
Identification of an Intravenous Injectable NK1 Receptor Antagonist for Use in Traumatic Brain Injury


Abstract
:1. Introduction
2. Results
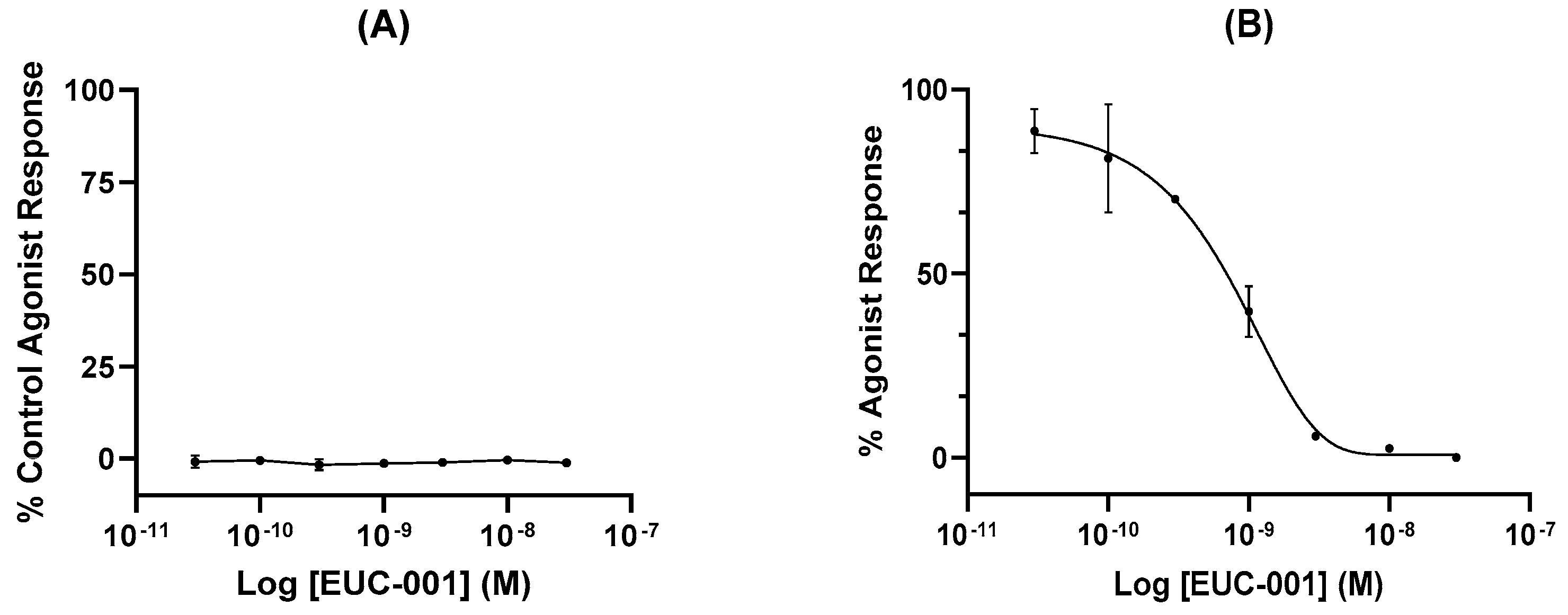
2.1. In Vitro Characterization of EUC-001
2.2. In Vivo Characterization of EUC-001
2.3. Effect of EUC-001 in a Rodent Model of TBI
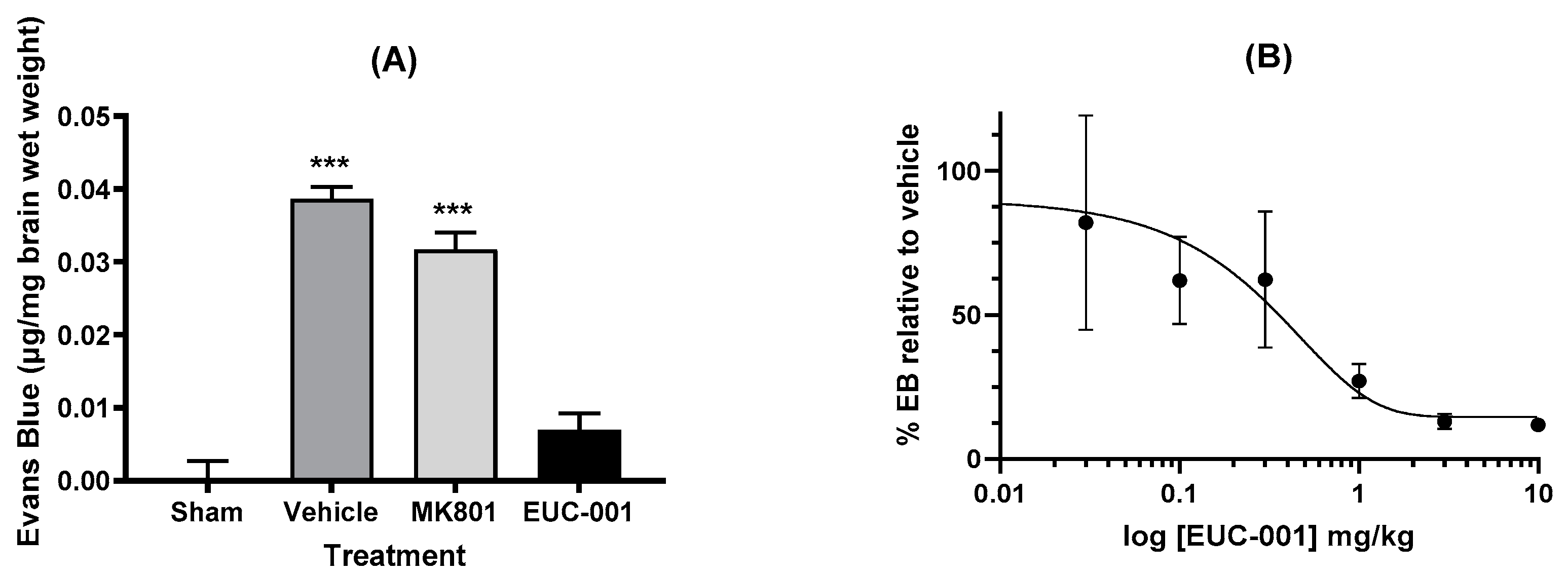
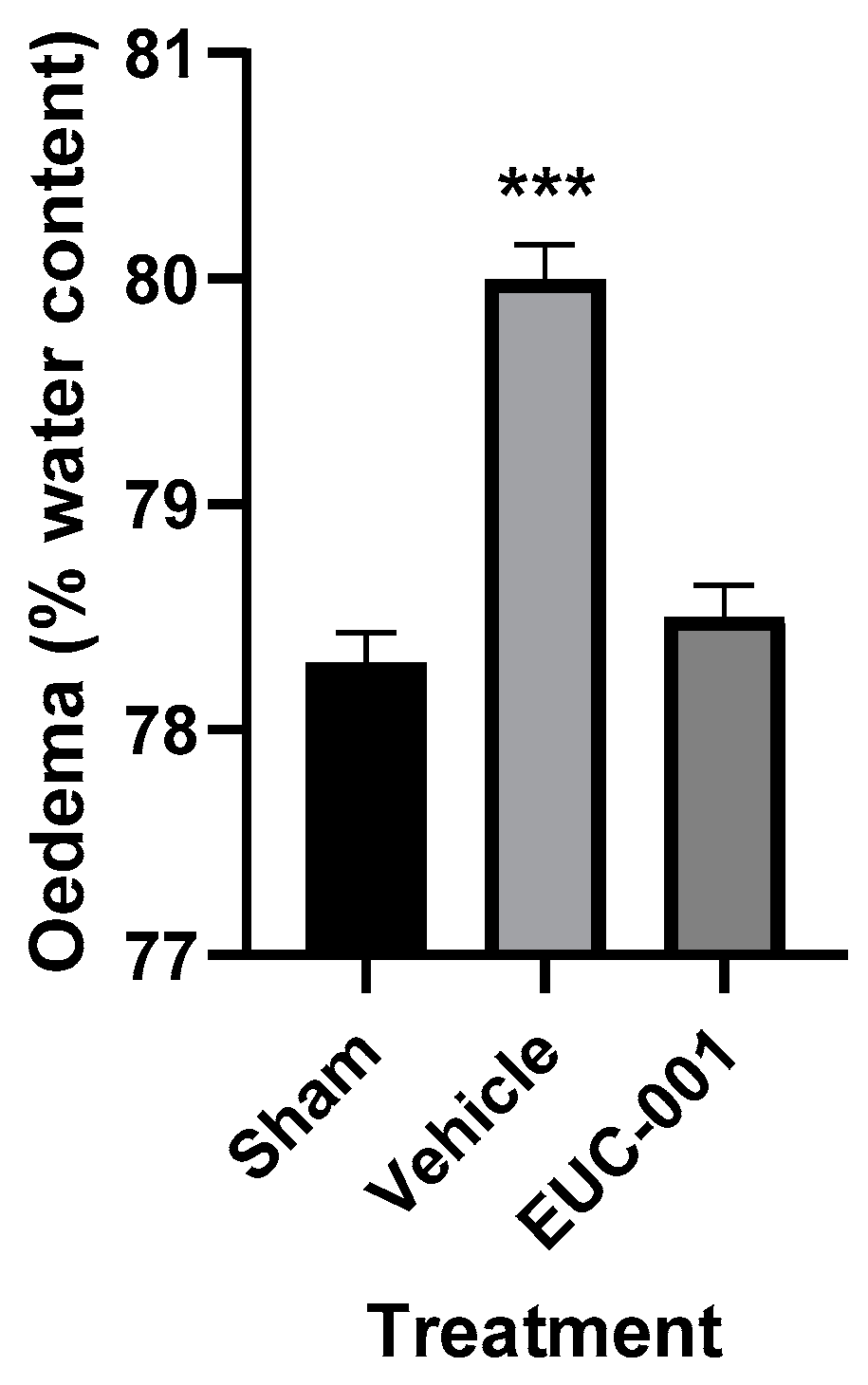
2.3.1. Effect of EUC-001 on BBB Permeability following TBI
2.3.2. Effect of Treatment with EUC-001 on Functional Outcomes following TBI
3. Discussion
4. Materials and Methods
4.1. In Vitro Radioligand Binding Assays
4.2. Pharmacological Selectivity Studies
4.3. In Vitro Functional Assay
4.4. Gerbil Foot-Tapping Respon
4.5. Induction of Traumatic Brain Injury
4.6. Drug Treatment
4.7. Dose Response for BBB Permeability
4.8. Oedema Determination
4.9. Functional Outcomes
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babikian, T.; Asarnow, R. Neurocognitive outcomes and recovery after pediatric TBI: Meta-analytic review of the literature. Neuropsychology 2009, 23, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Peeters, W.; Feigin, V.; Theadom, A.; Holkovic, L.; et al. Epidemiology of Traumatic Brain Injury in Europe: A Living Systematic Review. J. Neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Frati, A.; Santoro, A.; Frati, P.; Fineschi, V.; Pesce, A. Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances. Int. J. Mol. Sci. 2021, 22, 10865. [Google Scholar] [CrossRef] [PubMed]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 1993, 34, 216–222. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [PubMed]
- Morganti-Kossmann, M.C.; Satgunaseelan, L.; Bye, N.; Kossmann, T. Modulation of immune response by head injury. Injury 2007, 38, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg. Focus 2007, 22, E1. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef]
- Sahuquillo, J.; Dennis, J.A. Decompressive craniectomy for the treatment of high intracranial pressure in closed traumatic brain injury. Cochrane Database Syst. Rev. 2019, 12, Cd003983. [Google Scholar] [CrossRef]
- Visser, K.; Koggel, M.; Blaauw, J.; van der Horn, H.J.; Jacobs, B.; van der Naalt, J. Blood-based biomarkers of inflammation in mild traumatic brain injury: A systematic review. Neurosci. Biobehav. Rev. 2022, 132, 154–168. [Google Scholar] [CrossRef]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145 Pt B, 230–246. [Google Scholar] [CrossRef]
- Chiu, I.M.; von Hehn, C.A.; Woolf, C.J. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 2012, 15, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Jacques, L.; Couture, R.; Drapeau, G.; Regoli, D. Capillary permeability induced by intravenous neurokinins. Receptor characterization and mechanism of action. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1989, 340, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Valente, J.; Brain, S.D. A historical perspective on the role of sensory nerves in neurogenic inflammation. Semin. Immunopathol. 2018, 40, 229–236. [Google Scholar] [CrossRef]
- Nimmo, A.J.; Cernak, I.; Heath, D.L.; Hu, X.; Bennett, C.J.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47. [Google Scholar] [CrossRef]
- Hu, D.E.; Easton, A.S.; Fraser, P.A. TRPV1 activation results in disruption of the blood-brain barrier in the rat. Br. J. Pharmacol. 2005, 146, 576–584. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The Role of Neurogenic Inflammation in Blood-Brain Barrier Disruption and Development of Cerebral Oedema Following Acute Central Nervous System (CNS) Injury. Int. J. Mol. Sci. 2017, 18, 1788. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.L.; Mendes, J.; Leitão, R.; Silva, A.P.; Pinto, A.M. A multi-staged neuropeptide response to traumatic brain injury. Eur. J. Trauma Emerg. Surg. 2022, 48, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Almeida, T.; Hernández, M.; Ramos, L.; Argueso, M.; Cáceres, J.J.; Solé-Violán, J.; Jiménez, A. Serum substance P levels are associated with severity and mortality in patients with severe traumatic brain injury. Crit. Care 2015, 19, 192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ye, H.; Lu, W. Serum Substance P Concentration in Children with Traumatic Brain Injury: A First Report. World Neurosurg. 2021, 147, e200–e205. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chapman, K.; Clark, L.D.; Shao, Z.; Borek, D.; Xu, Q.; Wang, J.; Rosenbaum, D.M. Crystal structure of the human NK(1) tachykinin receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 13264–13269. [Google Scholar] [CrossRef] [PubMed]
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Gabrielian, L.; Helps, S.C.; Thornton, E.; Turner, R.J.; Leonard, A.V.; Vink, R. Substance P antagonists as a novel intervention for brain edema and raised intracranial pressure. Acta Neurochir. Suppl. 2013, 118, 201–204. [Google Scholar] [PubMed]
- Vink, R.; Donkin, J.J.; Cruz, M.I.; Nimmo, A.J.; Cernak, I. A substance P antagonist increases brain intracellular free magnesium concentration after diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 538S–540S. [Google Scholar] [CrossRef]
- Cernak, I.; Vink, R.; Zapple, D.N.; Cruz, M.I.; Ahmed, F.; Chang, T.; Fricke, S.T.; Faden, A.I. The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol. Dis. 2004, 17, 29–43. [Google Scholar] [CrossRef]
- Li, Q.; Wu, X.; Yang, Y.; Zhang, Y.; He, F.; Xu, X.; Zhang, Z.; Tao, L.; Luo, C. Tachykinin NK1 receptor antagonist L-733,060 and substance P deletion exert neuroprotection through inhibiting oxidative stress and cell death after traumatic brain injury in mice. Int. J. Biochem. Cell. Biol. 2019, 107, 154–165. [Google Scholar] [CrossRef]
- Donkin, J.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. A substance P antagonist reduces axonal injury and improves neurologic outcome when administered up to 12 hours after traumatic brain injury. J. Neurotrauma 2011, 28, 217–224. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.M.; Merchant, K.J.; Brookfield, F.; Kelleher, F.; Stevenson, G.; Owens, A.P.; Swain, C.J.; Casiceri, M.A.; Sadowski, S.; Ber, E.; et al. Identification of L-tryptophan derivatives with potent and selective antagonist activity at the NK1 receptor. J. Med. Chem. 1994, 37, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Matalinska, J.; Lipinski, P.F.J. Correcting a widespread error: Neuroprotectant N-acetyl-L-tryptophan does not bind to the neurokinin-1 receptor. Mol. Cell. Neurosci. 2022, 120, 103728. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Maity, S.; Mudgal, J.; Nampoothiri, M. In silico screening of neurokinin receptor antagonists as a therapeutic strategy for neuroinflammation in Alzheimer’s disease. Mol. Divers. 2022, 26, 443–466. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, A.J.; Vink, R. Recent patents in CNS drug discovery: The management of inflammation in the central nervous system. Recent Pat. CNS Drug Discov. 2009, 4, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Sorby-Adams, A.J.; Leonard, A.V.; Hoving, J.W.; Yassi, N.; Vink, R.; Wells, A.J.; Turner, R.J. NK1-r Antagonist Treatment Comparable to Decompressive Craniectomy in Reducing Intracranial Pressure Following Stroke. Front. Neurosci. 2019, 13, 681. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Nimmo, A.; Sleight, A.; Vankan, P.; Vink, R. Method of Treatment and/or Prevention of Brain, Spinal or Nerve Injury. U.S. Patent 20030083345A1, 1 May 2003. [Google Scholar]
- Almeida, T.A.; Rojo, J.; Nieto, P.M.; Pinto, F.M.; Hernandez, M.; Martin, J.D.; Candenas, M.L. Tachykinins and tachykinin receptors: Structure and activity relationships. Curr. Med. Chem. 2004, 11, 2045–2081. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Macdonald, D.M.; Hronowski, X.; Costello, C.E.; Leeman, S.E.; Boyd, N.D. Further definition of the substance P (SP)/neurokinin-1 receptor complex. MET-174 is the site of photoinsertion p-benzoylphenylalanine4 SP. J. Biol. Chem. 2001, 276, 10589–10593. [Google Scholar] [CrossRef]
- Swain, C.J.; Seward, E.M.; Cascieri, M.A.; Fong, T.M.; Herbert, R.; MacIntyre, D.E.; Merchant, K.J.; Owen, S.N.; Owens, A.P.; Sabin, V.; et al. Identification of a series of 3-(benzyloxy)-1-azabicyclo[2.2.2]octane human NK1 antagonists. J. Med. Chem. 1995, 38, 4793–4805. [Google Scholar] [CrossRef]
- Patel, L.; Lindley, C. Aprepitant--A novel NK1-receptor antagonist. Expert Opin. Pharmacother. 2003, 4, 2279–2296. [Google Scholar] [CrossRef]
- Aapro, M.; Carides, A.; Rapoport, B.L.; Schmoll, H.J.; Zhang, L.; Warr, D. Aprepitant and fosaprepitant: A 10-year review of efficacy and safety. Oncologist 2015, 20, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.S.; Cutler, N.; Feighner, J.; Shrivastava, R.; Carman, J.; Sramek, J.J.; Reines, S.A.; Liu, G.; Snavely, D.; Wyatt-Knowles, E.; et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 1998, 281, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A.; Deeks, E.D. Rolapitant: A Review in Chemotherapy-Induced Nausea and Vomiting. Drugs 2017, 77, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, S.; Feng, J.; Dechaphunkul, A.; Chang, J.; Wang, D.; Chessari, S.; Lanzarotti, C.; Jordan, K.; Aapro, M. A randomized phase III study evaluating the efficacy of single-dose NEPA, a fixed antiemetic combination of netupitant and palonosetron, versus an aprepitant regimen for prevention of chemotherapy-induced nausea and vomiting (CINV) in patients receiving highly emetogenic chemotherapy (HEC). Ann. Oncol. 2018, 29, 452–458. [Google Scholar] [PubMed]
- Colon-Gonzalez, F.; Kraft, W.K. Pharmacokinetic evaluation of fosaprepitant dimeglumine. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem. Biodivers. 2009, 6, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, S.; Chua, D.; Maru, A.; Dinis, J.; DeVandry, S.; Boice, J.A.; Hardwick, J.S.; Beckford, E.; Taylor, A.; Carides, A.; et al. Single-dose fosaprepitant for the prevention of chemotherapy-induced nausea and vomiting associated with cisplatin therapy: Randomized, double-blind study protocol—EASE. J. Clin. Oncol. 2011, 29, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Navari, R.M. Fosaprepitant (MK-0517): A neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Expert Opin. Investig. Drugs 2007, 16, 1977–1985. [Google Scholar] [CrossRef]
- Shirley, M. Netupitant/Palonosetron: A Review in Chemotherapy-Induced Nausea and Vomiting. Drugs 2021, 81, 1331–1342. [Google Scholar] [CrossRef]
- Cass, A.S.; Odinet, J.S.; Valgus, J.M.; Crona, D.J. Infusion reactions following administration of intravenous rolapitant at an academic medical center. J. Oncol. Pharm. Pract. 2019, 25, 1776–1783. [Google Scholar] [CrossRef]
- In brief: Severe hypersensitivity reactions with rolapitant IV emulsion (Varubi). Med. Lett. Drugs Ther. 2018, 60, 24.
- Peltz, E.D.; D’Alessandro, A.; Moore, E.E.; Chin, T.; Silliman, C.C.; Sauaia, A.; Hansen, K.C.; Banerjee, A. Pathologic metabolism: An exploratory study of the plasma metabolome of critical injury. J. Trauma Acute Care Surg. 2015, 78, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.C.; Couch, Y.; Losey, P.; Evans, M.C. The systemic response to brain injury and disease. Brain Behav. Immun. 2012, 26, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Hughes, P.M.; Iredale, J.P.; Wilcockson, D.C.; Waters, S.; Docagne, F.; Perry, V.H.; Anthony, D.C. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. 2003, 17, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Bös, M.; Stadler, H.; Schnider, P.; Hunkeler, W.; Godel, T.; Galley, G.; Ballard, T.M.; Higgins, G.A.; Poli, S.M.; et al. Design and synthesis of a novel, achiral class of highly potent and selective, orally active neurokinin-1 receptor antagonists. Bioorganic Med. Chem. Lett. 2006, 16, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Saria, A. The tachykinin NK1 receptor in the brain: Pharmacology and putative functions. Eur. J. Pharmacol. 1999, 375, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Varty, G.B.; Cohen-Williams, M.E.; Morgan, C.A.; Pylak, U.; Duffy, R.A.; Lachowicz, J.E.; Carey, G.J.; Coffin, V.L. The gerbil elevated plus-maze II: Anxiolytic-like effects of selective neurokinin NK1 receptor antagonists. Neuropsychopharmacology 2002, 27, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ballard, T.M.; Sanger, S.; Higgins, G.A. Inhibition of shock-induced foot tapping behaviour in the gerbil by a tachykinin NK1 receptor antagonist. Eur. J. Pharmacol. 2001, 412, 255–264. [Google Scholar] [CrossRef]
- Foda, M.A.; Marmarou, A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J. Neurosurg. 1994, 80, 301–313. [Google Scholar] [CrossRef]
- Cigel, A.; Sayin, O.; Gurgen, S.G.; Sonmez, A. Long term neuroprotective effects of acute single dose MK-801treatment against traumatic brain injury in immature rats. Neuropeptides 2021, 88, 102161. [Google Scholar] [CrossRef]
- Pitts, M.W. Barnes Maze Procedure for Spatial Learning and Memory in Mice. Bio-Protocol 2018, 8, e2744. [Google Scholar] [CrossRef] [PubMed]
- Hamm, R.J.; Pike, B.R.; O’Dell, D.M.; Lyeth, B.G.; Jenkins, L.W. The rotarod test: An evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma 1994, 11, 187–196. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Heath, D.L.; Cernak, I.; Nimmo, A.J.; Vink, R. Effects of daily versus weekly testing and pre-training on the assessment of neurologic impairment following diffuse traumatic brain injury in rats. J. Neurotrauma 2003, 20, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Vink, R.; Nimmo, A.J. Multifunctional drugs for head injury. Neurotherapeutics 2009, 6, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Postolache, T.T.; Wadhawan, A.; Can, A.; Lowry, C.A.; Woodbury, M.; Makkar, H.; Hoisington, A.J.; Scott, A.J.; Potocki, E.; Benros, M.E.; et al. Inflammation in Traumatic Brain Injury. J. Alzheimers Dis. 2020, 74, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol. Sci. 2017, 38, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.M.; Saria, A.; Brodin, E.; Rosell, S.; Folkers, K. A substance P antagonist inhibits vagally induced increase in vascular permeability and bronchial smooth muscle contraction in the guinea pig. Proc. Natl. Acad. Sci. USA 1983, 80, 1120–1124. [Google Scholar] [CrossRef]
- Saria, A.; Lundberg, J.M.; Skofitsch, G.; Lembeck, F. Vascular protein linkage in various tissue induced by substance P, capsaicin, bradykinin, serotonin, histamine and by antigen challenge. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1983, 324, 212–218. [Google Scholar] [CrossRef]
- Douglas, S.D.; Leeman, S.E. Neurokinin-1 receptor: Functional significance in the immune system in reference to selected infections and inflammation. Ann. N. Y. Acad. Sci. 2011, 1217, 83–95. [Google Scholar] [CrossRef]
- Baluk, P.; Hirata, A.; Thurston, G.; Fujiwara, T.; Neal, C.R.; Michel, C.C.; McDonald, D.M. Endothelial gaps: Time course of formation and closure in inflamed venules of rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997, 272, L155–L170. [Google Scholar] [CrossRef]
- Leroux, A.; Paiva Dos Santos, B.; Leng, J.; Oliveira, H.; Amedee, J. Sensory neurons from dorsal root ganglia regulate endothelial cell function in extracellular matrix remodelling. Cell Commun. Signal. 2020, 18, 162. [Google Scholar] [CrossRef] [PubMed]
- Suvas, S. Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J. Immunol. 2017, 199, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Hill, R. NK1 (substance P) receptor antagonists--why are they not analgesic in humans? Trends Pharmacol. Sci. 2000, 21, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Rupniak, N.M.J.; Kramer, M.S. NK1 receptor antagonists for depression: Why a validated concept was abandoned. J. Affect. Disord. 2017, 223, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhao, G.; Yang, S.; Chen, J. Efficacy, Tolerability and Pharmacokinetic Impact of Aprepitant in Sarcoma Patients Receiving Ifosfamide and Doxorubicin Chemotherapy: A Randomized Controlled Trial. Adv. Ther. 2019, 36, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Perkovich, B.; Atayee, R.S.; Kim, J.S.; Rubenzik, T. Use of Fosaprepitant for Management of Suspected Antimicrobial-Associated Nausea: A Case Report. J. Pain Palliat. Care Pharmacother. 2023, 38, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Uehara, S.; Kondo, N.; Takahashi, Y.; Yamamoto, S.; Kameda, A.; Kawagoe, S.; Inoue, N.; Yamada, M.; Yoshimura, N.; et al. Peptide-to-Small Molecule: A Pharmacophore-Guided Small Molecule Lead Generation Strategy from High-Affinity Macrocyclic Peptides. J. Med. Chem. 2022, 65, 10655–10673. [Google Scholar] [CrossRef]
- Snider, R.M.; Constantine, J.W.; Lowe, J.A., 3rd; Longo, K.P.; Lebel, W.S.; Woody, H.A.; Drozda, S.E.; Desai, M.C.; Vinick, F.J.; Spencer, R.W.; et al. A potent nonpeptide antagonist of the substance P (NK1) receptor. Science 1991, 251, 435–437. [Google Scholar] [CrossRef]
- McLean, S.; Ganong, A.; Seymour, P.A.; Snider, R.M.; Desai, M.C.; Rosen, T.; Bryce, D.K.; Longo, K.P.; Reynolds, L.S.; Robinson, G.; et al. Pharmacology of CP-99,994; a nonpeptide antagonist of the tachykinin neurokinin-1 receptor. J. Pharmacol. Exp. Ther. 1993, 267, 472–479. [Google Scholar]
- Hargreaves, R.; Ferreira, J.C.; Hughes, D.; Brands, J.; Hale, J.; Mattson, B.; Mills, S. Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Ann. N. Y. Acad. Sci. 2011, 1222, 40–48. [Google Scholar] [CrossRef]
- Saria, A.; Lundberg, J.M. Evans blue fluorescence: Quantitative and morphological evaluation of vascular permeability in animal tissues. J. Neurosci. Methods 1983, 8, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.K.; Brain, S.D. Activity of tachykinin NK1 and bradykinin B2 receptor antagonists, and an opioid ligand at different stimulation parameters in neurogenic inflammation in the rat. Neurosci. Lett. 1998, 257, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Avila, M.; Gonzalez, J.; El-Bacha, R.S.; Baez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, A.; Sayin, O.; Gurgen, S.G.; Calisir, M. Neuroprotective effects of MK-801 against traumatic brain injury in immature rats. Neurosci. Lett. 2015, 597, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Biegon, A. Novel approach to the role of NMDA receptors in traumatic brain injury. CNS Neurol. Disord. Drug Targets 2014, 13, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Dixon, C.E.; Kraus, M.F.; Kline, A.E.; Ma, X.; Yan, H.Q.; Griffith, R.G.; Wolfson, B.M.; Marion, D.W. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restor Neurol. Neurosci. 1999, 14, 285–294. [Google Scholar]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 2005, 25, 3312–3322. [Google Scholar] [CrossRef]
- Capponi, A.M.; Lew, P.D.; Jornot, L.; Vallotton, M.B. Correlation between cytosolic free Ca2+ and aldosterone production in bovine adrenal glomerulosa cells. Evidence for a difference in the mode of action of angiotensin II and potassium. J. Biol. Chem. 1984, 259, 8863–8869. [Google Scholar] [CrossRef]
- Eistetter, H.R.; Mills, A.; Brewster, R.; Alouani, S.; Rambosson, C.; Kawashima, E. Functional characterization of neurokinin-1 receptors on human U373MG astrocytoma cells. Glia 1992, 6, 89–95. [Google Scholar] [CrossRef]
- Marmarou, A.; Foda, M.A.; van den Brink, W.; Campbell, J.; Kita, H.; Demetriadou, K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994, 80, 291–300. [Google Scholar] [CrossRef]
- Goldim, M.P.d.S.; Della Giustina, A.; Petronilho, F. Using Evans Blue Dye to Determine Blood-Brain Barrier Integrity in Rodents. Curr. Protoc. Immunol. 2019, 126, e83. [Google Scholar] [CrossRef]
- Kaya, M.; Ahishali, B. Assessment of permeability in barrier type of endothelium in brain using tracers: Evans blue, sodium fluorescein, and horseradish peroxidase. Methods Mol. Biol. 2011, 763, 369–382. [Google Scholar]
- O’Connor, C.A.; Cernak, I.; Vink, R. The temporal profile of edema formation differs between male and female rats following diffuse traumatic brain injury. Acta Neurochir. Suppl. 2006, 96, 121–124. [Google Scholar]
- Habgood, M.D.; Bye, N.; Dziegielewska, K.M.; Ek, C.J.; Lane, M.A.; Potter, A.; Morganti-Kossmann, C.; Saunders, N.R. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Subtype | IC50 (M) |
|---|---|---|
| Serotonin | 5-HT1B | 6.1 × 10−6 |
| 5-HT2A | 1.5 × 10−5 | |
| 5-HT5A | 4 × 10−6 | |
| Adenosine | A2A | 3 × 10−5 |
| A3 | Not calculable | |
| Dopamine | D1 | 1.6 × 10−5 |
| D2S | Not calculable | |
| Histamine | H2 | 1.6 × 10−5 |
| Melanocortin | MC4 | 1.3 × 10−5 |
| Muscarinic cholinergic | M1 | 3.3 × 10−6 |
| M2 | 3 × 10−5 | |
| M3 | 3.3 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vink, R.; Nimmo, A. Identification of an Intravenous Injectable NK1 Receptor Antagonist for Use in Traumatic Brain Injury. Int. J. Mol. Sci. 2024, 25, 3535. https://doi.org/10.3390/ijms25063535
Vink R, Nimmo A. Identification of an Intravenous Injectable NK1 Receptor Antagonist for Use in Traumatic Brain Injury. International Journal of Molecular Sciences. 2024; 25(6):3535. https://doi.org/10.3390/ijms25063535
Chicago/Turabian StyleVink, Robert, and Alan Nimmo. 2024. "Identification of an Intravenous Injectable NK1 Receptor Antagonist for Use in Traumatic Brain Injury" International Journal of Molecular Sciences 25, no. 6: 3535. https://doi.org/10.3390/ijms25063535
APA StyleVink, R., & Nimmo, A. (2024). Identification of an Intravenous Injectable NK1 Receptor Antagonist for Use in Traumatic Brain Injury. International Journal of Molecular Sciences, 25(6), 3535. https://doi.org/10.3390/ijms25063535