
Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms?

Abstract
:1. Introduction
2. L-ARG
3. Nitric Oxide Synthase
4. L-ARG Levels in MELAS

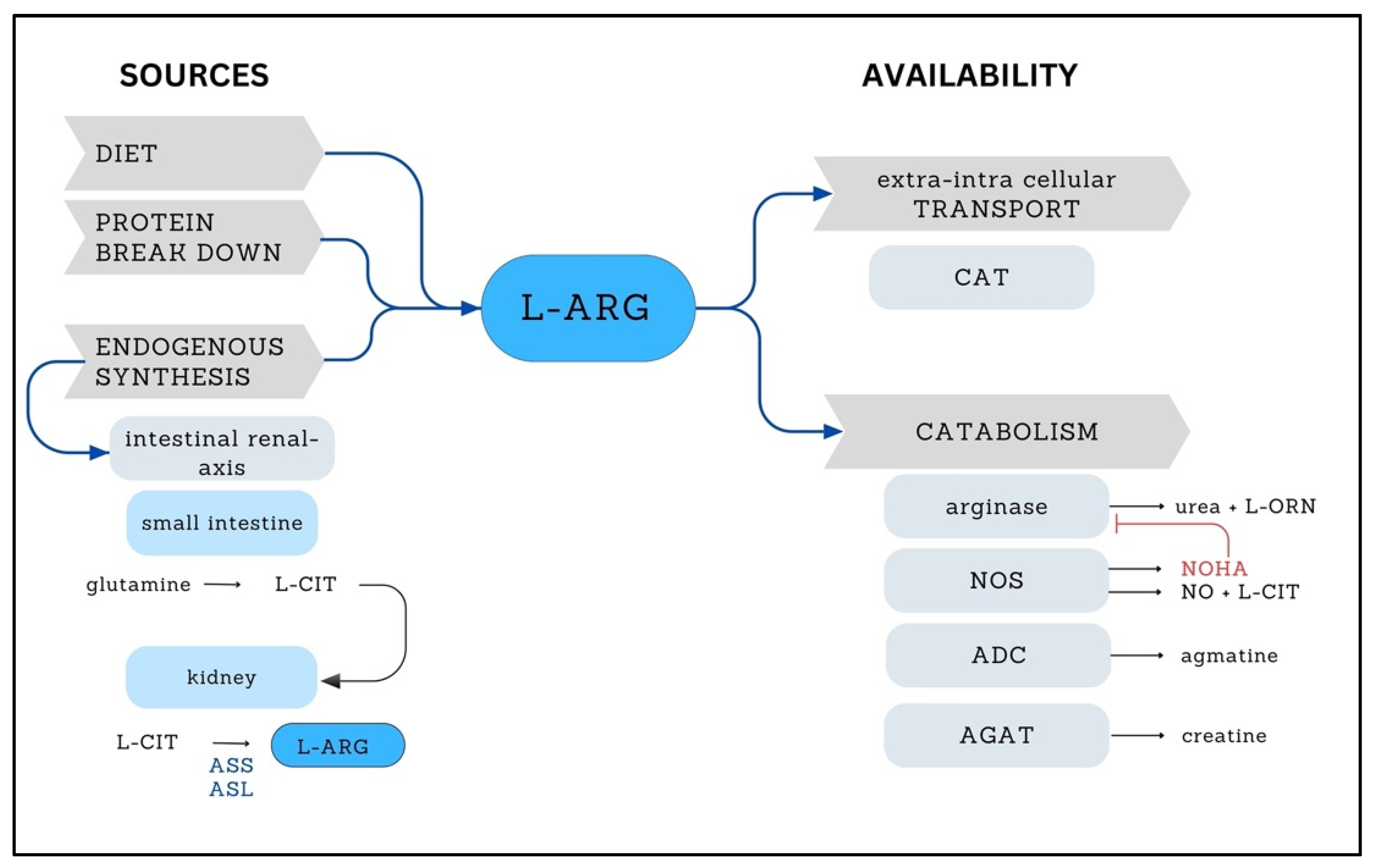{kind=link}
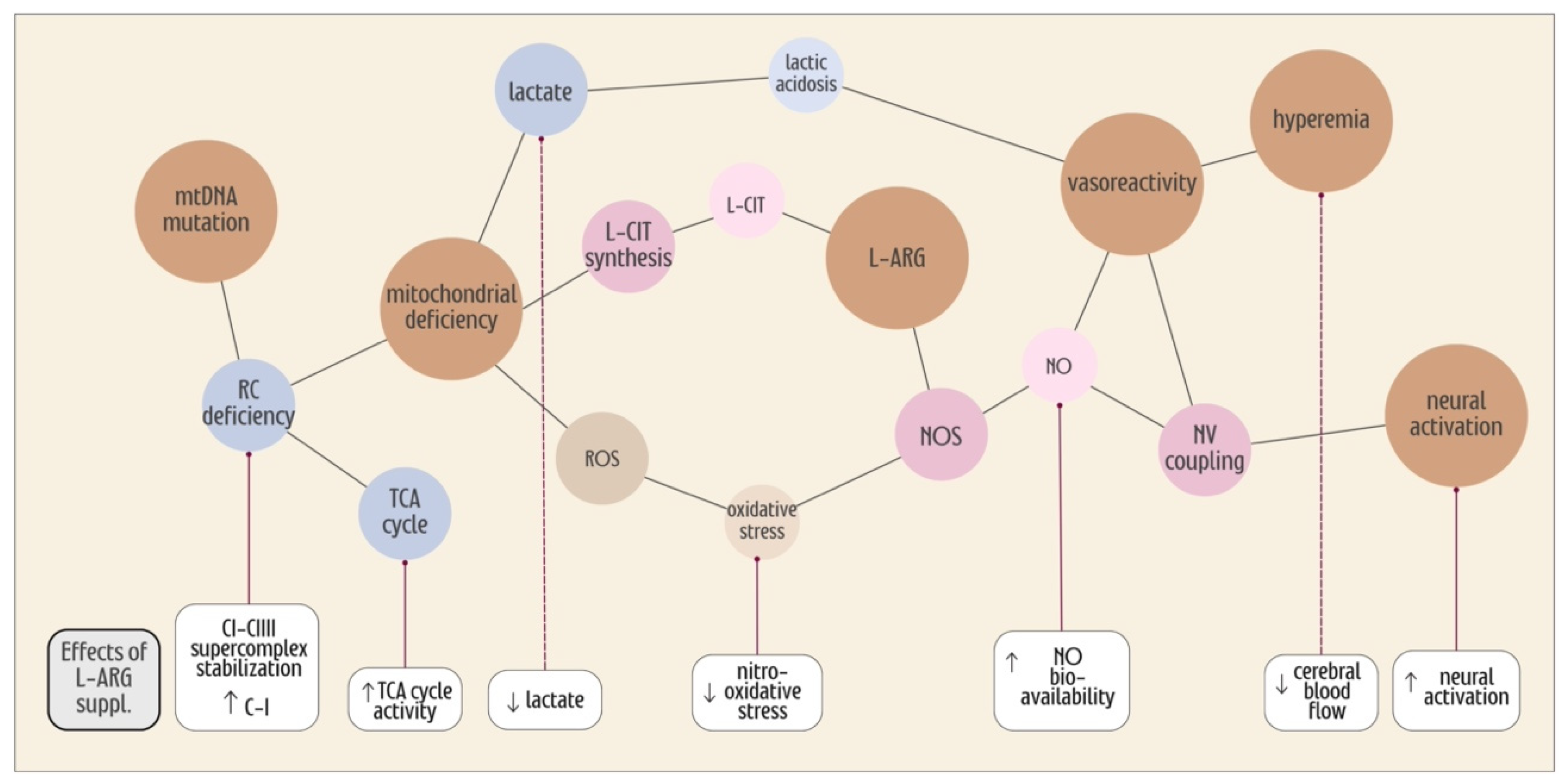{kind=link}
| Ref. | Arginine (µmol/L) | ||||||
|---|---|---|---|---|---|---|---|
| Acute Phase | Interictal Phase | Controls | |||||
| Pat. (n) | Ctrl (n) | Basal | L-ARG | Basal | L-ARG | Basal | |
| [10] | 1 | 0 | 60 ± 10 | 80 ± 10 | - | - | 100 ± 40 |
| [14] | 24 | 72 | 47 ± 13 a,b | 92.4±15.9 | 84 ± 26 | - | 108 ± 28 |
| [35] | 10 | 10 | 81 ± 14 | 85 ± 19 | |||
| [11] | 15 | 20 | 36.4 ± 11.6 a,b,c | 101.7 ± 19.8 | - | - | 102.6 ± 15.7 |
| [40] | 24 | 72 | 46.6 ± 12.7 a,b,c | - | 83.6 ± 25.8 | - | 108.1 ± 27.6 |
| [41] | 1 | 46 | 27–108 * | ||||
| [33] | 10 | 10 | 57.1 ± 3.2 a,c | 143.8 ± 9.9 | 77.8 ± 4.4 | ||
| [39] | 3 | 4 | - | - | 53 ± 11 a | 76 to 230 | 94 ± 18 |
| [34] | 5 | 5 | - | - | 59 ± 5 a,c | 184 ± 14 | 151 ± 18 |
| [42] | 1 | - | - | - | 62.4 ± 2.7 | - | - |
| [36] | 10 $; 13 # | - | 89.81 ± 76.25 | 143.44 ± 122.78 | 65.74 ± 16.74 | 162.36 ± 45.23 | - |
| [37] | 3 | 4 | - | - | 53 ± 11 a | 76 to 230 | 94 ± 18 |
5. Effects of L-ARG Supplementation on NO• Synthesis
6. Effects of L-ARG Supplementation on Vascular Regulation and Hemodynamics
7. L-ARG Supplementation, the TCA Cycle and Mitochondrial Efficiency
8. Oxidative Stress in MELAS
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Sproule, D.M.; Kaufmann, P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann. N. Y. Acad. Sci. 2008, 1142, 133–158. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A. Genetic bases of mitochondrial respiratory chain disorders. Diabetes Metab. 2010, 36, 97–107. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T. MELAS and L-arginine therapy: Pathophysiology of stroke-like episodes. Ann. N. Y. Acad. Sci. 2010, 1201, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Horai, S.; Matsuoka, T.; Koga, Y.; Nihei, K.; Kobayashi, M.; Nonaka, I. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): A correlative study of the clinical features and mitochondrial DNA mutation. Neurology 1992, 42 Pt 1, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Desquiret-Dumas, V.; Gueguen, N.; Barth, M.; Chevrollier, A.; Hancock, S.; Wallace, D.C.; Amati-Bonneau, P.; Henrion, D.; Bonneau, D.; Reynier, P.; et al. Metabolically induced heteroplasmy shifting and l-arginine treatment reduce the energetic defect in a neuronal-like model of MELAS. Biochim. Biophys. Acta 2012, 1822, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Ikawa, M.; Arakawa, K.; Kudo, T.; Kimura, H.; Fujibayashi, Y.; Okazawa, H. In vivo functional brain imaging and a therapeutic trial of L-arginine in MELAS patients. Biochim. Biophys. Acta 2012, 1820, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Sakai, F.; Suzuki, N.; Hata, T.; Tsukahara, S.; Fukuda, M.; Takiyama, Y. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology 2002, 59, 816–824. [Google Scholar] [CrossRef]
- Iizuka, T.; Sakai, F. Pathophysiology of stroke-like episodes in MELAS: Neuron–astrocyte uncoupling in neuronal hyperexcitability. Future Neurol. 2010, 5, 61–83. [Google Scholar] [CrossRef]
- Koga, Y.; Ishibashi, M.; Ueki, I.; Yatsuga, S.; Fukiyama, R.; Akita, Y.; Matsuishi, T. Effects of L-arginine on the acute phase of strokes in three patients with MELAS. Neurology 2002, 58, 827–828. [Google Scholar] [CrossRef]
- Koga, Y.; Akita, Y.; Junko, N.; Yatsuga, S.; Povalko, N.; Fukiyama, R.; Ishii, M.; Matsuishi, T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 2006, 66, 1766–1769. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Craigen, W.J.; Scaglia, F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012, 107, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.P., Jr.; Lambertucci, R.H. Effects of N-acetylcysteine and L-arginine in the antioxidant system of C2C12 cells. J. Sports Med. Phys. Fitness 2015, 55, 691–699. [Google Scholar] [PubMed]
- Koga, Y.; Akita, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Tanabe, Y.; Fujimoto, S.; Matsuishi, T. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology 2005, 64, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Falk, M.J. 8-year retrospective analysis of intravenous arginine therapy for acute metabolic strokes in pediatric mitochondrial disease. Mol. Genet. Metab. 2018, 123, 301–308. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Ng, Y.S.; Errington, L.; Blain, A.P.; McFarland, R.; Gorman, G.S. l-Arginine in Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes: A Systematic Review. Neurology 2022, 98, e2318–e2328. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380–1397. [Google Scholar] [CrossRef]
- Barcelos, I.; Shadiack, E.; Ganetzky, R.D.; Falk, M.J. Mitochondrial medicine therapies: Rationale, evidence, and dosing guidelines. Curr. Opin. Pediatr. 2020, 32, 707–718. [Google Scholar] [CrossRef]
- Morris, C.R.; Hamilton-Reeves, J.; Martindale, R.G.; Sarav, M.; Ochoa Gautier, J.B. Acquired Amino Acid Deficiencies: A Focus on Arginine and Glutamine. Nutr. Clin. Pract. 2017, 32 (Suppl. S1), 30S–47S. [Google Scholar] [CrossRef]
- Almannai, M.; El-Hattab, A.W. Nitric Oxide Deficiency in Mitochondrial Disorders: The Utility of Arginine and Citrulline. Front. Mol. Neurosci. 2021, 14, 682780. [Google Scholar] [CrossRef]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef]
- Elms, S.; Chen, F.; Wang, Y.; Qian, J.; Askari, B.; Yu, Y.; Pandey, D.; Iddings, J.; Caldwell, R.B.; Fulton, D.J. Insights into the arginine paradox: Evidence against the importance of subcellular location of arginase and eNOS. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H651–H666. [Google Scholar] [CrossRef] [PubMed]
- Popolo, A.; Adesso, S.; Pinto, A.; Autore, G.; Marzocco, S. L-Arginine and its metabolites in kidney and cardiovascular disease. Amino Acids 2014, 46, 2271–2286. [Google Scholar] [CrossRef]
- Coman, D.; Yaplito-Lee, J.; Boneh, A. New indications and controversies in arginine therapy. Clin. Nutr. 2008, 27, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Jahoor, F. Assessment of Nitric Oxide Production in Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes Syndrome with the Use of a Stable Isotope Tracer Infusion Technique. J. Nutr. 2017, 147, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Lipton, S.A. Nitric oxide signaling regulates mitochondrial number and function. Cell Death Differ. 2003, 10, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Tengan, C.H.; Rodrigues, G.S.; Godinho, R.O. Nitric oxide in skeletal muscle: Role on mitochondrial biogenesis and function. Int. J. Mol. Sci. 2012, 13, 17160–17184. [Google Scholar] [CrossRef] [PubMed]
- Bombicino, S.S.; Iglesias, D.E.; Zaobornyj, T.; Boveris, A.; Valdez, L.B. Mitochondrial nitric oxide production supported by reverse electron transfer. Arch. Biochem. Biophys. 2016, 607, 8–19. [Google Scholar] [CrossRef]
- Zaobornyj, T.; Valdez, L.B. Heart mitochondrial nitric oxide synthase: A strategic enzyme in the regulation of cellular bioenergetics. Vitam. Horm. 2014, 96, 29–58. [Google Scholar]
- Brown, G.C.; Borutaite, V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc. Res. 2007, 75, 283–290. [Google Scholar] [CrossRef]
- Rodan, L.H.; Wells, G.D.; Banks, L.; Thompson, S.; Schneiderman, J.E.; Tein, I. L-Arginine Affects Aerobic Capacity and Muscle Metabolism in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-Like Episodes) Syndrome. PLoS ONE 2015, 10, e0127066. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.J.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol. Genet. Metab. 2012, 105, 607–614. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Almannai, M.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol. Genet. Metab. 2016, 117, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Naini, A.; Kaufmann, P.; Shanske, S.; Engelstad, K.; De Vivo, D.C.; Schon, E.A. Hypocitrullinemia in patients with MELAS: An insight into the “MELAS paradox”. J. Neurol. Sci. 2005, 229–230, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Povalko, N.; Inoue, E.; Nakamura, H.; Ishii, A.; Suzuki, Y.; Yoneda, M.; Kanda, F.; Kubota, M.; Okada, H.; et al. Therapeutic regimen of L-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J. Neurol. 2018, 265, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Rodan, L.H.; Poublanc, J.; Fisher, J.A.; Sobczyk, O.; Mikulis, D.J.; Tein, I. L-arginine effects on cerebrovascular reactivity, perfusion and neurovascular coupling in MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) syndrome. PLoS ONE 2020, 15, e0238224. [Google Scholar] [CrossRef]
- Evans, R.W.; Fernstrom, J.D.; Thompson, J.; Morris, S.M., Jr.; Kuller, L.H. Biochemical responses of healthy subjects during dietary supplementation with L-arginine. J. Nutr. Biochem. 2004, 15, 534–539. [Google Scholar] [CrossRef]
- Rodan, L.H.; Poublanc, J.; Fisher, J.A.; Sobczyk, O.; Wong, T.; Hlasny, E.; Mikulis, D.; Tein, I. Cerebral hyperperfusion and decreased cerebrovascular reactivity correlate with neurologic disease severity in MELAS. Mitochondrion 2015, 22, 66–74. [Google Scholar] [CrossRef]
- Koga, Y.; Akita, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Katayama, K.; Matsuishi, T. MELAS and L-arginine therapy. Mitochondrion 2007, 7, 133–139. [Google Scholar] [CrossRef]
- Lekoubou, A.; Kouame-Assouan, A.E.; Cho, T.H.; Luaute, J.; Nighoghossian, N.; Derex, L. Effect of long-term oral treatment with L-arginine and idebenone on the prevention of stroke-like episodes in an adult MELAS patient. Rev. Neurol. 2011, 167, 852–855. [Google Scholar] [CrossRef]
- Hanff, E.; Kayacelebi, A.A.; Herrmann, C.; Obermann, M.; Das, A.M.; Tsikas, D. Unaltered l-arginine/NO pathway in a MELAS patient: Is mitochondrial NO synthase involved in the MELAS syndrome? Int. J. Cardiol. 2016, 223, 479–481. [Google Scholar] [CrossRef]
- Li, H.; Uittenbogaard, M.; Navarro, R.; Ahmed, M.; Gropman, A.; Chiaramello, A.; Hao, L. Integrated proteomic and metabolomic analyses of the mitochondrial neurodegenerative disease MELAS. Mol. Omics 2022, 18, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Sodja, C.; McRae, K.; Li, Y.; Rippstein, P.; Wei, Y.H.; Lach, B.; Lee, F.; Bucurescu, S.; Harper, M.E.; et al. Effects of nitric oxide donors on cybrids harbouring the mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) A3243G mitochondrial DNA mutation. Biochem. J. 2005, 391 Pt 2, 191–202. [Google Scholar] [CrossRef]
- Gamba, J.; Gamba, L.T.; Rodrigues, G.S.; Kiyomoto, B.H.; Moraes, C.T.; Tengan, C.H. Nitric oxide synthesis is increased in cybrid cells with m.3243A>G mutation. Int. J. Mol. Sci. 2012, 14, 394–410. [Google Scholar] [CrossRef]
- Barros, C.D.S.; Livramento, J.B.; Mouro, M.G.; Higa, E.M.S.; Moraes, C.T.; Tengan, C.H. L-Arginine Reduces Nitro-Oxidative Stress in Cultured Cells with Mitochondrial Deficiency. Nutrients 2021, 13, 534. [Google Scholar] [CrossRef]
- Tengan, C.H.; Kiyomoto, B.H.; Godinho, R.O.; Gamba, J.; Neves, A.C.; Schmidt, B.; Oliveira, A.S.; Gabbai, A.A. The role of nitric oxide in muscle fibers with oxidative phosphorylation defects. Biochem. Biophys. Res. Commun. 2007, 359, 771–777. [Google Scholar] [CrossRef]
- Rodrigues, G.S.; Godinho, R.O.; Kiyomoto, B.H.; Gamba, J.; Oliveira, A.S.; Schmidt, B.; Tengan, C.H. Integrated analysis of the involvement of nitric oxide synthesis in mitochondrial proliferation, mitochondrial deficiency and apoptosis in skeletal muscle fibres. Sci. Rep. 2016, 6, 20780. [Google Scholar] [CrossRef] [PubMed]
- Gropen, T.I.; Prohovnik, I.; Tatemichi, T.K.; Hirano, M. Cerebral hyperemia in MELAS. Stroke 1994, 25, 1873–1876. [Google Scholar] [CrossRef] [PubMed]
- Narai, H.; Manabe, Y.; Deguchi, K.; Iwatsuki, K.; Sakai, K.; Abe, K. Serial MRI findings in patient with chronic cryptococcus meningo-encephalitis. Neurol. Res. 2001, 23, 810–812. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Nakamoto, Y.; Yoneda, M. PET Imaging for Oxidative Stress in Neurodegenerative Disorders Associated with Mitochondrial Dysfunction. Antioxidants 2020, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Nariai, T.; Ohno, K.; Ohta, Y.; Hirakawa, K.; Ishii, K.; Senda, M. Discordance between cerebral oxygen and glucose metabolism, and hemodynamics in a mitochondrial encephalomyopathy, lactic acidosis, and strokelike episode patient. J. Neuroimaging 2001, 11, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hu, B.; Sun, C.; Geng, D.; Lin, J.; Li, Y. Metabolic abnormality in acute stroke-like lesion and its relationship with focal cerebral blood flow in patients with MELAS: Evidence from proton MR spectroscopy and arterial spin labeling. Mitochondrion 2021, 59, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, X.; Sun, C.; Hu, B.; Yang, L.; Liu, Y.; Geng, D.; Lin, J.; Li, Y. Altered Neurovascular Coupling in Patients with Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS): A Combined Resting-State fMRI and Arterial Spin Labeling Study. J. Magn. Reson. Imaging 2023. [Google Scholar] [CrossRef] [PubMed]
- Stackhouse, T.L.; Mishra, A. Neurovascular Coupling in Development and Disease: Focus on Astrocytes. Front. Cell Dev. Biol. 2021, 9, 702832. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.A.; Chan, F.H.; Zheng, M.M.; Krassioukov, A.V.; Ainslie, P.N. Neurovascular coupling in humans: Physiology, methodological advances and clinical implications. J. Cereb. Blood Flow. Metab. 2016, 36, 647–664. [Google Scholar] [CrossRef]
- Alkayed, N.J.; Cipolla, M.J. The Ever-Evolving Concept of the Neurovascular Unit. Stroke 2023, 54, 2178–2180. [Google Scholar] [CrossRef]
- Tiedt, S.; Buchan, A.M.; Dichgans, M.; Lizasoain, I.; Moro, M.A.; Lo, E.H. The neurovascular unit and systemic biology in stroke—Implications for translation and treatment. Nat. Rev. Neurol. 2022, 18, 597–612. [Google Scholar] [CrossRef]
- Lia, A.; Di Spiezio, A.; Speggiorin, M.; Zonta, M. Two decades of astrocytes in neurovascular coupling. Front. Netw. Physiol. 2023, 3, 1162757. [Google Scholar] [CrossRef]
- Ouellette, J.; Lacoste, B. From Neurodevelopmental to Neurodegenerative Disorders: The Vascular Continuum. Front. Aging Neurosci. 2021, 13, 749026. [Google Scholar] [CrossRef]
- Katwal, S.B.; Gore, J.C.; Gatenby, J.C.; Rogers, B.P. Measuring relative timings of brain activities using fMRI. Neuroimage 2013, 66, 436–448. [Google Scholar] [CrossRef]
- Petcharunpaisan, S.; Ramalho, J.; Castillo, M. Arterial spin labeling in neuroimaging. World J. Radiol. 2010, 2, 384–398. [Google Scholar] [CrossRef]
- Lourenco, C.F.; Laranjinha, J. Nitric Oxide Pathways in Neurovascular Coupling Under Normal and Stress Conditions in the Brain: Strategies to Rescue Aberrant Coupling and Improve Cerebral Blood Flow. Front. Physiol. 2021, 12, 729201. [Google Scholar] [CrossRef]
- Arakawa, K.; Kudo, T.; Ikawa, M.; Morikawa, N.; Kawai, Y.; Sahashi, K.; Lee, J.D.; Kuriyama, M.; Miyamori, I.; Okazawa, H.; et al. Abnormal myocardial energy-production state in mitochondrial cardiomyopathy and acute response to L-arginine infusion. C-11 acetate kinetics revealed by positron emission tomography. Circ. J. 2010, 74, 2702–2711. [Google Scholar] [CrossRef]
- Morris, S.M., Jr. Arginine metabolism: Boundaries of our knowledge. J. Nutr. 2007, 137 (Suppl. S2), 1602S–1609S. [Google Scholar] [CrossRef]
- Xu, W.; Ghosh, S.; Comhair, S.A.; Asosingh, K.; Janocha, A.J.; Mavrakis, D.A.; Bennett, C.D.; Gruca, L.L.; Graham, B.B.; Queisser, K.A.; et al. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J. Clin. Investig. 2016, 126, 2465–2481. [Google Scholar] [CrossRef]
- Distelmaier, F.; Klopstock, T. Neuroimaging in mitochondrial disease. Handb. Clin. Neurol. 2023, 194, 173–185. [Google Scholar] [PubMed]
- Koga, Y.; Povalko, N.; Nishioka, J.; Katayama, K.; Yatsuga, S.; Matsuishi, T. Molecular pathology of MELAS and L-arginine effects. Biochim. Biophys. Acta 2012, 1820, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Sakakihara, Y.; Mori, M.; Yamagata, T.; Momoi-Yoshida, M. Beneficial effect of L-arginine for stroke-like episode in MELAS. Brain Dev. 2004, 26, 481–483; discussion 480. [Google Scholar] [CrossRef] [PubMed]
- Hovsepian, D.A.; Galati, A.; Chong, R.A.; Mazumder, R.; DeGiorgio, C.M.; Mishra, S.; Yim, C. MELAS: Monitoring treatment with magnetic resonance spectroscopy. Acta Neurol. Scand. 2019, 139, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Cortopassi, G. Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 2015, 88 Pt A, 10–17. [Google Scholar] [CrossRef]
- Katayama, Y.; Maeda, K.; Iizuka, T.; Hayashi, M.; Hashizume, Y.; Sanada, M.; Kawai, H.; Kashiwagi, A. Accumulation of oxidative stress around the stroke-like lesions of MELAS patients. Mitochondrion 2009, 9, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.Y.; Lee, H.C.; Wei, Y.H. Enhanced oxidative damage in human cells harboring A3243G mutation of mitochondrial DNA: Implication of oxidative stress in the pathogenesis of mitochondrial diabetes. Diabetes Res. Clin. Pract. 2001, 54 (Suppl. S2), S45–S56. [Google Scholar] [CrossRef]
- Santacatterina, F.; Torresano, L.; Nunez-Salgado, A.; Esparza-Molto, P.B.; Olive, M.; Gallardo, E.; Garcia-Arumi, E.; Blazquez, A.; Gonzalez-Quintana, A.; Martin, M.A.; et al. Different mitochondrial genetic defects exhibit the same protein signature of metabolism in skeletal muscle of PEO and MELAS patients: A role for oxidative stress. Free Radic. Biol. Med. 2018, 126, 235–248. [Google Scholar] [CrossRef]
- Vattemi, G.; Mechref, Y.; Marini, M.; Tonin, P.; Minuz, P.; Grigoli, L.; Guglielmi, V.; Klouckova, I.; Chiamulera, C.; Meneguzzi, A.; et al. Increased protein nitration in mitochondrial diseases: Evidence for vessel wall involvement. Mol. Cell Proteom. 2011, 10, M110.002964. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Arakawa, K.; Kudo, T.; Kimura, H.; Fujibayashi, Y.; Kuriyama, M.; Yoneda, M. PET imaging of redox and energy states in stroke-like episodes of MELAS. Mitochondrion 2009, 9, 144–148. [Google Scholar] [CrossRef]
- Jabecka, A.; Ast, J.; Bogdaski, P.; Drozdowski, M.; Pawlak-Lemaska, K.; Cielewicz, A.R.; Pupek-Musialik, D. Oral L-arginine supplementation in patients with mild arterial hypertension and its effect on plasma level of asymmetric dimethylarginine, L-citruline, L-arginine and antioxidant status. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1665–1674. [Google Scholar] [PubMed]
- Fazelian, S.; Hoseini, M.; Namazi, N.; Heshmati, J.; Sepidar Kish, M.; Mirfatahi, M.; Some Olia, A.S. Effects of L- Arginine Supplementation on Antioxidant Status and Body Composition in Obese Patients with Pre-diabetes: A Randomized Controlled Clinical Trial. Adv. Pharm. Bull. 2014, 4 (Suppl. S1), 449–454. [Google Scholar] [PubMed]
- Jablecka, A.; Bogdanski, P.; Balcer, N.; Cieslewicz, A.; Skoluda, A.; Musialik, K. The effect of oral L-arginine supplementation on fasting glucose, HbA1c, nitric oxide and total antioxidant status in diabetic patients with atherosclerotic peripheral arterial disease of lower extremities. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 342–350. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, C.D.S.; Coutinho, A.; Tengan, C.H. Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms? Int. J. Mol. Sci. 2024, 25, 3629. https://doi.org/10.3390/ijms25073629
Barros CDS, Coutinho A, Tengan CH. Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms? International Journal of Molecular Sciences. 2024; 25(7):3629. https://doi.org/10.3390/ijms25073629
Chicago/Turabian StyleBarros, Camila D. S., Aryane Coutinho, and Celia H. Tengan. 2024. "Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms?" International Journal of Molecular Sciences 25, no. 7: 3629. https://doi.org/10.3390/ijms25073629
APA StyleBarros, C. D. S., Coutinho, A., & Tengan, C. H. (2024). Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms? International Journal of Molecular Sciences, 25(7), 3629. https://doi.org/10.3390/ijms25073629