, A.; Bonhomme, S.; Goormachtig, S.; Struk, S. Masks Start to Drop: Suppressor of MAX2 1-Like Proteins Reveal Their Many Faces. Front. Plant Sci. 2022, 13, 887232. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ward, S.; Li, P.; Bennett, T.; Leyser, O. SMAX1-LIKE7 signals from the nucleus to regulate shoot development in Arabidopsis via partially EAR motif-independent mechanisms. Plant Cell 2016, 28, 1581–1601. [Google Scholar] [CrossRef]
- Walker, C.H.; Siu-Ting, K.; Taylor, A.; O’Connell, M.J.; Bennett, T. Strigolactone synthesis is ancestral in land plants, but canonical strigolactone signalling is a fowering plant innovation. BMC Biol. 2019, 17, 70. [Google Scholar] [CrossRef]
- Kerr, S.C.; Patil, S.B.; de Saint Germain, A.; Pillot, J.P.; Saffar, J.; Ligerot, Y.; Aubert, G.; Citerne, S.; Bellec, Y.; Dun, E.A.; et al. Integration of the SMXL/D53 strigolactone signalling repressors in the model of shoot branching regulation in Pisum sativum. Plant J. 2021, 107, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Lian, Y.; Kang, J.; Bian, Z.; Xuan, L.; Gao, Z.; Wang, X.; Deng, J.; Wang, C. The SUPPRESSOR of MAX2 1 (SMAX1)-Like SMXL6, SMXL7 and SMXL8 Act as Negative Regulators in Response to Drought Stress in Arabidopsis. Plant Cell Physiol. 2020, 61, 1477–1492. [Google Scholar] [CrossRef] [PubMed]
- Soundappan, I.; Bennett, T.; Morffy, N.; Liang, Y.; Stanga, J.P.; Abbas, A.; Leyser, O.; Nelson, D.C. SMAX1-LIKE/D53 Family Members Enable Distinct MAX2-Dependent Responses to Strigolactones and Karrikins in Arabidopsis. Plant Cell 2015, 27, 3143–3159. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; Yu, H.; Guo, H.; Lin, T.; Kou, L.; Wang, A.; Shao, N.; Ma, H.; Xiong, G.; et al. Transcriptional regulation of strigolactone signalling in Arabidopsis. Nature 2020, 583, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; An, J.P.; You, C.X.; Wang, X.F.; Hao, Y.J. Genome-wide analysis and identifcation of the SMXL gene family in apple (Malus x domestica). Tree Genet. Genomes 2018, 14, 1–3. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed]
- Wang, L.; Wang, B.; Jiang, L.; Liu, X.; Li, X.; Lu, Z.; Meng, X.; Wang, Y.; Smith, S.M.; Li, J. Strigolactone signaling in Arabidopsis regulates shoot development by targeting D53-Like SMXL repressor proteins for ubiquitination and degradation. Plant Cell 2015, 27, 3128–3142. [Google Scholar] [CrossRef] [PubMed]
- Bennett, T.; Liang, Y.; Seale, M.; Ward, S.; Müller, D.; Leyser, O. Strigolactone regulates shoot development through a core signalling pathway. Biol. Open 2016, 5, 1806–1820. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Hou, B.H.; Lee, W.C.; Lu, S.H.; Yang, C.J.; Vaucheret, H.; Chen, H.M. DCL2- and RDR6-dependent transitive silencing of SMXL4 and SMXL5 in Arabidopsis dcl4 mutants causes defective phloem transport and carbohydrate over-accumulation. Plant J. 2017, 90, 1064–1078. [Google Scholar] [CrossRef]
- Zheng, X.; Yang, X.; Chen, Z.; Xie, W.; Yue, X.; Zhu, H.; Chen, S.; Sun, X. Arabidopsis SMAX1 overaccumulation suppresses rosette shoot branching and promotes leaf and petiole elongation. Biochem. Biophys. Res. Commun. 2021, 553, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, Z.; Tian, L.; Zhang, Y.; Qi, D.; Huo, H.; Xu, J.; Li, Z.; Liao, R.; Shi, M.; et al. De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol. J. 2020, 18, 581–595. [Google Scholar] [CrossRef]
- Shirasawa, K.; Itai, A.; Isobe, S. Chromosome-scale genome assembly of Japanese pear (Pyrus pyrifolia) variety ‘Nijisseiki’. DNA Res. 2021, 28, dsab001. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.W.; Donoghue, P.C.J. Whole-genome duplication and plant macroevolution. Trends Plant Sci. 2018, 23, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidizationdiploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- The International Peach Genome Initiative. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Ireland, H.S.; Wu, C.; Deng, C.H.; Hilario, E.; Saei, A.; Erasmuson, S.; Crowhurst, R.N.; David, K.M.; Schaffer, R.J.; Chagné, D. The Gillenia trifoliata genome reveals dynamics correlated with growth and reproduction in Rosaceae. Hortic. Res. 2021, 8, 233. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Jing, Y.; Lin, S.; Yue, Z.; Yang, X.; Xu, J.; Wu, J.; Zhang, Z.; Xia, R.; Zhu, J.; et al. Polyploidy underlies co-option and diversification of biosynthetic triterpene pathways in the apple tribe. Proc. Natl. Acad. Sci. USA. 2021, 118, e2101767118. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, F.; Zhang, X.; Li, Z.; Zhao, Y.; Lohaus, R.; Chang, X.; Dong, W.; Ho, S.Y.W.; Liu, X.; et al. The water lily genome and the early evolution of flowering plants. Nature 2020, 577, 79–84. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef]
- Iorizzo, M.; Ellison, S.; Senalik, D.; Zeng, P.; Satapoomin, P.; Huang, J.; Bowman, M.; Iovene, M.; Sanseverino, W.; Cavagnaro, P.; et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asteroid genome evolution. Nat. Genet. 2016, 48, 657–666. [Google Scholar] [CrossRef]
- Vision, T.J.; Brown, D.G.; Tanksley, S.D. The origins of genomic duplications in Arabidopsis. Science 2000, 290, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Simillion, C.; Vandepoele, K.; Van Montagu, M.C.; Zabeau, M.; Van de Peer, Y. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2002, 99, 13627–13632. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Hokamp, K.; Wolfe, K.H. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003, 13, 137–144. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009; Volume 3, pp. 361–362. [Google Scholar]
- Stanga, J.P.; Smith, S.M.; Briggs, W.R.; Nelson, D.C. SUPPRESSOR OF MORE AXILLARY GROWTH2 1 controls seed germination and seedling development in Arabidopsis. Plant Physiol. 2013, 163, 318–330. [Google Scholar] [CrossRef]
- Stanga, J.P.; Morffy, N.; Nelson, D.C. Functional redundancy in the control of seedling growth by the karrikin signaling pathway. Planta 2016, 243, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Bythell-Douglas, R.; Rothfels, C.J.; Stevenson, D.W.D.; Graham, S.W.; Wong, G.K.S.; Nelson, D.C.; Bennett, T. Evolution of strigolactone receptors by gradual neo-functionalization of KAI2 paralogues. BMC Biol. 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Bunsick, M.; Toh, S.; Wong, C.; Xu, Z.; Ly, G.; McErlean, C.S.P.; Pescetto, G.; Nemrish, K.E.; Sung, P.; Li, J.D.; et al. SMAX1- dependent seed germination bypasses GA signalling in Arabidopsis and Striga. Nat. Plants 2020, 6, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, L.; Gao, Y.; Guo, Y.; Zheng, N.; Xu, X.; Xu, M.; Wang, W.; Liu, C.; Liu, W.; et al. Genome-Wide Identification of SMXL Gene Family in Soybean and Expression Analysis of GmSMXLs under Shade Stress. Plants 2022, 11, 2410. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, T. Expansion and stress responses of the AP2/EREBP superfamily in cotton. BMC Genom. 2017, 18, 118. [Google Scholar] [CrossRef]
- Muiño, J.M.; de Bruijn, S.; Pajoro, A.; Geuten, K.; Vingron, M.; Angenent, G.C.; Kaufmann, K. Evolution of DNA-binding sites of a floral master regulatory transcription factor. Mol. Biol. Evol. 2016, 33, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Martinez, J.A.; Poza-Carrión, C.; Cubas, P. Arabidopsis BRANCHED1 acts as an integrator of branching signals within axillary buds. Plant Cell 2007, 19, 458–472. [Google Scholar] [CrossRef]
- Martin-Trillo, M.; Cubas, P. TCP genes: A family snapshot ten years later. Trends Plant Sci. 2010, 15, 31–39. [Google Scholar] [CrossRef]
- Wang, X.B.; Wang, Q.P.; Yan, L.X.; Hao, Y.H.; Lian, X.D.; Zhang, H.P.; Zheng, X.B.; Cheng, J.; Wang, W.; Zhang, L.L.; et al. PpTCP18 is upregulated by lncRNA5 and controls branch number in peach (Prunus persica) through positive feedback regulation of strigolactone biosynthesis. Hortic. Res. 2022, 10, uhac224. [Google Scholar] [CrossRef]
- Gao, R.; Gruber, M.Y.; Amyot, L.; Hannoufa, A. SPL13 regulates shoot branching and flowering time in medicago sativa. Plant Mol. Biol. 2018, 96, 119–133. [Google Scholar] [CrossRef]
- Cheng, Y.; Liang, C.; Qiu, Z.; Zhou, S.; Liu, J.; Yang, Y.; Wang, R.; Yin, J.; Ma, C.; Cui, Z.; et al. Jasmonic acid negatively regulates branch growth in pear. Front. Plant Sci. 2023, 14, 1105521. [Google Scholar] [CrossRef]
- Sun, M.; Wang, D.; Liu, C.; Liu, Y.; Niu, M.; Wang, J.; Li, J. Genome-wide identification and analysis of the SUPPRESSOR of MAX2 1-LIKE gene family and its interaction with DWARF14 in poplar. BMC Plant Biol. 2023, 23, 105. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Angiosperm Phylogeny Group. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, NY, USA, 1965; pp. 97–166. [Google Scholar]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Adamcsek, B.; Palla, G.; Farkas, I.J.; Derenyi, I.; Vicsek, T. CFinder: Locating cliques and overlapping modules in biological networks. Bioinformatics 2006, 22, 1021–1023. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Jiang, X.; Liu, S.; Liu, Y.; Li, H.; Wang, Z.; Kan, J.; Yang, Q.; Li, X. Comprehensive Evolutionary Analysis of the SMXL Gene Family in Rosaceae: Further Insights into Its Origin, Expansion, Diversification, and Role in Regulating Pear Branching. Int. J. Mol. Sci. 2024, 25, 2971. https://doi.org/10.3390/ijms25052971
Liu C, Jiang X, Liu S, Liu Y, Li H, Wang Z, Kan J, Yang Q, Li X. Comprehensive Evolutionary Analysis of the SMXL Gene Family in Rosaceae: Further Insights into Its Origin, Expansion, Diversification, and Role in Regulating Pear Branching. International Journal of Molecular Sciences. 2024; 25(5):2971. https://doi.org/10.3390/ijms25052971
Chicago/Turabian StyleLiu, Chunxiao, Xianda Jiang, Susha Liu, Yilong Liu, Hui Li, Zhonghua Wang, Jialiang Kan, Qingsong Yang, and Xiaogang Li. 2024. "Comprehensive Evolutionary Analysis of the SMXL Gene Family in Rosaceae: Further Insights into Its Origin, Expansion, Diversification, and Role in Regulating Pear Branching" International Journal of Molecular Sciences 25, no. 5: 2971. https://doi.org/10.3390/ijms25052971
APA StyleLiu, C., Jiang, X., Liu, S., Liu, Y., Li, H., Wang, Z., Kan, J., Yang, Q., & Li, X. (2024). Comprehensive Evolutionary Analysis of the SMXL Gene Family in Rosaceae: Further Insights into Its Origin, Expansion, Diversification, and Role in Regulating Pear Branching. International Journal of Molecular Sciences, 25(5), 2971. https://doi.org/10.3390/ijms25052971